
2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization




Abstract
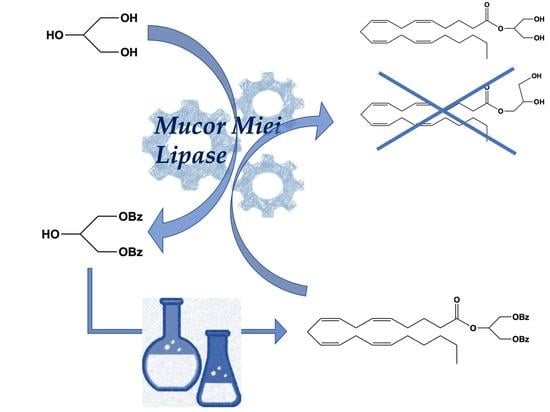
:
1. Introduction
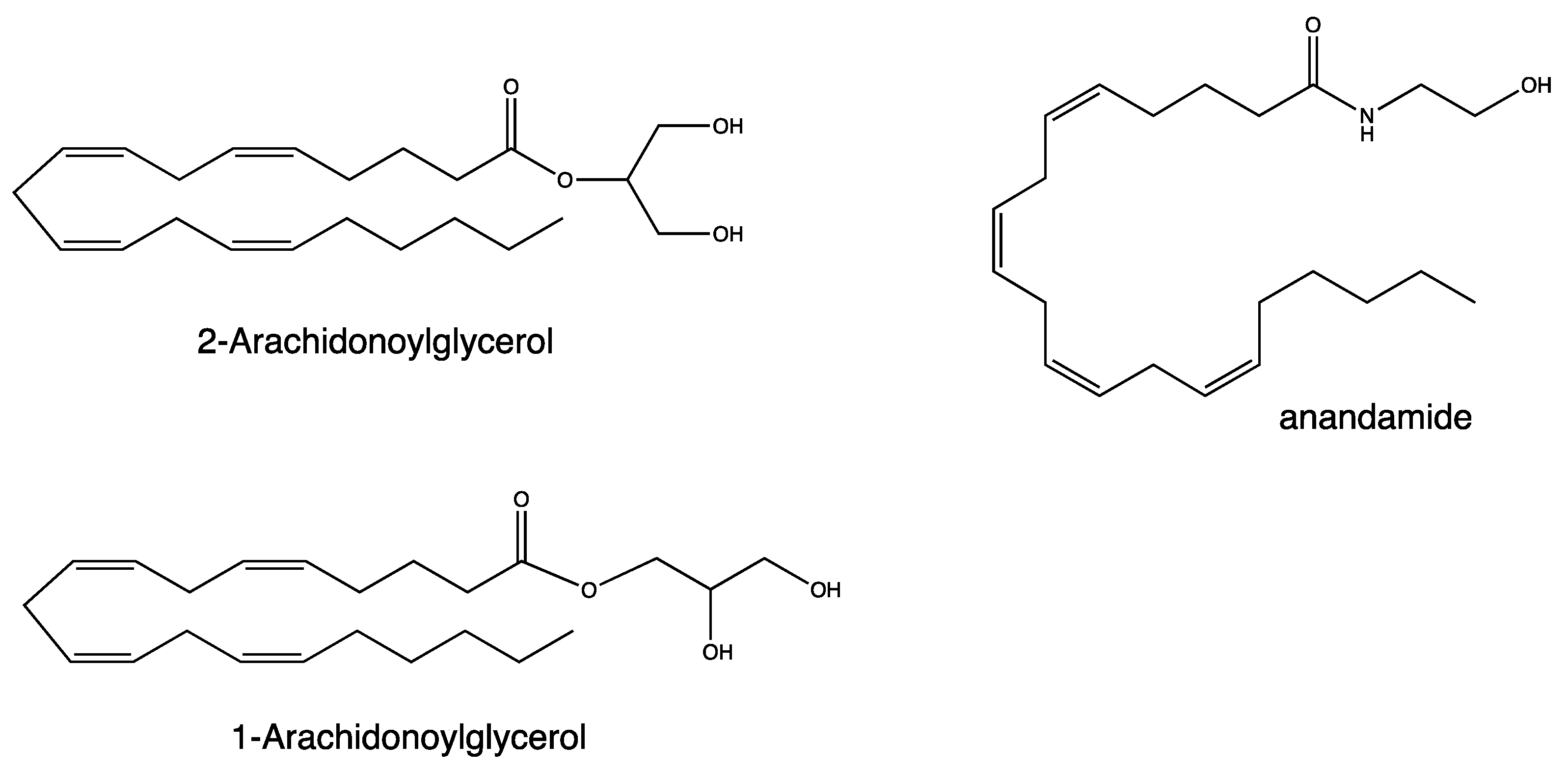
2. Results and Discussion
3. Material and Methods
3.1. General Information

3.2. NMR Analysis
3.3. Synthesis and Purification of Glycerol Mono-, Di-, and Tri-Benzoate as Standard for Gas Chromatography Analyses
3.4. Gas Chromatography Separation of Glycerol Mono-, Di-, and Tri-Benzoate
3.5. Synthesis of 2-Hydroxypropane-1,3-diyl dibenzoate (1c)
3.6. Synthesis of 2-((5Z,8Z,11Z,14Z)-Icosa-5,8,11,14-tetraenoyloxy)propane-1,3-diyl dibenzoate (2)
3.7. Synthesis of (5Z,8Z,11Z,14Z)-1,3-Dihydroxypropan-2-yl Icosa-5,8,11,14-tetraenoate (3)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pertwee, R.G. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict. Biol. 2008, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Vago, R.; Ravelli, A.; Bettiga, A.; Casati, S.; Lavorgna, G.; Benigni, F.; Salonia, A.; Montorsi, F.; Orioli, M.; Ciuffreda, P.; et al. Urine Endocannabinoids as Novel Non-Invasive Biomarkers for Bladder Cancer at Early Stage. Cancers 2020, 12, 870. [Google Scholar] [CrossRef] [PubMed]
- Pezzilli, R.; Ciuffreda, P.; Ottria, R.; Ravelli, A.; Melzi d’Eril, G.; Barassi, A. Serum endocannabinoids in assessing pain in patients with chronic pancreatitis and in those with pancreatic ductal adenocarcinoma. Scand. J. Gastroenterol. 2017, 52, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Bersani, G.; Pacitti, F.; Iannitelli, A.; Caroti, E.; Quartini, A.; Xenos, D.; Marconi, M.; Cuoco, V.; Bigio, B.; Bowles, N.P.; et al. Inverse correlation between plasma 2-arachidonoylglycerol levels and subjective severity of depression. Hum. Psychopharmacol. 2021, 36, e2779. [Google Scholar] [CrossRef]
- Marchioni, C.; de Souza, I.D.; Acquaro, V.R.; de Souza, C.J.A.; Tumas, V.; Queiroz, M.E.C. Recent advances in LC-MS/MS methods to determine endocannabinoids in biological samples: Application in neurodegenerative diseases. Anal. Chim. Acta 2018, 1044, 12–28. [Google Scholar] [CrossRef]
- Ottria, R.; Cappelletti, L.; Ravelli, A.; Mariotti, M.; Gigli, F.; Romagnosi, S.; Ciuffreda, P.; Banfi, G.; Drago, L. Plasma Endocannabinoids behavior in Total Knee and Hip Arthroplasty. J. Biol. Regul. Homeost. Agent 2016, 30, 1147–1152. [Google Scholar] [CrossRef]
- Lauria, S.; Perrotta, C.; Casati, S.; Di Renzo, I.; Ottria, R.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Design, synthesis, molecular modelling and in vitro cytotoxicity analysis of novel carbamate derivatives as inhibitors of Monoacylglycerol lipase. Bioorg. Med. Chem. 2018, 26, 2561–2572. [Google Scholar] [CrossRef]
- Vago, R.; Bettiga, A.; Salonia, A.; Ciuffreda, P.; Ottria, R. Development of new inhibitors for N-acylethanolamine-hydrolyzing acid amidase as promising tool against bladder cancer. Bioorg. Med. Chem. 2017, 25, 1242–1249. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cordaro, M.; Siracusa, R.; Casili, G.; Evangelista, M.; Cuzzocrea, S. N-palmitoylethanolamide Prevents Parkinsonian Phenotypes in Aged Mice. Mol. Neurobiol. 2018, 55, 8455–8472. [Google Scholar] [CrossRef]
- Finn, D.P.; Haroutounian, S.; Hohmann, A.G.; Krane, E.; Soliman, N.; Rice, A.S.C. Cannabinoids, the endocannabinoid system, and pain: A review of preclinical studies. Pain 2021, 162, S5–S25. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; Grenald, S.A.; Ciccone, H.A.; BassiriRad, N.; Niphakis, M.J.; Cravatt, B.F.; Largent-Milnes, T.M.; Vanderah, T.W. The endocannabinoid system alleviates pain in a murine model of cancer-induced bone pain. J. Pharmacol. Exp. Ther. 2020, 373, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Giannasi, C.; Minoli, M.; Niada, S.; Ravelli, A.; Angeli, I.; Mergenthaler, V.; Ottria, R.; Ciuffreda, P.; Orioli, M.; et al. Quantitative lipidomic analysis of osteosarcoma cell-derived products by UHPLC-MS/MS. Biomolecules 2020, 10, 1302. [Google Scholar] [CrossRef]
- Compton, D.L.; Vermillion, K.E.; Laszlo, J.A. Acyl migration kinetics of 2-monoacylglycerols from soybean oil via 1H NMR. J. Amer. Oil Chem. Soc. 2007, 84, 343–348. [Google Scholar] [CrossRef]
- Boswinkel, G.; Derksen, J.T.P.; van’t Riet, K.; Cuperus, F.P. Kinetics of acyl migration in monoglycerides and dependence on acyl chainlength. J. Amer. Oil Chem. Soc. 1996, 73, 707–711. [Google Scholar] [CrossRef]
- Stamatov, S.D.; Stawinski, J. Novel, regioselective transformation of an oxirane system. An efficient approach to the synthesis of endocannabinoid 2-arachidonoylglycerol. Tetrahedron Lett. 2002, 43, 1759–1761. [Google Scholar] [CrossRef]
- Cartoni, A.; Margonelli, A.; Angelini, G.; Finazzi-Agrò, A.; Maccarrone, M. Simplified chemical and radiochemical synthesis of 2-arachidonoyl-glycerol, an endogenous ligand of cannabinoid receptors. Tetrahedron Lett. 2004, 45, 2723–2726. [Google Scholar] [CrossRef]
- Suhara, Y.; Takayama, H.; Nakane, S.; Miyashita, T.; Waku, K.; Sugiura, T. Synthesis and biological activities of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, and its metabolically stable ether-linked analogues. Chem. Pharm. Bull. 2000, 48, 903–907. [Google Scholar] [CrossRef]
- Roche, M.J.; Madren, S.M.; Tallent, C.R.; Carroll, F.I.; Seltzman, H.H. Mild acetal cleavage using B-chlorocatecholborane in the synthesis of rearrangement-sensitive 2-arachidonoylglycerol. Tetrahedron Lett. 2012, 53, 3825–3827. [Google Scholar] [CrossRef]
- Whitten, K.M.; Makriyannis, A.; Vadivel, S.K. Application of chemoenzymatic hydrolysis in the synthesis of 2-monoacylglycerols. Tetrahedron 2012, 68, 5422–5428. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, S.K.; Whitten, K.M.; Makriyannis, A. Chemoenzymatic synthesis of 2-arachidonoylglycerol, an endogenous ligand for cannabinoid receptors. Tetrahedron Lett. 2011, 52, 1149–1150. [Google Scholar] [CrossRef]
- Duclos, R.I.; Johnston, M.; Vadivel, S.K.; Makriyannis, A.; Glaser, S.T.; Gatley, S.J. A methodology for radiolabeling of the endocannabinoid 2-arachidonoylglycerol (2-AG). J. Org. Chem. 2011, 76, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, M.; Wang, T.; Jin, Q.; Wang, X. An improved method for the synthesis of 2-arachidonoylglycerol. Process Biochem. 2014, 49, 1415–1421. [Google Scholar] [CrossRef]
- Han, L.; Razdan, R.K. Total synthesis of 2-Arachidonylglycerol (2-Ara-Gl). Tetrahedron Lett. 1999, 40, 1631–1634. [Google Scholar] [CrossRef]
- Martin, J.B. Preparation of saturated and unsaturated symmetrical monoglycerides. J. Am. Chem. Soc. 1953, 75, 5482–5483. [Google Scholar] [CrossRef]
- Seltzman, H.H.; Fleming, D.N.; Hawkins, G.D.; Carroll, F. Facile synthesis and stabilization of 2-arachidonylglycerol via its 1,3-phenylboronate ester. Tetrahedron Lett. 2000, 41, 3589–3592. [Google Scholar] [CrossRef]
- Thangaraj, B.; Solomon, P.R. Immobilization of Lipases—A ReviewPart I: Enzyme Immobilization. ChemBioEng Rev. 2019, 6, 157–166. [Google Scholar] [CrossRef]
- Mulinari, J.; Oliveira, J.V.; Hotza, D. Lipase immobilization on ceramic supports: An overview on techniques and Materials. Biotechnol. Adv. 2020, 42, 107581. [Google Scholar] [CrossRef]
- Rafiee, F.; Rezaee, M. Different strategies for the lipase immobilization on the chitosan based supports and their applications. Int. J. Bio. Macromol. 2021, 179, 170–195. [Google Scholar] [CrossRef] [PubMed]
- Rodriguesa, R.C.; Virgen-Ortízb, J.J.; dos Santosc, J.C.; Berenguer-Murciad, Á.; Alcantarae, A.R.; Barbosaf, O.; Ortizg, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Advan. 2019, 37, 746–770. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Rota, P.; Allevi, P.; Mingione, A.; Ottria, R.; Ciuffreda, P. Clarifying the use of benzylidene protecting group for d-(+)-ribono-1,4-lactone, an essential building block in the synthesis of c-nucleosides. Molecules 2021, 26, 6447. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley: Hoboken, NJ, USA, 2006; ISBN 978-0-470-05348-5. [Google Scholar]
- Santaniello, E.; Casati, S.; Ciuffreda, P.; Gamberoni, L. Lipase-catalyzed alcoholysis of diol dibenzoates: Selective enzymatic access to the 2-benzoyl ester of 1,2-propanediol and preparation of the enantiomerically pure (R)-1-O-benzoyl-2-methylpropane-1,3-diol. Tetrahedron Asymmetry 2005, 16, 1705–1708. [Google Scholar] [CrossRef]
- Ciuffreda, P.; Casati, S.; Santaniello, E. Lipase-catalyzed monoprotection of 1,4-diols in an organic solvent using vinyl benzoate as acyl transfer agent. Tetrahedron Lett. 2003, 44, 3663–3665. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Jin, Q.; Wang, T. Improved synthesis of monopalmitin on a large scale by two enzymatic methods. J. Amer. Oil Chem. Soc. 2013, 90, 1455–1463. [Google Scholar] [CrossRef]
- Li, L.; Du, W.; Liu, D.; Wang, L.; Li, Z. Lipase-catalyzed transesterification of rapeseed oils for biodiesel production with a novel organic solvent as the reaction medium. J. Mol. Catal. B-Enzym 2006, 43, 58–62. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Derewenda, U.; Derewenda, Z.S.; Dodson, G.G.; Lawson, D.M.; Turkenburg, J.P.; Bjorkling, F.; Huge-Jensen, B.; Patkar, S.A.; Thim, L. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature 1991, 351, 491–494. [Google Scholar] [CrossRef]
- Huge-Jensen, B.; Galluzzo, D.R.; Jensen, R.G. Partial purification and characterization of free and immobilized lipases from Mucor miehei. Lipids 1987, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Handayani, N.; Loos, K.; Wahyuningrum, D.; Zulfikar, M.A. Immobilization of Mucor miehei lipase onto macroporous aminated polyethersulfone membrane for enzymatic reactions. Membranes 2012, 2, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, J.; Šmelcerović, A.; Zvezdanović, J.; Yancheva, D.; Casati, S.; Ottria, R.; Ciuffreda, P. Lipid peroxidation inhibition study: A promising case of 1,3-di(1,1’-biphenyl-3-yl)urea. Chem. Biol. Interact. 2020, 326, 109137. [Google Scholar] [CrossRef]
- Ottria, R.; Casati, S.; Ciuffreda, P. Optimized synthesis and characterization of N-acylethanolamines and O-acylethanolamines, important family of lipid-signalling molecules. Chem. Phys. Lip. 2012, 165, 705–711. [Google Scholar] [CrossRef]
- Miceli, M.; Casati, S.; Ottria, R.; Di Leo, S.; Eberini, I.; Palazzolo, L.; Parravicini, C.; Ciuffreda, P. Set-Up and validation of a high throughput screening method for Human Monoacylglycerol Lipase (MAGL) Based on a new red fluorescent probe. Molecules 2019, 24, 2241. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Ottria, R.; Ciuffreda, P. Simple Synthesis of 17-β-O-hemisuccinate of stanozolol for immunoanalytical methods. Molecules 2020, 25, 2019. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DIOX | DIOX-DCM | THF | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1a | 1b | 1c | 1d | 1a | 1b | 1c | 1d | |
| MML | 7 | 80 | 13 | - | 8 | 90 | 2 | - | 10 | 51 | 39 | - |
| CCL | 16 | 82 | 2 | - | 21 | 76 | 2 | - | 18 | 58 | 25 | - |
| CAL | 100 | - | - | - | 54 | 38 | 8 | - | 36 | 52 | 18 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottria, R.; Casati, S.; Rota, P.; Ciuffreda, P. 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules 2022, 27, 5190. https://doi.org/10.3390/molecules27165190
Ottria R, Casati S, Rota P, Ciuffreda P. 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules. 2022; 27(16):5190. https://doi.org/10.3390/molecules27165190
Chicago/Turabian StyleOttria, Roberta, Silvana Casati, Paola Rota, and Pierangela Ciuffreda. 2022. "2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization" Molecules 27, no. 16: 5190. https://doi.org/10.3390/molecules27165190
APA StyleOttria, R., Casati, S., Rota, P., & Ciuffreda, P. (2022). 2-Arachidonoylglycerol Synthesis: Facile and Handy Enzymatic Method That Allows to Avoid Isomerization. Molecules, 27(16), 5190. https://doi.org/10.3390/molecules27165190