
In Silico Analysis of the Effects of Omicron Spike Amino Acid Changes on the Interactions with Human Proteins

 , , and
, , and 
Abstract
:1. Introduction
2. Results
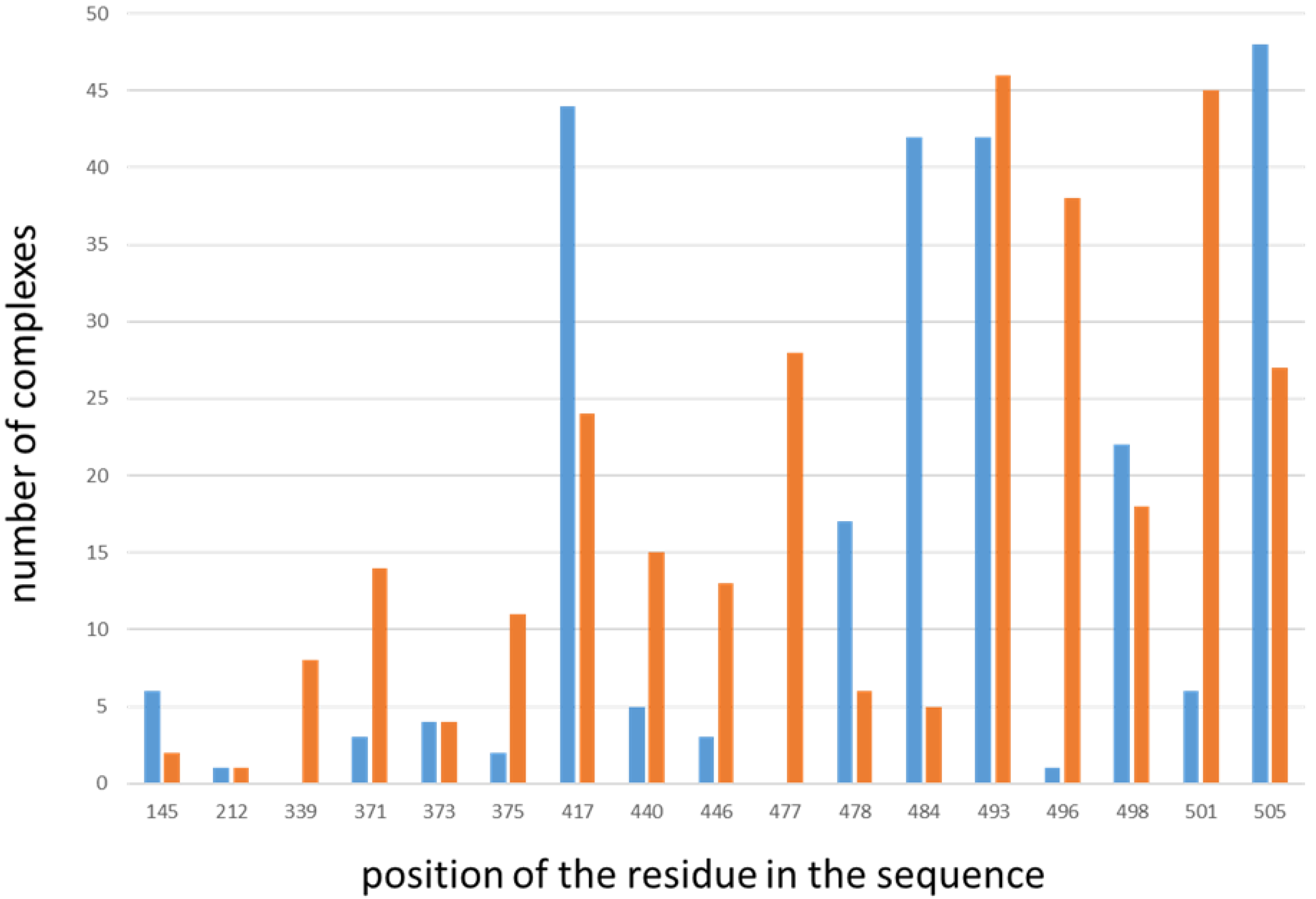
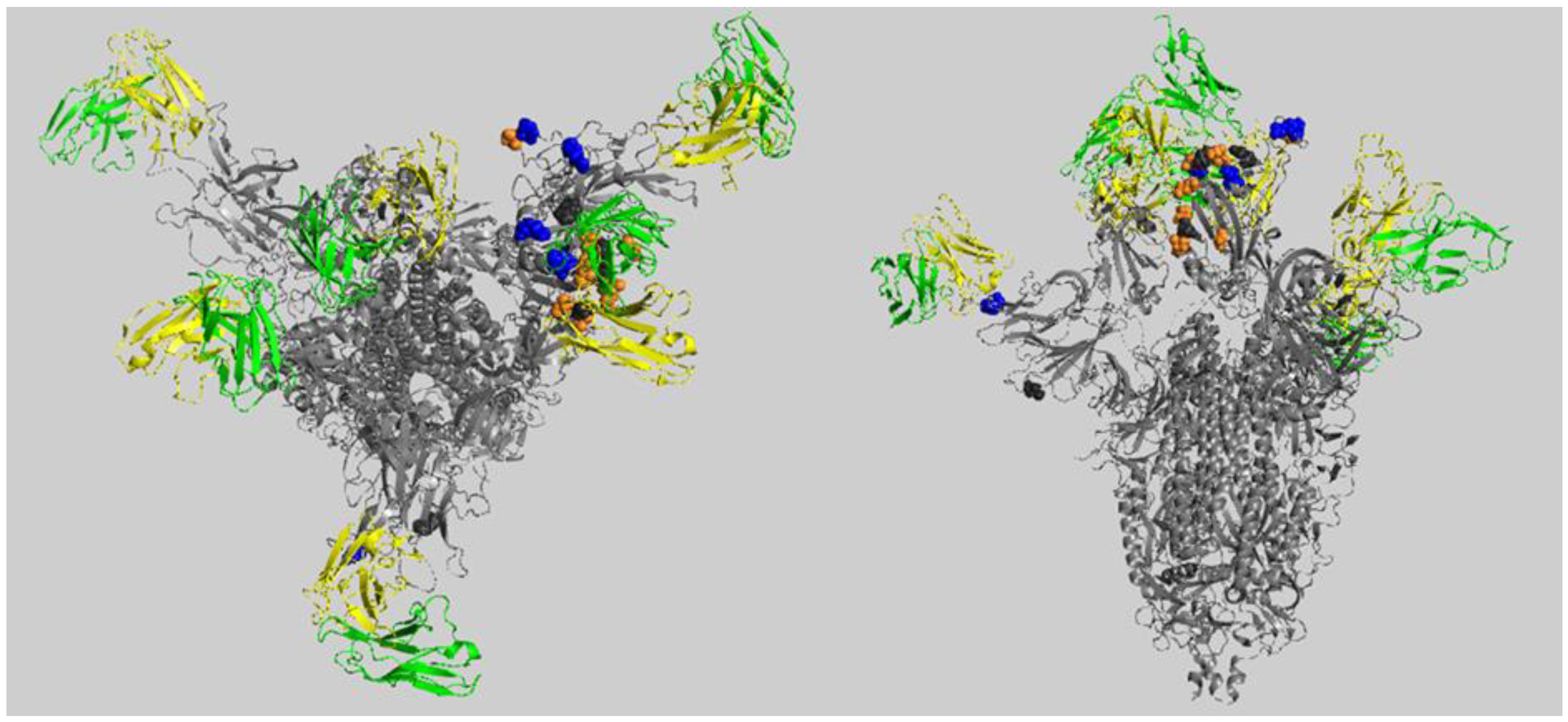
2.1. Predicted Impact of Omicron Variant on Spike–Antibodies Interfaces
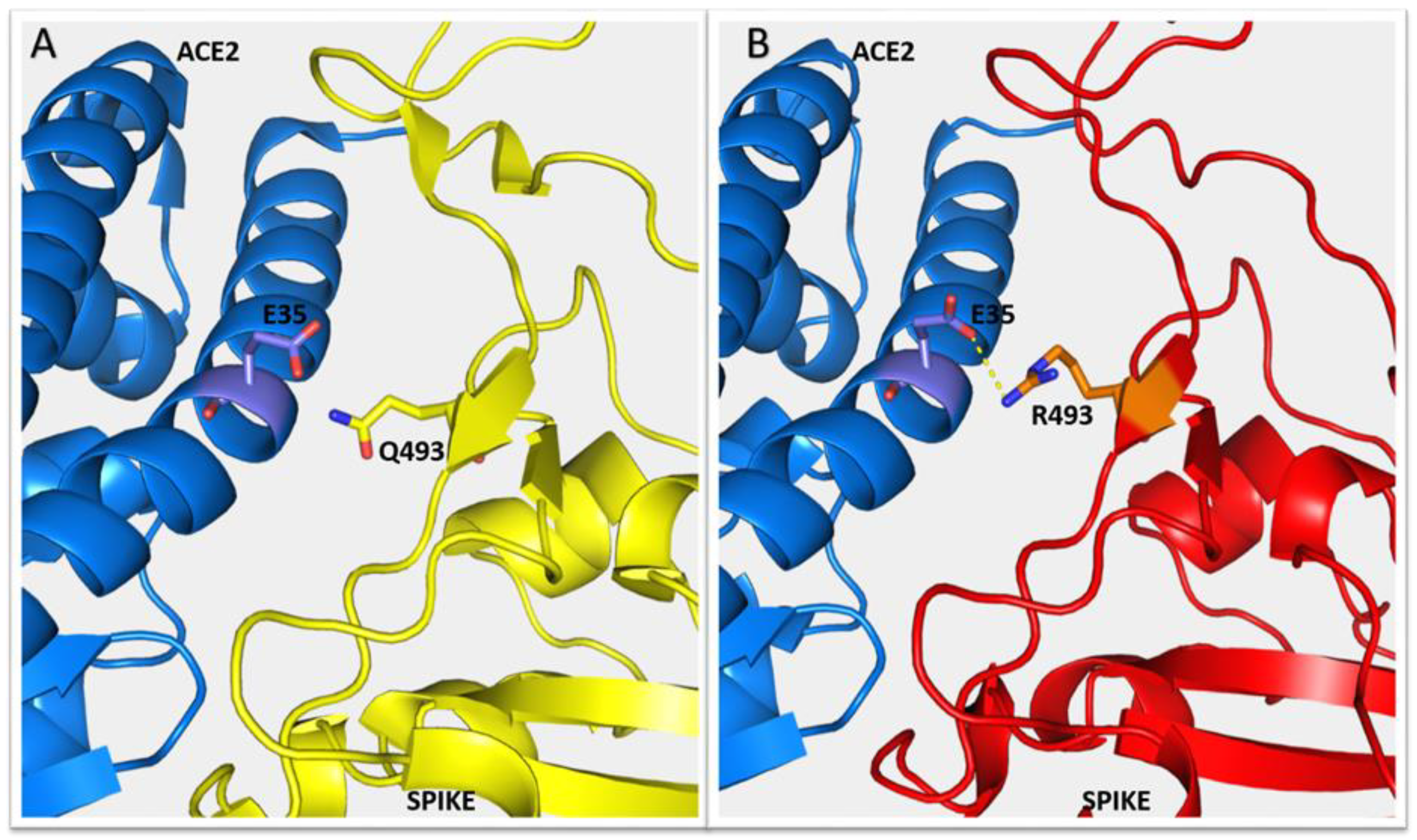
2.2. Predicted Impact of Omicron Variant on Spike-ACE2 Interface
3. Discussion
4. Materials and Methods
4.1. Definition of the Set of Omicron Amino Acid Changes
4.2. Datasets Used for Structural Analyses
4.3. Modeling and Analysis of the Effects of Mutations Associated with the Spike Protein in the Omicron Variant
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-Omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 25 July 2022).
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef]
- Callaway, E.; Ledford, H. How bad is Omicron? What scientists know so far. Nature 2021, 600, 197–199. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Xia, X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, J.T.; Jastrzebska, B.; Rangel, H.R. Omicron SARS-CoV-2 Variant Spike Protein Shows an Increased Affinity to the Human ACE2 Receptor: An In Silico Analysis. Pathogens 2022, 11, 45. [Google Scholar] [CrossRef]
- Xie, Y.; Karki, C.B.; Du, D.; Li, H.; Wang, J.; Sobitan, A.; Teng, S.; Tang, Q.; Li, L. Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. Front. Mol. Biosci. 2020, 7, 591873. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.; De Masi, L.; Argenio, M.A.; Facchiano, A. Structural Dissection of Viral Spike-Protein Binding of SARS-CoV-2 and SARS-CoV-1 to the Human Angiotensin-Converting Enzyme 2 (ACE2) as Cellular Receptor. Biomedicines 2021, 9, 1038. [Google Scholar] [CrossRef]
- Guérin, P.; Yahi, N.; Azzaz, F.; Chahinian, H.; Sabatier, J.M.; Fantini, J. Structural Dynamics of the SARS-CoV-2 Spike Protein: A 2-Year Retrospective Analysis of SARS-CoV-2 Variants (from Alpha to Omicron) Reveals an Early Divergence between Conserved and Variable Epitopes. Molecules 2022, 27, 3851. [Google Scholar] [CrossRef]
- Facchiano, A.; Facchiano, F.; Facchiano, A. An investigation into the molecular basis of cancer comorbidities in coronavirus infection. FEBS Open Bio 2020, 10, 2363–2374. [Google Scholar] [CrossRef]
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Cauda, R.; Cassone, A. Shortening Epitopes to Survive: The Case of SARS-CoV-2 Lambda Variant. Biomolecules 2021, 11, 1494. [Google Scholar] [CrossRef]
- Contractor, D.; Globisch, C.; Swaroop, S.; Jain, A. Structural basis of Omicron immune evasion: A comparative computational study. Comput. Biol. Med. 2022, 147, 105758. [Google Scholar] [CrossRef]
- Xiong, D.; Zhao, X.; Luo, S.; Cong, Y.; Zhang, J.Z.H.; Duan, L. Immune Escape Mechanisms of SARS-CoV-2 Delta and Omicron Variants against Two Monoclonal Antibodies That Received Emergency Use Authorization. J. Phys. Chem. Lett. 2022, 13, 6064–6073. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Marabotti, A. Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions. Molecules 2021, 26, 5941. [Google Scholar] [CrossRef] [PubMed]
- Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Catapano, L.; Marabotti, A. Simulation of the Interactions of Arginine with Wild-Type GALT Enzyme and the Classic Galactosemia-Related Mutant p.Q188R by a Computational Approach. Molecules 2021, 26, 6061. [Google Scholar] [CrossRef] [PubMed]
- d’Acierno, A.; Scafuri, B.; Facchiano, A.; Marabotti, A. The evolution of a Web resource: The Galactosemia Proteins Database 2.0. Hum. Mutat. 2018, 39, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Biancaniello, C.; D’Argenio, A.; Giordano, D.; Dotolo, S.; Scafuri, B.; Marabotti, A.; d’Acierno, A.; Tagliaferri, R.; Facchiano, A. Investigating the Effects of Amino Acid Variations in Human Menin. Molecules 2022, 27, 1747. [Google Scholar] [CrossRef] [PubMed]
- Modeller—Mutate_Model Script. Available online: https://salilab.org/modeller/wiki/Mutate%20model (accessed on 25 July 2022).
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- PyMOL Molecular Graphics System. Available online: https://sourceforge.net/projects/pymol/ (accessed on 25 July 2022).
- European Commission. Technical Working Group on COVID-19 Diagnostic Tests. Available online: https://health.ec.europa.eu/health-security-and-infectious-diseases/crisis-management/covid-19-diagnostic-tests_en (accessed on 25 July 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue Position in the Spike Sequence | Number of Complexes with Loss of Interactions Involving the Residue | Number of Complexes with Gain of Interactions Involving the Residue | Number of Spike Chains Including the Residue in the Starting Dataset (Total Number of Spike Chains in the Dataset: 342) |
|---|---|---|---|
| 145 | 6 | 2 | 69 |
| 212 | 1 | 1 | 204 |
| 339 | 0 | 8 | 321 |
| 371 | 3 | 14 | 318 |
| 373 | 4 | 4 | 319 |
| 375 | 2 | 11 | 321 |
| 417 | 44 | 24 | 322 |
| 440 | 5 | 15 | 321 |
| 446 | 3 | 13 | 258 |
| 477 | 0 | 28 | 270 |
| 478 | 17 | 6 | 270 |
| 484 | 42 | 5 | 279 |
| 493 | 42 | 46 | 321 |
| 496 | 1 | 38 | 321 |
| 498 | 22 | 18 | 310 |
| 501 | 6 | 45 | 307 |
| 505 | 48 | 27 | 323 |
| Residues Involved in H-Bonds in the Spike-ACE2 Interface | Reference Spike-ACE2 Complex | Omicron Spike-ACE2 Complex | |||
|---|---|---|---|---|---|
| Spike Residue (in Parenthesis, the Replacement in Omicron Variant) | ACE2 Residue | 6LZG | 6M0J | 6LZG mut | 6M0J mut |
| LYS 417 (ASN) | ASP 30 | + | + | ||
| GLY 446 (SER) | GLN 42 | + | + (SER 446) | ||
| TYR 449 | ASP 38 | + | + | + | |
| TYR 449 | GLN 42 | + | + | + | |
| ALA 475 | SER 19 | + | |||
| ASN 487 | GLN 24 | + | + | ||
| ASN 487 | TYR 83 | + | + | + | + |
| GLN 493 (ARG) | GLU 35 | + | |||
| GLY 496 (SER) | LYS 353 | + | + | ||
| GLN 498 (ARG) | GLN 42 | + | |||
| THR 500 | TYR 41 | + | + | + | + |
| THR 500 | ASP 355 | + | |||
| THR 500 | ARG 358 | + | |||
| ASN 501 (TYR) | LYS 353 | + (TYR 500) | |||
| GLY 502 | LYS 353 | + | + | + | + |
| Total number of H-bonds | 10 | 10 | 3 | 9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Arminio, N.; Giordano, D.; Scafuri, B.; Biancaniello, C.; Petrillo, M.; Facchiano, A.; Marabotti, A. In Silico Analysis of the Effects of Omicron Spike Amino Acid Changes on the Interactions with Human Proteins. Molecules 2022, 27, 4827. https://doi.org/10.3390/molecules27154827
D’Arminio N, Giordano D, Scafuri B, Biancaniello C, Petrillo M, Facchiano A, Marabotti A. In Silico Analysis of the Effects of Omicron Spike Amino Acid Changes on the Interactions with Human Proteins. Molecules. 2022; 27(15):4827. https://doi.org/10.3390/molecules27154827
Chicago/Turabian StyleD’Arminio, Nancy, Deborah Giordano, Bernardina Scafuri, Carmen Biancaniello, Mauro Petrillo, Angelo Facchiano, and Anna Marabotti. 2022. "In Silico Analysis of the Effects of Omicron Spike Amino Acid Changes on the Interactions with Human Proteins" Molecules 27, no. 15: 4827. https://doi.org/10.3390/molecules27154827
APA StyleD’Arminio, N., Giordano, D., Scafuri, B., Biancaniello, C., Petrillo, M., Facchiano, A., & Marabotti, A. (2022). In Silico Analysis of the Effects of Omicron Spike Amino Acid Changes on the Interactions with Human Proteins. Molecules, 27(15), 4827. https://doi.org/10.3390/molecules27154827