3.1. Synthesis and Characterization
The IR spectra were recorded on a Spectrum 100 FT-IR spectrometer (Perkin-Elmer, Shelton, CT, USA) using an ATR technique. The
1H,
13C, and
15N NMR (400, 100, and 40 MHz, respectively) and COSY,
1H–
13C HSQC,
1H–
13C HMBC, DEPT, and
1H–
15N HSQC,
1H–
15N HMBC spectra were acquired on a Bruker Avance DRX 400 spectrometer (Bruker BioSpin, Rheinstetten, Germany) in CDCl
3 (NMR spectra for all the compounds are available online, see the
Supplementary Materials). The
1H NMR chemical shifts were reported relative to the residual solvent protons as internal standards (7.26 ppm). The solvent carbon atoms served as internal standard for the
13C NMR spectra (77.0 ppm). The
15N NMR spectra were obtained using MeNO
2 (380.5 ppm) and urea (73.4 ppm) as internal standards. Optical rotations measurements were performed on a Jasco DIP-370 polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA) with a 10 cm microcell. Melting points were determined on a Boetius (VEB Analytik, DDR) hot stage apparatus and were not uncorrected. The progress of reactions and purity of products were examined by TLC on Merck silica gel 60 plates, eluent CH
2Cl
2 or a mixture of CH
2Cl
2–MeOH, 99:1; 49:1. Visualization was achieved by the treatment with conc. H
2SO
4 and heating at 80 °C or using an UV lamp (254 or 365 nm). All solvents were purified and dried by standard techniques prior to use.
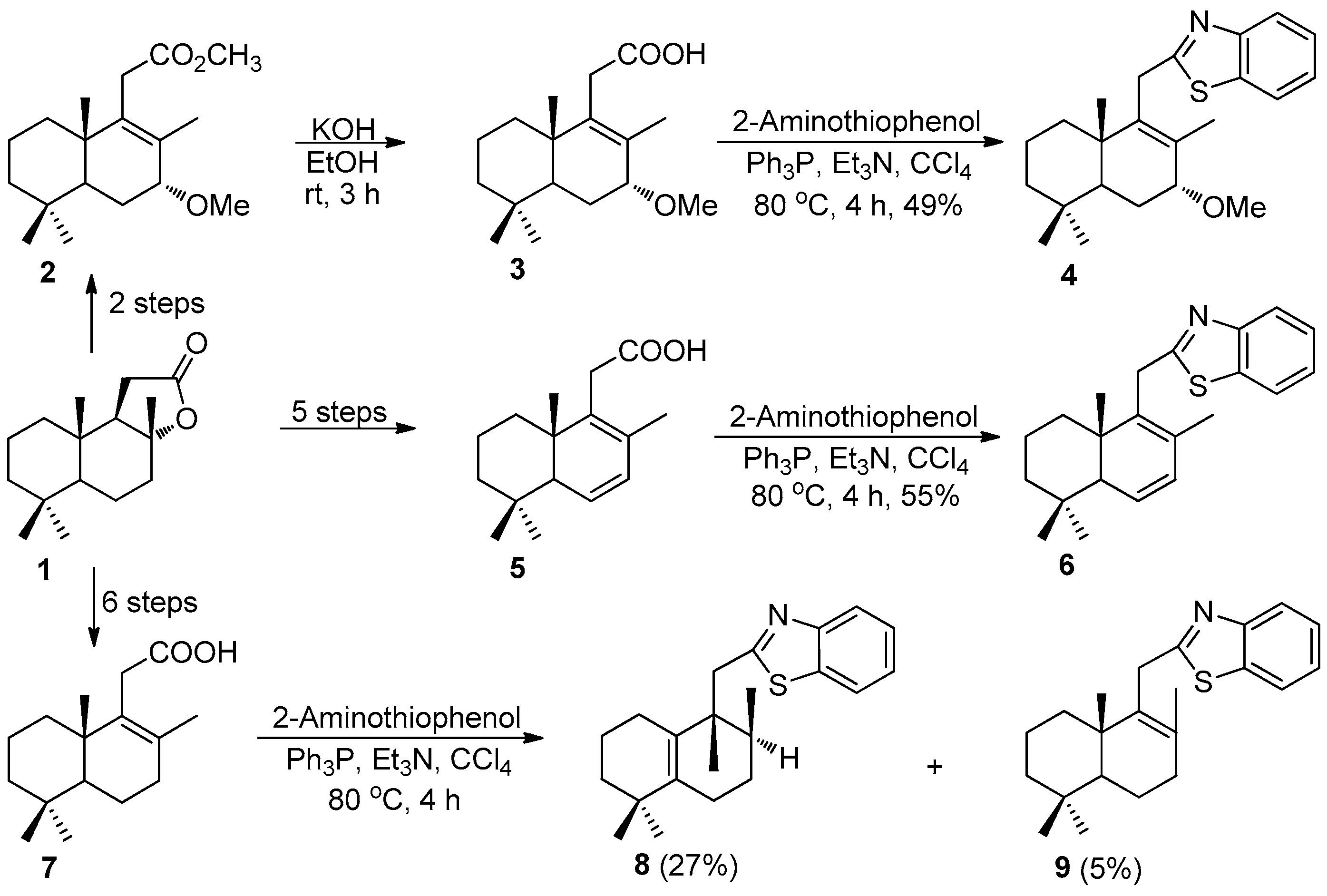
Compound 3: Compound 2 (294 mg, 1 mmol) was dissolved in EtOH (10 mL) and solid KOH (615 mg, 11 mmol) was added. The resulting mixture was heated at 50 °C for 3 h, and then, 2/3 of alcohol was distilled under reduced pressure on a rotary evaporator. The residue was diluted with H2O (20 mL), acidified with 40% HCl (20 mL) and extracted with Et2O (3 × 20 mL). The organic layer was washed with H2O (30 mL), dried over anhydrous Na2SO4 and concentrated, giving compound 3 (249 mg, 89% yield) as a colorless oil. +50.6 (c 2.4, CHCl3). IR spectrum, ν, cm−1: 736, 1070, 1376, 1459, 1626, 1704, 2927. 1H NMR (400 MHz, CDCl3) δ 0.82 (3H, s, 10-CH3), 0.87 (3H, s, 4-CH3), 0.88 (3H, s, 4-CH3), 1.09–1.15 (2H, m, CH2), 1.16–1.19 (1H, m, CH2), 1.35–1.58 (5H, m, H-5, 2CH2), 1.64 (3H, s, 8-CH3), 1.91–1.94 (1H, m, CH2), 3.01 (2H, d, J = 7.0 Hz, H-11), 3.32 (3H, s, 7-CH3), 3.43 (1H, d, J = 2.6 Hz, H-7). 13C NMR (100 MHz, CDCl3) δ 17.7 (C-17), 17.9 (C-20), 18.7 (C-2), 21.5 (C-18), 22.5 (C-6), 32.7 (C-19), 32.8 (C-4), 33.0 (C-11), 35.9 (C-1), 39.3 (C-10), 41.1 (C-3), 45.7 (7-OCH3), 56.4 (C-5), 79.4 (C-7), 130.0 (C-8), 139.1 (C-9), 171.9 (C-12). HRMS (ESI) calculated for C17H28O3 [M + H]+, 280.4035. Found: 280.4127.
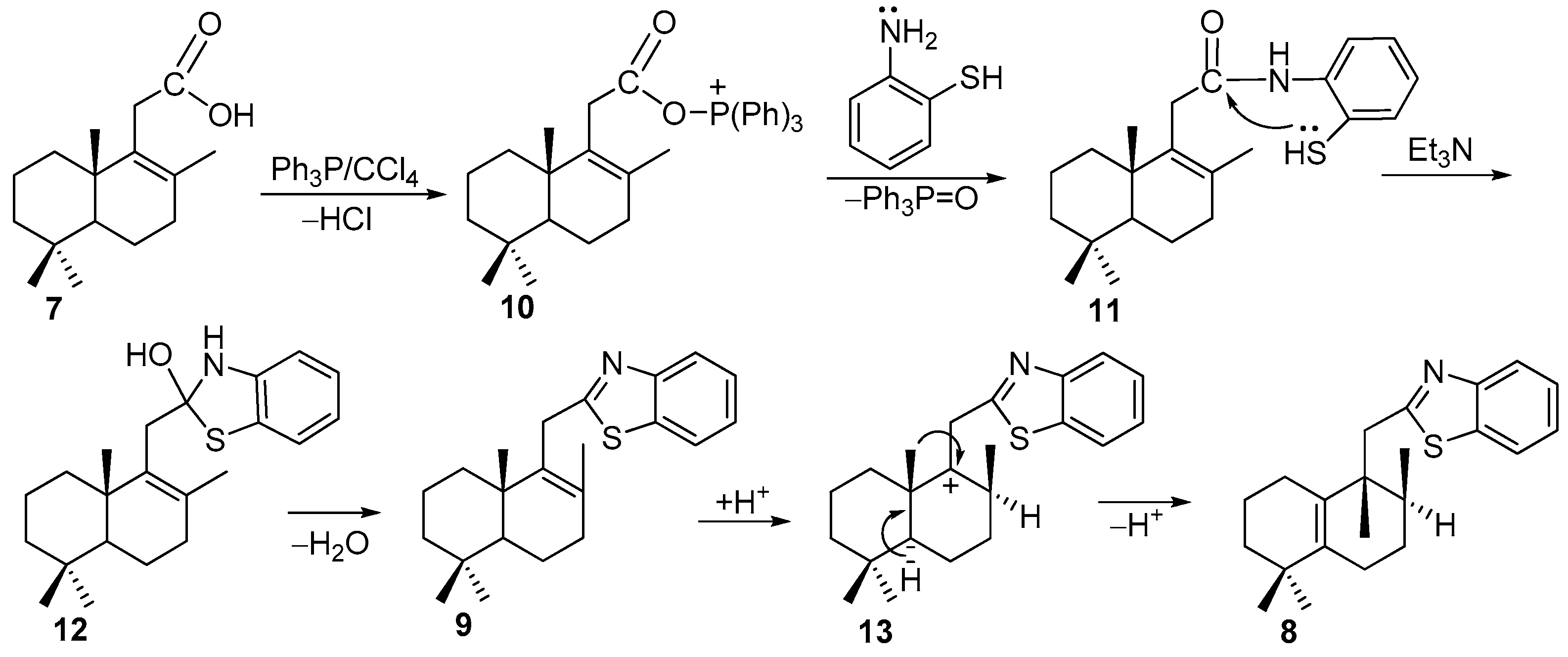
Compounds 4, 6, 8, and 9 (General method).
To an ice bath-cooled solution of Ph3P (786 mg, 3 mmol) and Et3N (0.16 mL, 1.2 mmol) dissolved in CCl4 (7 mL), one of the acids 3 (280 mg, 1 mmol), 5 (248 mg, 1 mmol) or 7 (250 mg, 1 mmol) was added. After 10 min of stirring, the solution of 2-aminothiophenol (150 mg, 1.2 mmol) dissolved in CCl4 (3 mL) was added and the reaction mixture was refluxed under stirring for 4 h. The solvents were removed under a reduced pressure on a rotary evaporator to dryness and crude reaction products were subjected to silica gel flash chromatography (CH2Cl2).
Compound 4. (180 mg, 49%), colorless oil. 78.3 (c 0.6, CHCl3). IR spectrum, ν, cm−1: 729, 758, 1080, 1373, 1437, 1456, 1509, 1707, 1759, 2926. 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, s, 10-CH3), 0.93 (3H, s, 4-CH3), 0.99 (3H, s, 4-CH3), 1.09–1.16 (2H, m, CH2), 1.23–1.27 (1H, m, CH2), 1.35–1.40 (2H, m, CH2), 1.51–1.55 (3H, m, H-5, CH2), 1.83 (3H, s, 8-CH3), 2.01–2.05 (1H, m, CH2), 3.43 (3H, s, 7-CH3), 3.51 (1H, t, J = 2.7 Hz, 7-CH), 3.88 (2H, t, J = 17.4 Hz, H-11), 7.31 (1H, dt, J = 7.5, 1.0 Hz, H-6′), 7.43 (1H, dt, J = 7.7, 1.0 Hz, H-7′), 7.78 (1H, d, J = 7.8 Hz, H-5′), 7.93 (1H, d, J = 8.0 Hz, C-8′). 13C NMR (100 MHz, CDCl3) δ 18.6 (C-20 and C-17), 18.7 (C-2), 21.7 (C-18), 22.7 (C-6), 32.8 (C-19), 32.8 (C-11), 32.9 (C-4), 36.0 (C-1), 39.9 (C-10), 41.2 (C-3), 46.0 (7-OCH3), 56.9 (C-5), 79.3 (C-7), 121.5 (C-5′), 122.3 (C-8′), 124.4 (C-6′), 125.7 (C-7′), 131.1 (C-8), 135.1 (C-9), 142.5 (C-4′), 153.4 (C-9′), 173.6 (C-2′). 15N NMR (400 MHz, CDCl3) δ 301. HRMS (ESI) calculated for C23H31NOS [M + H]+, 369.5667. Found: 369.5693.
Compound 6. (185 mg, 55%), yellow oil. −260.94 (c 0.59, CHCl3). IR spectrum, ν, cm−1: 729, 757, 1369, 1456, 1508, 1726, 2924. 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, s, 10-CH3), 0.95 (3H, s, 4-CH3), 0.96 (3H, s, 4-CH3), 1.10–1.82 (6H, m, 3CH2), 1.86 (3H, s, 8-CH3), 2.15 (1H, t, J = 2.9 Hz, H-5), 3.85 (1H, d, J = 16.7 Hz, H-11), 3.96 (1H, d, J = 16.7 Hz, H-11), 5.88 (1H, dd, J = 9.5 Hz, J = 2.5 Hz, H-6), 5.95 (1H, dd, J = 9.5, 3.0 Hz, H-7); 7.33 (1H, ddd, J = 7.5, 7.2 Hz, J = 1.1 Hz, H-6′), 7.44 (1H, ddd, J = 8.1, 7.2, 1.2 Hz, H-7′), 7.82 (1H, dm, J = 8.3 Hz, H-5′), 7.96 (1H, dm, J = 8.1 Hz, H-8′). 13C NMR (100 MHz, CDCl3) δ 15.5 (C-20), 18.5 (C-8), 18.8 (C-2), 22.7 (C-18), 32.1 (C-11), 32.3 (C-19), 32.9 (C-4), 35.1 (C-1), 39.2 (C-10), 40.8 (C-3), 52.9 (C-5), 121.4 (C-5′), 122.4 (C-8′), 124.5 (C-6′), 125.7 (C-7′), 128.7 (C-6), 129.1 (C-7), 135.4 (C-8), 139.9 (C-9), 139.9 (C-9′), 153.6 (C-4′), 174.0 (C-2′). 15N NMR (400 MHz, CDCl3) δ 303. HRMS (ESI) calculated for C22H27NS [M + H]+, 337.1864. Found: 337.1949.
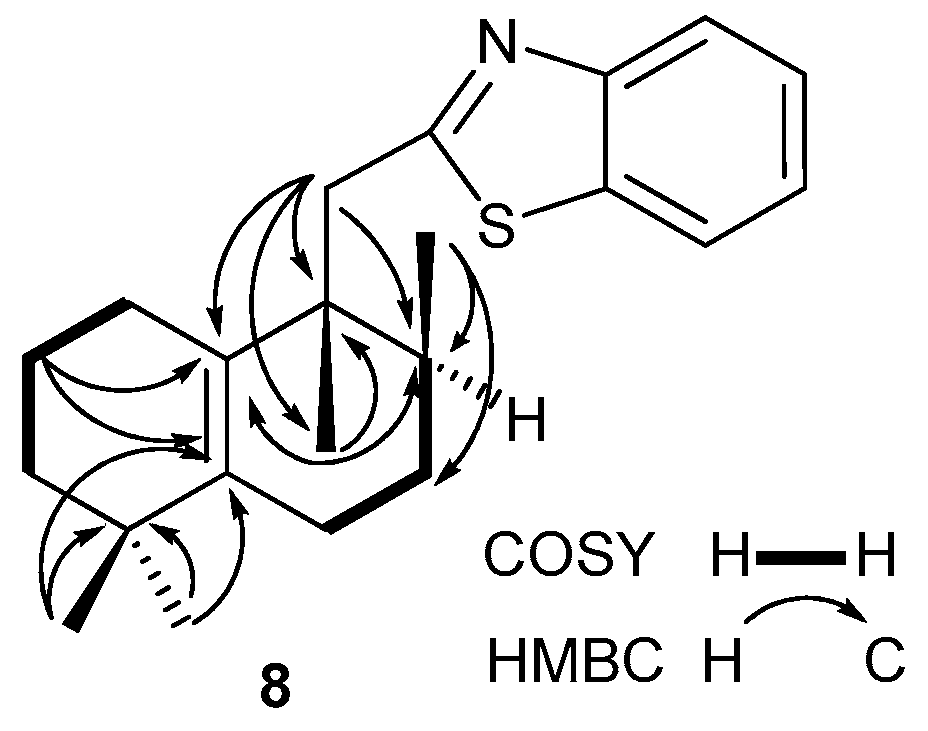
Compound 8. (92 mg, 27%), mp 58–59 °C, 7.63 (c 3.2, CHCl3). IR spectrum, ν, cm−1: 737, 763, 1122, 1311, 1377, 1433, 1454, 1505, 1713, 2920, 3059. 1H NMR (400 MHz, CDCl3) δ 0.92 (3H, d, J = 6.8 Hz, 8-CH3), 1.01 (3H, s, 4-CH3), 1.02 (3H, s, 4-CH3), 1.05 (3H, s, 9-CH3), 1.30–1.40 (2H, m, CH2), 1.46–1.63 (5H, m, H-8, 2CH2), 1.87–2.04 (2H, m, H-6), 2.17–2.22 (2H, m, CH2), 3.27 (2H, d, J = 3.9 Hz, H-11), 7.32 (1H, td, J = 7.8, 1.2 Hz, H-6′), 7.42 (1H, td, J = 7.0, 1.2 Hz, H-7′), 7.83 (1H, dm, J = 8.0 Hz, H-5′), 7.96 (1H, d, J = 8.0 Hz, H-8′). 13C NMR (100 MHz, CDCl3) δ 16.2 (C-17), 20.1 (C-2), 21.4 (C-20), 24.9 (C-6), 26.6 (C-1), 27.0 (C-7), 27.8 (C-18), 28.3 (C-19), 34.5 (C-8); 34.7 (C-4), 39.6 (C-3), 42.1 (C-11, C-9), 121.3 (C-5′), 122.5 (C-8′), 124.5 (C-6′), 125.6 (C-7′), 130.8 (C-10), 135.8 (C-9′), 139.4 (C-5), 152.1 (C-4′), 169.5 (C-2′). 15N NMR (400 MHz, CDCl3) δ 305. HRMS (ESI) calculated for C22H29NS [M + H]+, 339.2021. Found: 339.2106.
Compound 9. (17 mg, 5%), yellow oil. +28.62 (c 0.2, CHCl3). IR spectrum, ν, cm−1: 729, 757, 1014, 1124, 1146, 1293, 1376, 1435, 1456, 1506, 1577, 1694, 2865, 2926. 1H NMR (400 MHz, CDCl3) δ 0.84 (3H, s, 4-CH3), 0.90 (3H, s, 4-CH3), 1.02 (3H, s, 10-CH3), 1.05–1.11 (2H, m, CH2), 1.25–1.40 (3H, m, H-5, CH2), 1.45–1.60 (4H, m, 2CH2), 1.70 (3H, s, 8-CH3), 2.09–2.21 (2H, m, H-7), 3.83 (1H, d, J = 17.2 Hz, H-11), 3.90 (1H, d, J = 17.2 Hz, H-11), 7.32 (1H, ddd, J = 7.9 Hz, J = 7.3, 1.1 Hz, H-6′), 7.43 (1H, ddd, J = 8.2, 7.3, 1.3 Hz, H-7′), 7.81 (1H, ddd, J = 7.9, 1.3, 0.5 Hz, H-5′), 7.94 (1H, d, J = 8.2 Hz, H-8′). 13C NMR (100 MHz, CDCl3) δ 18.8 (C-6), 18.9 (C-2), 20.3 (C-20), 20.7 (C-17), 21.6 (C-18), 32.8 (C-11), 33.2 (C-19), 33.3 (C-4); 33.6 (C-7), 36.6 (C-1), 39.1 (C-10), 41.6 (C-3), 51.8 (C-5), 121.3 (C-5′), 122.4 (C-8′), 124.3 (C-6′), 125.6 (C-7′), 131.2 (C-8), 135.4 (C-9′), 137.7 (C-9), 153.6 (C-4′), 175.4 (C-2′). 15N NMR (400 MHz, CDCl3) δ 299. HRMS (ESI) calculated for C22H29NS [M + H]+, 339.2021. Found: 339.2107.
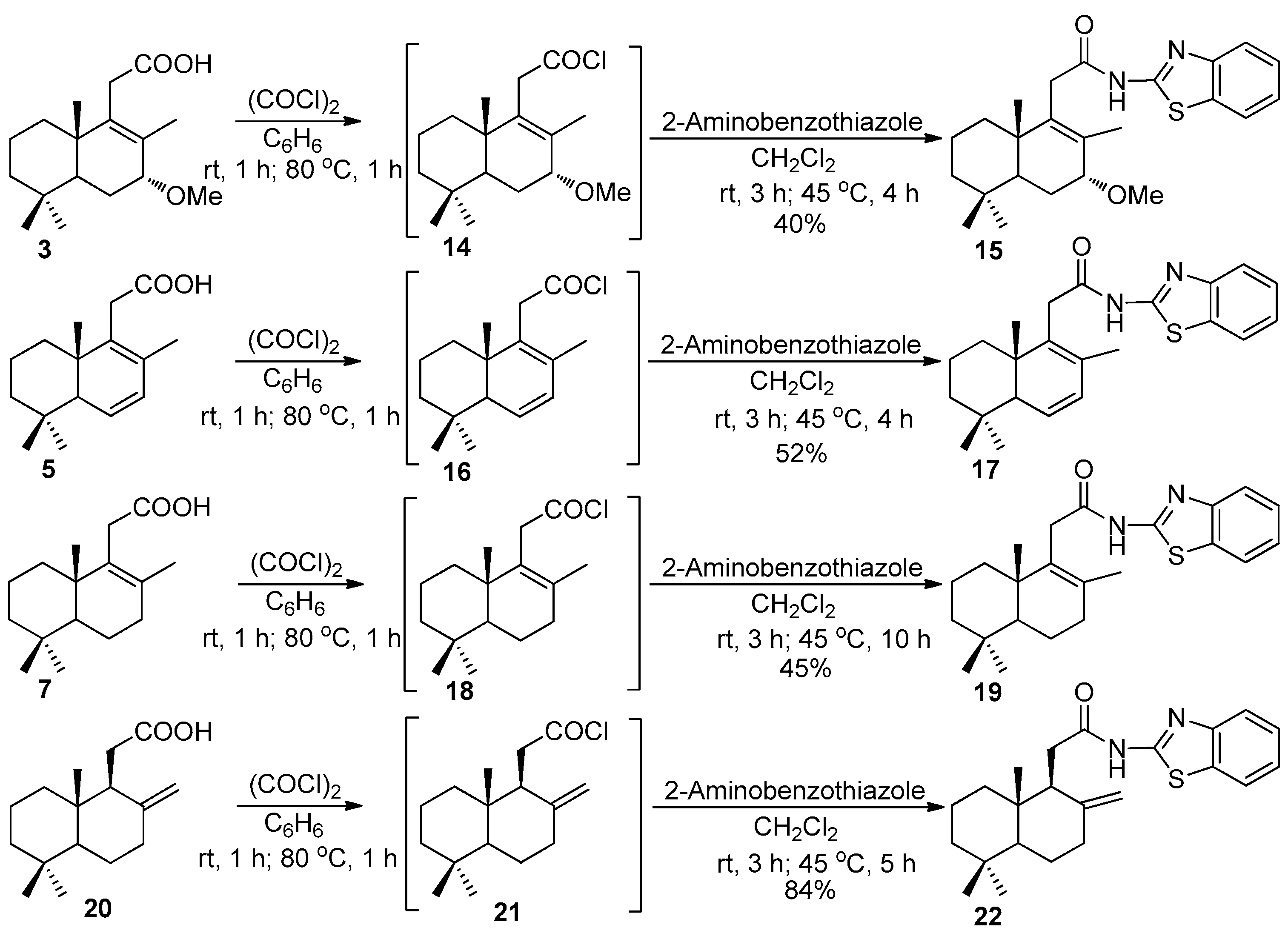
Compounds 15, 17, 19, and 22 (General method).
The solution of one of the acids 3 (280 mg, 1 mmol), 5 (248 mg, 1 mmol), 7 (250 mg, 1 mmol) or 20 (250 mg, 1 mmol) dissolved in anhydrous C6H6 (5 mL) was treated with a solution of (COCl)2 (0.95 mL, 11 mmol) dissolved in C6H6 (2.5 mL). The reaction mixture was stirred at room temperature for 1 h and subsequently refluxed for 1 h. The C6H6 and excess of (COCl)2 were removed at a reduced pressure on a rotary evaporator. Next, 2-aminobenzothiazole (225 mg, 1.5 mmol) was added to the solution of an acyl chloride 14, 16, 18 or 21 in CH2Cl2 (10 mL), and the resulting mixtures were stirred at r.t. for 3 h, then refluxed for 4–10 h. After cooling, the precipitates were filtered off, washed with CH2Cl2, and the filtrates were concentrated to dryness at a reduced pressure on a rotary evaporator. The crude reaction products were purified by silica gel flash chromatography (1 → 2% MeOH-CH2Cl2).
Compound 15. (164 mg, 40%), white crystals, mp 93–94 °C. 99.96 (c 1.4, CHCl3). IR spectrum, ν, cm−1: 728, 755, 1076, 1341, 1264, 1441, 1538, 1600, 1697, 2926, 3182. 1H NMR (400 MHz, CDCl3) δ 0.85 (3H, s, 10-CH3), 0.91 (3H, s, 4-CH3), 0.93 (3H, s, 4-CH3); 1.09–1.20 (2H, m, CH2), 1.37–1.58 (5H, m, H-5, 2CH2), 1.62–1.66 (1H, m, CH2), 1.77 (3H, s, 8-CH3), 2.01 (1H, d, J = 13.0 Hz, CH2), 3.22 (1H, d, J = 17.4 Hz, H-11), 3.32 (1H, d, J = 17.4 Hz, H-11), 3.40 (3H, s, 7-CH3), 3.50 (1H, d, J = 5.6 Hz, H-7), 7.30 (1H, dt, J = 7.5, 0.9 Hz, H-6′), 7.42 (1H, dt, J = 7.6, 1.0 Hz, H-7′), 7.77 (1H, d, J = 8.0 Hz, H-5′), 7.80 (1H, d, J = 7.7 Hz, H-8′), 9.91 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 18.2 (C-20), 18.3 (C-17), 18.7 (C-2), 21.6 (C-18), 22.4 (C-6), 32.7 (C-19), 32.8 (C-4), 35.7 (C-11), 35.8 (C-1), 39.6 (C-10), 40.9 (C-3), 45.5 (7-CH3), 56.8 (C-5), 78.9 (C-7), 120.8 (C-5′), 121.3 (C-8′), 123.9 (C-6′), 126.2 (C-7′), 132.1 (C-4′), 132.7 (C-8), 138.9 (C-9), 148.1 (C-9′), 158.2 (C-2′), 169.6 (C = O). 15N NMR (400 MHz, CDCl3) δ 140, 259. HRMS (ESI) calculated for C24H32N2O2S [M-H]-, 412.2184. Found: 412.2112.
Compound 17. (23 mg, 52%), white crystals, mp 83–84 °C. −172.5 (c 0.1, CHCl3). IR spectrum, ν, cm−1: 728, 755, 908, 1147, 1267, 1343, 1442, 1536, 1599, 1702, 2925, 3178. 1H NMR (400 MHz, CDCl3) δ 0.84 (3H, s, 10-CH3), 0.96 (6H, s, 4-CH3, 4-CH3), 1.15–1.80 (6H, m, 3CH2), 1.83 (3H, s, 8-CH3), 2.10 (1H, t, J = 2.2 Hz, H-5), 3.21 (1H, d, J = 17.3 Hz, H-11), 3.42 (1H, d, J = 17.0 Hz, H-11), 5.93 (1H, d, J = 1.8 Hz, H-6), 5.94 (1H, d, J = 2.2 Hz, H-6), 7.31 (1H, dt, J = 1.0, 0.8 Hz, H-6′), 7.44 (1H, dt, J = 1.2, 1.0 Hz, H-7′), 7.75 (1H, d, J = 8.2 Hz, H-5′), 7.82 (1H, dd, J = 7.87, 0.47 Hz, H-8′), 9.74 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 15.1 (C-20), 18. (C-17), 18.7 (C-2), 22.8 (C-18), 32.2 (C-19), 33.0 (C-4), 35.0 (C-1), 35.3 (C-11), 39.1 (C-10), 40.6 (C-3), 52.9 (C-5), 120.7 (C-5′), 121.4 (C-8′), 124.0 (C-6′), 126.3 (C-7′), 128.8 (C-6), 129.6 (C-7), 130.9 (C-9), 132.1 (C-4′), 135.7 (C-8), 148.0 (C-9′), 157.9 (C-2′), 169.9 (C-12). 15N NMR (400 MHz, CDCl3) δ 139, 254. HRMS (ESI) calculated for C23H28N2OS [M + H]+, 380.1922. Found: 380.2011.
Compound 19. (171 mg, 45%), white crystals, mp 84–85 °C. +102.5 (c 2.0, CHCl3). IR spectrum, ν, cm−1: 727, 755, 883, 907, 975, 1018, 1152, 1267, 1334, 1379, 1443, 1533, 1600, 1704, 1773, 2927, 3175, 3365. 1H NMR (400 MHz, CDCl3) δ 0.80 (3H, s, 4-CH3), 0.84 (3H, s, 4-CH3), 0.88 (3H, s, 10-CH3), 0.95–1.18 (4H, m, 2CH2), 1.27 (1H, dd, J = 12.6 Hz, J = 1.9 Hz, H-5), 1.33–1.57 (4H, m, 2CH2), 1.67 (3H, s, 8-CH3), 2.09 (1H, dd, J = 18.0 Hz, J = 6.4 Hz, H-7), 2.27 (1H, ddd, J = 18.4 Hz, J = 11.2 Hz, J = 7.4 Hz, H-7), 3.12 (1H, d, J = 17.6 Hz, H-11), 3.33 (1H, d, J = 17.6 Hz, H-11), 7.30 (1H, ddd, J = 8.0 Hz, J = 7.3 Hz, J = 1.0 Hz, H-7′), 7.42 (1H, ddd, J = 8.0 Hz, J = 7.3 Hz, J = 1.2 Hz, H-6′), 7.79 (1H, d, J = 8.0 Hz, H-5′), 7.81 (1H, ddd, J = 8.0 Hz, J = 1.2 Hz, J = 0.6 Hz, H-8′), 9.73 (1H, br.s., NH). 13C NMR (100 MHz, CDCl3) δ 18.7 (C-6), 18.8 (C-2), 19.7 (C-20), 20.4 (C-17), 21.6 (C-18), 33.1 (C-19), 33.3 (C-4), 33.5 (C-7), 35.9 (C-11), 36.3 (C-1), 38.8 (C-10), 41.3 (C-3), 51.5 (C-5), 120.7 (C-5′), 121.5 (C-8′), 124.0 (C-7′), 126.3 (C-6′), 132.1 (C-9′), 133.0 (C-9), 134.2 (C-8), 148.0 (C-4′), 158.4 (C-2′), 170.4 (C-12). 15N NMR (400 MHz, CDCl3) δ 137, 257. HRMS (ESI) calculated for C23H30N2OS [M + H]+, 382.2079. Found: 382.2165.
Compound 22. (320 mg, 84%), white crystals, mp 99–100 °C. −34.2 (c 2.0, CHCl3). IR spectrum, ν, cm−1: 750, 884, 998, 1018, 1135, 1157, 1215, 1270, 1292, 1324, 1332, 1365, 1383, 1443, 1457, 1542, 1599, 1644, 1697, 2932, 3063, 3178. 1H NMR (400 MHz, CDCl3) δ 0.46 (3H, s, 10-CH3), 0.76 (3H, s, 4-CH3), 0.84 (3H, s, 4-CH3), 0.99–1.18 (3H, m, H-5, CH2), 1.25 (1H, dd, J = 13.0, 4.4 Hz, H-6), 1.30–1.54 (4H, m, 2CH2), 1.68 (1H, dm, J = 13.0 Hz, H-6), 2.05 (1H, td, J = 13.0, 5.1 Hz, H-7), 2.33 (1H, ddd, J = 13.0, 4.0, 2.1 Hz, H-7), 2.45 (1H, dd, J = 11.2, 10.3 Hz, H-9), 2.47 (1H, dd, J = 6.2, 10.3 Hz, H-11), 2.63 (1H, dd, J = 26.2, 11.2 Hz, H-11), 4.39 (1H, s, 8-CH2), 4.73 (1H, s, 8-CH2), 7.33 (1H, ddd, J = 8.1, 7.1, 1.0 Hz, H-7′), 7.44 (1H, ddd, J = 8.2, 7.1, Hz, 1.1 Hz, H-6′), 7.81 (1H, dd, J = 8.2, 1.0 Hz, H-5′), 7.84 (1H, dd, J = 8.1, 1.1 Hz, H-8′), 11.26 (1H, br.s., NH). 13C NMR (100 MHz, CDCl3) δ 14.3 (C-20), 19.2 (C-2); 21.6 (C-18), 23.9 (C-6), 32.8 (C-11), 33.4 (C-4), 33.5 (C-19), 37.5 (C-7), 38.9 (C-1), 39.0 (C-10), 41.8 (C-3), 52.2 (C-9), 55.0 (C-5), 106.5 (8-CH2), 120.6 (C-5′), 121.7 (C-8′), 123.9 (C-7′), 126.3 (C-6′), 132.1 (C-9′), 147.8 (C-4′), 148.7 (C-8), 159.6 (C-2′), 171.8 (C-12). 15N NMR (400 MHz, CDCl3) δ 251 (C = N). HRMS (ESI) calculated for C23H30N2OS [M + H]+, 382.2079. Found: 382.2166.
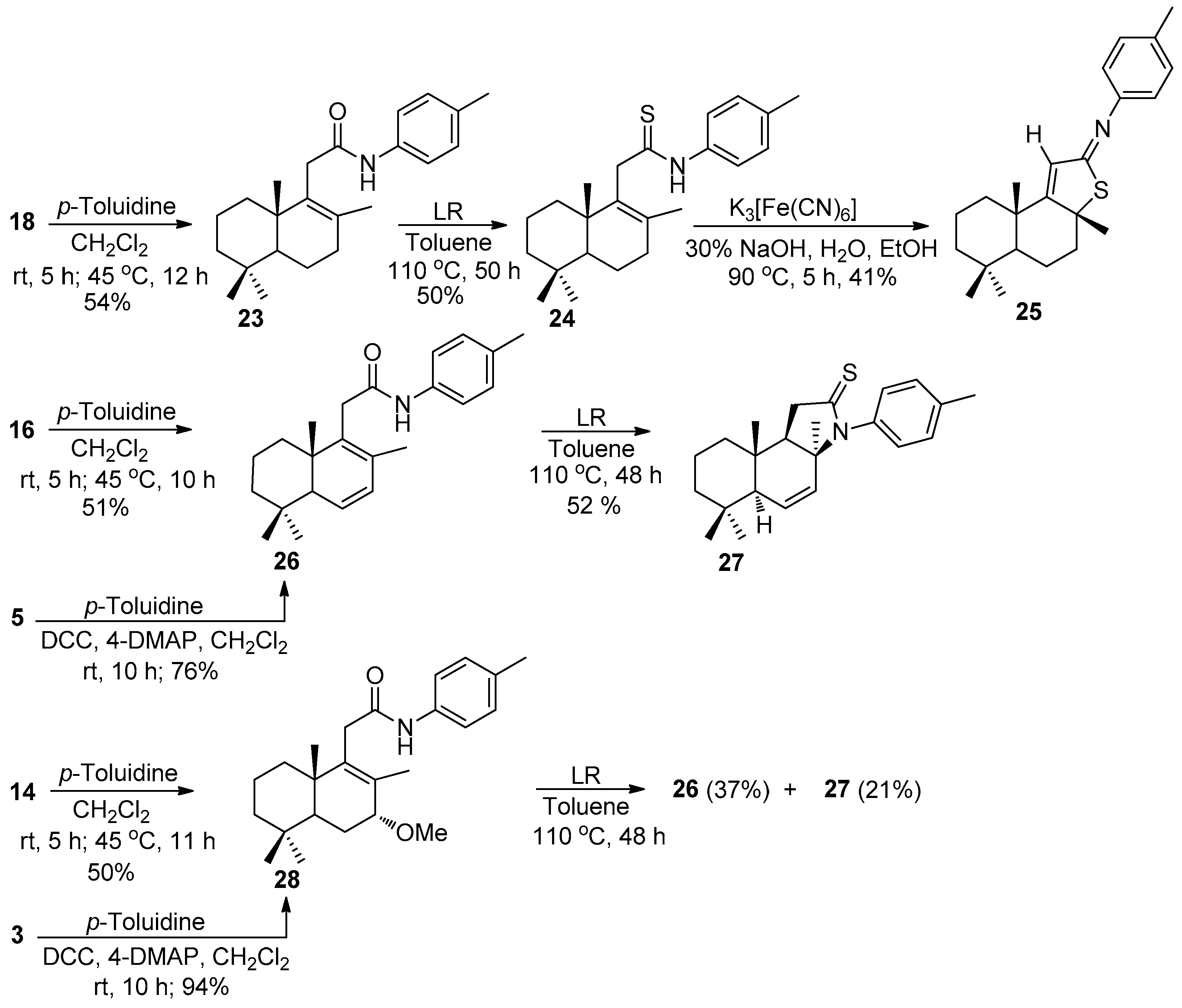
Compounds 23, 26, and 28. (Typical procedure).
Method 1. The solution of one of the acids 3 (280 mg, 1 mmol), 5 (248 mg, 1 mmol) or 7 (250 mg, 1 mmol) dissolved in anhydrous C6H6 (5 mL) was treated with a solution of (COCl)2 (0.95 mL, 11 mmol) dissolved in C6H6 (2.5 mL). The reaction mixture was stirred at room temperature for 1 h, and additionally refluxed for 1 h. The C6H6 and excess of (COCl)2 were removed at reduced pressure on a rotary evaporator. Next, p-toluidine (160 mg, 1.5 mmol) was added to the solutions of acyl chlorides 14, 16 or 18 obtained in situ in CH2Cl2 (10 mL), and the resulting mixtures were stirred at r.t. for 5 h, and refluxed for 10–12 h. After cooling, the precipitate was filtered off, washed with CH2Cl2, and the filtrate was concentrated to dryness on a rotary evaporator. The crude reaction products were purified by silica gel flash chromatography (1→2% MeOH-CH2Cl2) to give products 23, 26, and 28.
Method 2. A solution of DCC (412 mg, 2 mmol), 4-DMAP (244 mg, 2 mmol), p-toluidine (214 mg, 2 mmol) and an acid 3 (280 mg, 1 mmol) or 5 (248 mg, 1 mmol) dissolved in CH2Cl2 (8 mL) was stirred for 10 h at room temperature. After the reaction period, the mixture was filtered, and the solvent was removed under a reduced pressure on a rotary evaporator to give the crude product which was purified by silica gel flash chromatography (CH2Cl2) to give compounds 26 and 28.
Compound 23. (183 mg, 54%), white crystals, mp 152–153 °C. +132.64 (c 1.0, CHCl3). IR spectrum, ν, cm−1: 733, 818, 908, 1171, 1248, 1346, 1405, 1458, 1516, 1603, 1661, 2867, 2927, 3293. 1H NMR (400 MHz, CDCl3) σσ 0.85 (3H, s, 4-CH3), 0.92 (3H, s, 4-CH3), 0.99 (3H, s, 10-CH3), 1.04–1.10 (2H, m, CH2), 1.17 (1H, dd, J = 12.6, 1.9 Hz, H-5), 1.40–1.62 (4H, m, 2CH2), 1.68 (3H, s, 8-CH3), 1.71–1.82 (2H, m, CH2), 2.08–2.26 (2H, m, H-7), 2.31 (3H, s, 4′-CH3), 3.04 (1H, d, J = 17.5 Hz, H-11), 3.22 (1H, d, J = 17.5 Hz, H-11), 7.12 (2H, d, J = 8.2 Hz, H-3′ and H-5′), 7.35 (2H, d, J = 8.3 Hz, H-2′ and H-6′), 7.53 (1H, br.s, NH). 13C NMR (100 MHz, CDCl3) δ 18.8 (C-6), 18.9 (C-2), 20.1 (C-20), 20.3 (C-17), 21.7 (C-18), 29.9 (C-7′), 33.3 (C-19), 33.4 (C-4), 33.7 (C-7), 36δ.3 (C-1), 37.2 (C-11), 39.0 (C-10), 41.6 (C-3), 52.4 (C-5), 119.9 (C-2′ and C-6′), 129.5 (C-3′ and C-5′), 132.1 (C-8), 133.9 (C-4′), 135.3 (C-1′), 136.4 (C-9), 169.6 (C-12). 15N NMR (400 MHz, CDCl3) δ 127. HRMS (ESI) calculated for C23H33NO [M + H]+, 339.2562. Found: 339.2649.
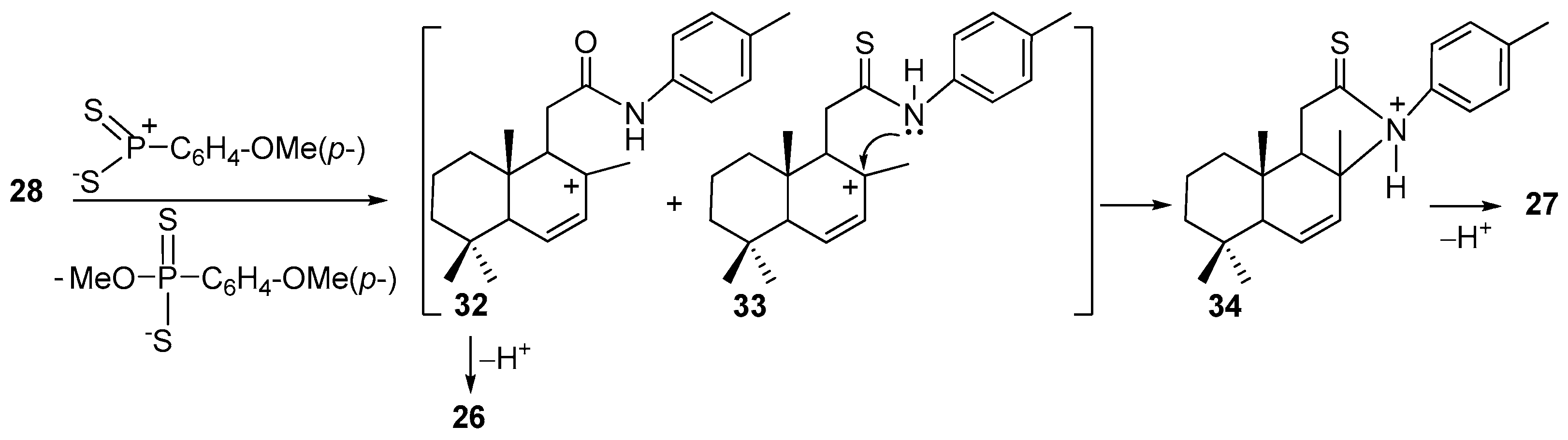
Compound 26. (256 mg, 76%), white crystals, mp 69–70 °C, −155.56 (c 0.69, CHCl3). IR spectrum, ν, cm−1: 817, 1177, 1607, 1245, 1351, 1454, 1513, 1542, 1653, 2927, 3292. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, s, 10-CH3), 0.97 (3H, s, 4-CH3), 0.99 (3H, s, 4-CH3), 1.10–1.62 (6H, m, 3CH2), 1.83 (3H, s, 8-CH3), 2.04 (1H, t, J = 2.5 Hz, H-5), 2.32 (3H, s, 4′-CH3), 3.08 (1H, d, J = 16.9 Hz, H-11), 3.31 (1H, d, J = 16.9 Hz, H-11),5.92 (1H, dd, J = 9.4, 2.4 Hz, H-6), 5.97 (1H, dd, J = 9.5, 2.7 Hz, H-7), 7.14 (2H, d, J = 8.2 Hz, H-2′ and H-6′), 7.37 (2H, d, J = 8.4 Hz, H-3′ and H-5′), 7.66 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 15.0 (C-17), 18.4 (C-20), 18.7 (C-2); 20.8 (C-7′); 22.7 (C-18), 32.4 (C-19), 33.0 (C-4), 34.8 (C-1), 36.5 (C-11), 39.1 (C-10), 40.8 (C-3), 53.6 (C-5), 119.9 (C-2′ and C-6′), 128.9 (C-6), 129.4 (C-7), 129.7 (C-3′and C-5′), 129.9 (C-8), 135.1 (C-9), 138.1 (C-1′), 169.0 (C-12). 15N NMR (400 MHz, CDCl3) δ 125. HRMS (ESI) calculated for C23H31NO [M + H]+, 337.2406. Found: 337.2492.
Compound 28. (346 mg, 94%), white solid, mp 187–188 °C, +70.2 (c 0.39, CHCl3). IR spectrum, ν, cm−1: 816, 1086, 1243, 1310, 1448, 1536, 1571, 1624, 2928, 3322. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, s, 10-CH3), 0.93 (3H, s, 4-CH3), 0.95 (3H, s, 4-CH3), 1.10–1.19 (2H, m, CH2), 1.39–1.60 (5H, m, H-5 and 2CH2), 1.78 (3H, s, 8-CH3), 2.00–2.04 (2H, m, CH2), 2.30 (3H, s, 4′-CH3), 3.05 (1H, d, J = 17.6 Hz, H-11), 3.22 (1H, d, J = 17.6 Hz, H-11), 3.39 (3H, s, 7-CH3), 3.48 (1H, d, J = 2.6 Hz, H-7), 7.11 (2H, d, J = 8.2 Hz, H-2′ and H-6′), 7.35 (2H, d, J = 8.4 Hz, H-3′ and H-5′), 7.70 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 18.2 (C-20), 18.4 (C-2), 18.6 (C-17), 20.8 (C-7′), 21.6 (C-18), 22.4 (C-6), 32.8 (C-19), 32.9 (C-4), 35.4 (C-1), 37.0 (C-11), 39.7 (C-10), 41.2 (C-3), 45.9 (7-OCH3), 56.8 (C-5), 79.0 (C-7), 120.1 (C-2′ and C-6′), 129.3 (C-3′ and 5′), 132.0 (C-8), 133.3 (C-4′), 135.1 (C-9), 140.8 (C-1′), 168.6 (C-12). 15N NMR (400 MHz, CDCl3) δ 128. HRMS (ESI) calculated for C24H35NO2 [M-31]+, 369.2668. Found: 338.2492.
Compounds 24 and 27. (Typical procedure)
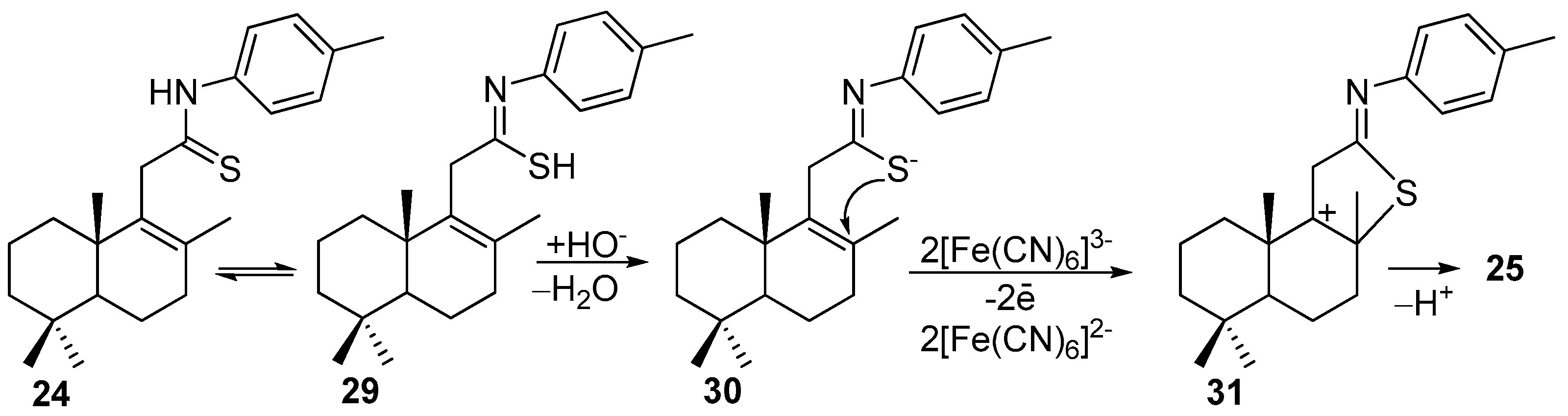
To a solution of one of the amides 23 (339 mg, 1 mmol), 26 (337 mg, 1 mmol) or 28 (369 mg, 1 mmol) dissolved in toluene (8 mL), Lawesson’s reagent (203 mg, 0.5 mmol) was added and the reaction mixture was refluxed for 48–50 h. Then, the mixture was filtered, and the solvent was removed under a reduced pressure on a rotary evaporator to afford the crude reaction product, which was purified by silica gel flash column chromatography (1% MeOH-CH2Cl2).
Compound 24. (177 mg, 50%), white solid, mp 104–105 °C. +45.63 (c 0.5, CHCl3). IR spectrum, ν, cm−1: 730, 826, 852, 908, 998, 1056, 1066, 1267, 1395, 1406, 1453, 1516, 1599, 2052, 2214, 2972, 2987, 3147, 3246. 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, s, 4-CH3), 0.91 (3H, s, 4-CH3), 1.00 (3H, s, 10-CH3), 1.03–1.14 (2H, m, CH2), 1.15 (1H, dd, J = 12.6, 2.0 Hz, H-5), 1.34–1.59 (4H, m, 2CH2), 1.67 (3H, s, 8-CH3), 1.72–1.88 (2H, m, CH2), 2.11–2.23 (2H, m, H-7), 2.36 (3H, s, 4′-CH3), 3.71 (2H, s, H-11), 7.22 (2H, d, J = 8.4 Hz, H-3′ and H-5′), 7.50 (2H, d, J = 8.4 Hz, H-2′ and H-6′), 9.03 (1H, s, NH). 13C NMR (100 MHz, CDCl3) δ 18.7 (C-2), 18.8 (C-6), 20.1 (C-20), 20.2 (C-17), 21.1 (4′-CH3), 21.6 (C-18), 33.2 (C-19), 33.4 (C-4), 33.6 (C-7), 36.2 (C-1), 39.2 (C-10), 41.6 (C-3), 47.8 (C-11), 52.5 (C-5), 123.8 (C-2′ and C-6′), 129.5 (C-3′ and C-5′), 134.2 (C-8), 136.2 (C-4′), 136.8 (C-9), 136.9 (C-1′), 200.9 (C = S). HRMS (ESI) calculated for C23H33NS [M + H]+, 355.2334. Found: 355.2419.
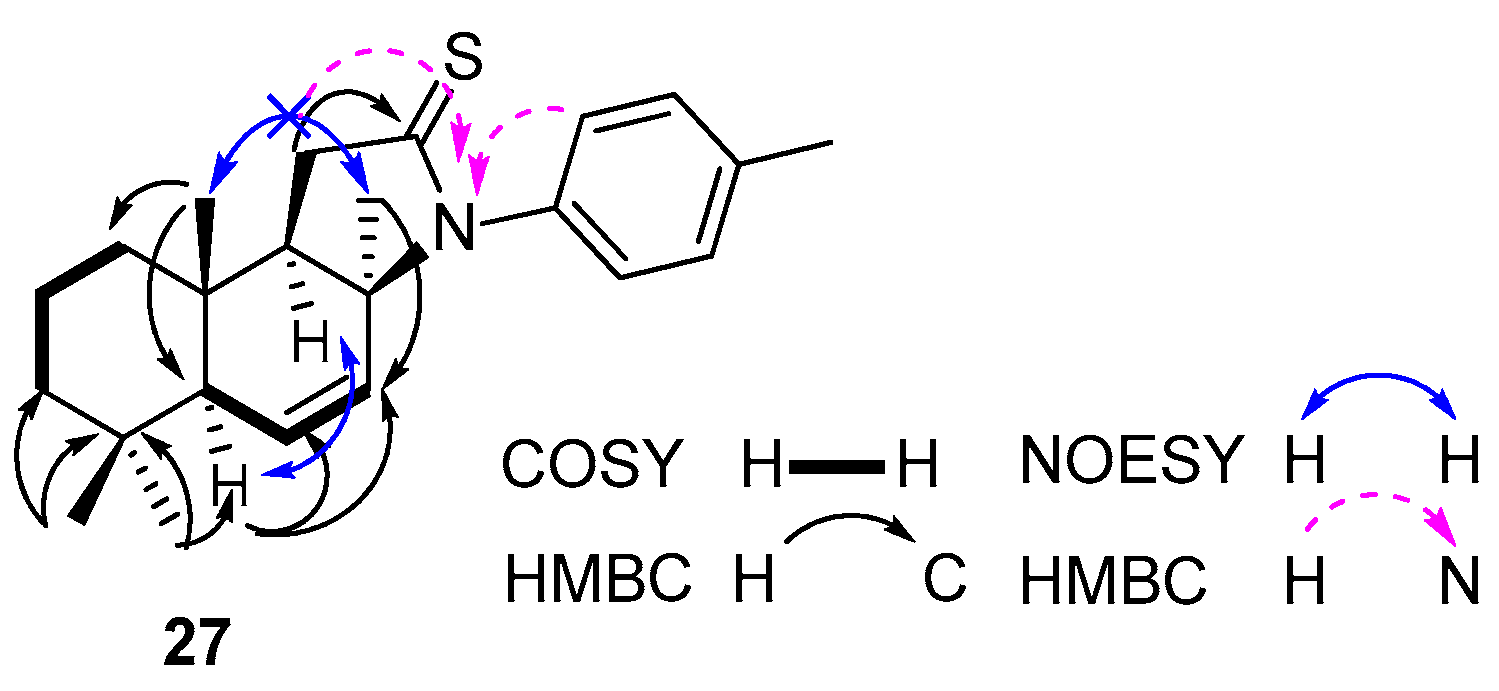
Compound 27. (176 mg, 52%), white solid, mp 103–105 °C. +155.73 (c 0.83, CHCl3). IR spectrum, ν, cm−1: 817, 1033, 1097, 1365, 1454, 1502, 1600, 1625, 2917. 1H NMR (400 MHz, CDCl3) δ 0.82 (3H, s, 4-CH3), 0.87 (3H, s, 4-CH3), 0.92 (3H, s, 10-CH3), 1.08–1.40 (2H, m, CH2), 1.46–1.55 (2H, m, CH2), 1.56 (3H, s, 8-CH3), 1.64–1.72 (2H, m, CH2), 1.88 (1H, d, J = 8.2 Hz, H-9), 2.23 (3H, s, H-7′), 3.07 (1H, d, J = 17.5 Hz, H-11), 3.19 (1H, dd, J = 17.5, 8.2 Hz, H-11), 5.62 (1H, dd, J = 10.1, 2.1 Hz, H-6), 5.66 (1H, dd, J = 10.1, 1.2 Hz, H-7), 6.75 (2H, d, J = 8.0 Hz, H-2′ and H-6′), 7.11 (2H, d, J = 8.0 Hz, H-3′ and H-5′). 13C NMR (100 MHz, CDCl3) δ 14.5 (C-20), 18.2 (C-2), 20.9 (C-7′), 21.6 (C-18), 32.5 (C-4), 33.9 (C-19), 33.9 (C-17), 37.6 (C-1), 37.7 (C-10), 40.7 (C-11), 40.9 (C-3), 51.9 (C-5), 58.2 (C-9), 59.2 (C-8), 119.9 (C-2′ and C-6′), 126.8 (C-6), 129.6 (C-3′ and C-5′), 130.6 (C-7), 133.4 (C-4′), 149.8 (C-1′), 175.9 (C-12). 15N NMR (400 MHz, CDCl3) δ 293. HRMS (ESI) calculated for C23H31NS [M + H]+, 353.5677. Found: 353.5748.
Compound 25. To a solution of carbothioamide 24 (355 mg, 1 mmol) dissolved in EtOH (9 mL), 30% NaOH (1 mL, 7.9 mmol) was added. The mixture was diluted with EtOH (20 mL) to give 10% NaOH. Portions of this mixture were added to a stirred solution of K3[Fe(CN)6] (1.3 g, 3.9 mmol) in H2O (2 mL) at 85 °C. The resultant mixture was further heated at 85 °C for 5 h and filtered to isolate a light yellow solid (720 mg) K4[Fe(CN)6]·3H2O. Then, the solvent was removed in vacuo from the filtrate. To the residue, H2O (20 mL) was added and the obtained mixture was extracted with CH2Cl2 (2 × 30 mL). The combined extracts were washed with H2O (2 × 20 mL), dried over MgSO4, and the solvent was removed to afford an orange oil. The crude reaction product was purified by flash column chromatography (SiO2, elution CH2Cl2) to give compound 25.
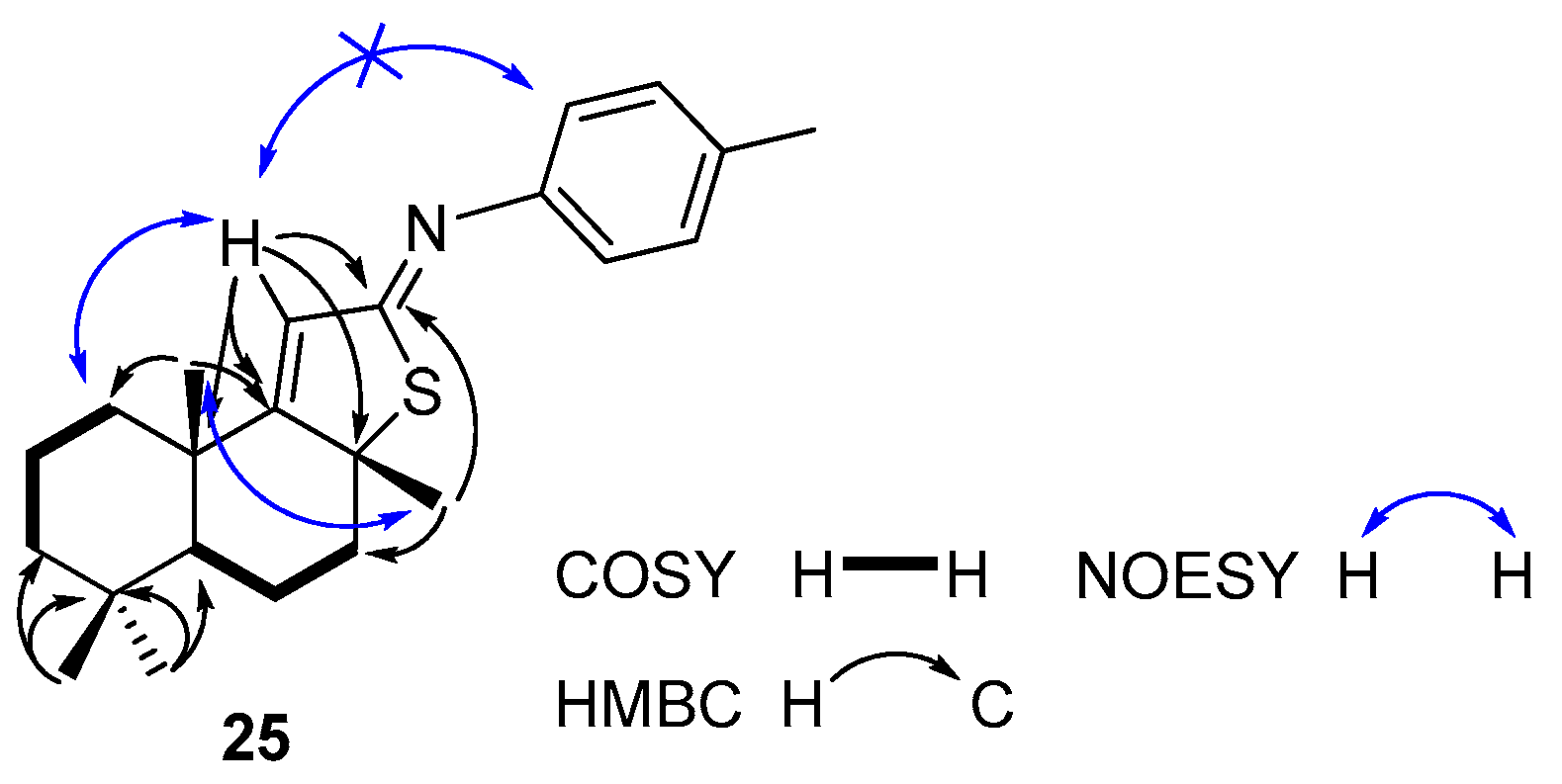
Compound 25. (145 mg, 41%), yellow oil. −92.68 (c 2.0, CHCl3). IR spectrum, ν, cm−1: 730, 824, 851, 909, 1004, 1127, 1147, 1169, 1201, 1249, 1306, 1379, 1451, 1504, 1594, 1618, 2868, 2927. 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, s, 4-CH3), 0.91 (3H, s, 4-CH3), 1.07 (1H, dd, J = 12.5 Hz, J = 2.5 Hz, H-5), 1.19–1.22 (1H, m, CH2), 1.23 (3H, s, 10-CH3), 1.42–1.50 (2H, m, CH2), 1.58–1.65 (2H, m, CH2), 1.72 (3H, s, 8-CH3), 1.78–1.95 (4H, m, 2CH2), 2.22–2.27 (1H, m, CH2), 2.33 (3H, s, 4′-CH3), 6.19 (1H, s, H-11), 6.98 (2H, d, J = 8.0 Hz, H-3′ and H-5′), 7.14 (2H, d, J = 8.0 Hz, H-2′ and H-6′). 13C NMR (100 MHz, CDCl3) δ 18.6 (C-6); 19.6 (C-20), 19.9 (C-2), 21.0 (C-7′), 21.6 (C-18), 29.4 (C-17), 33.3 (C-19), 34.0 (C-4), 39.0 (C-1), 41.2 (C-10), 41.7 (C-3), 43.2 (C-7), 55.2 (C-5), 62.9 (C-8), 120.5 (C-2′ and C-6′), 123.6 (C-11), 129.6 (C-3′ and C-5′), 134.0 (C-4′), 148.9 (C-1′), 170.4 (C-9), 176.6 (C-12). 15N NMR (400 MHz, CDCl3) δ 191. HRMS (ESI) calculated for C23H31NS [M + H]+, 353.5677. Found: 353.5725.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}