
Drought Stress Stimulates the Terpenoid Backbone and Triterpenoid Biosynthesis Pathway to Promote the Synthesis of Saikosaponin in Bupleurum chinense DC. Roots


Abstract
:1. Introduction
2. Results
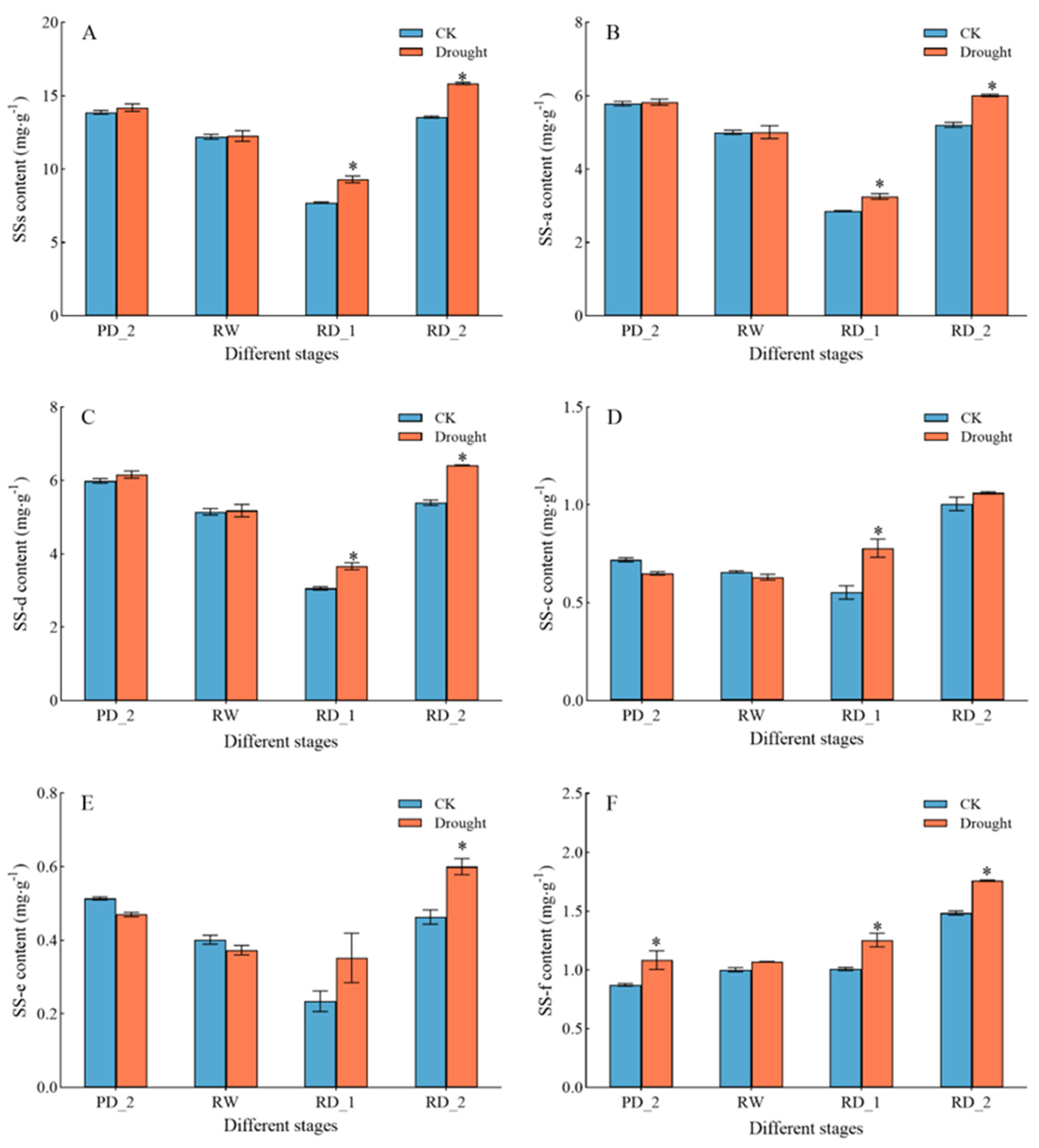
2.1. Saikosaponin Content
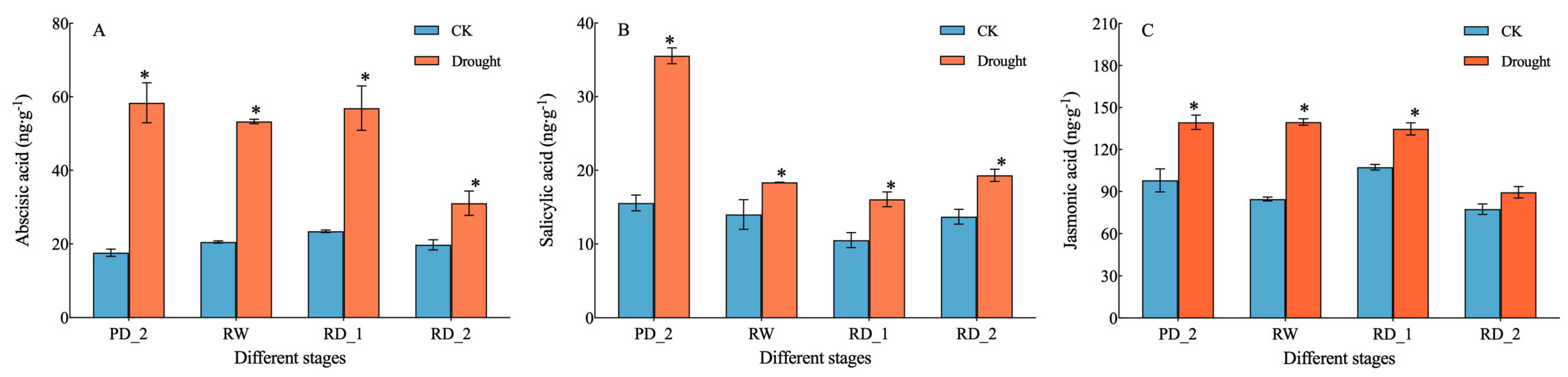
2.2. Endogenous Hormone Content
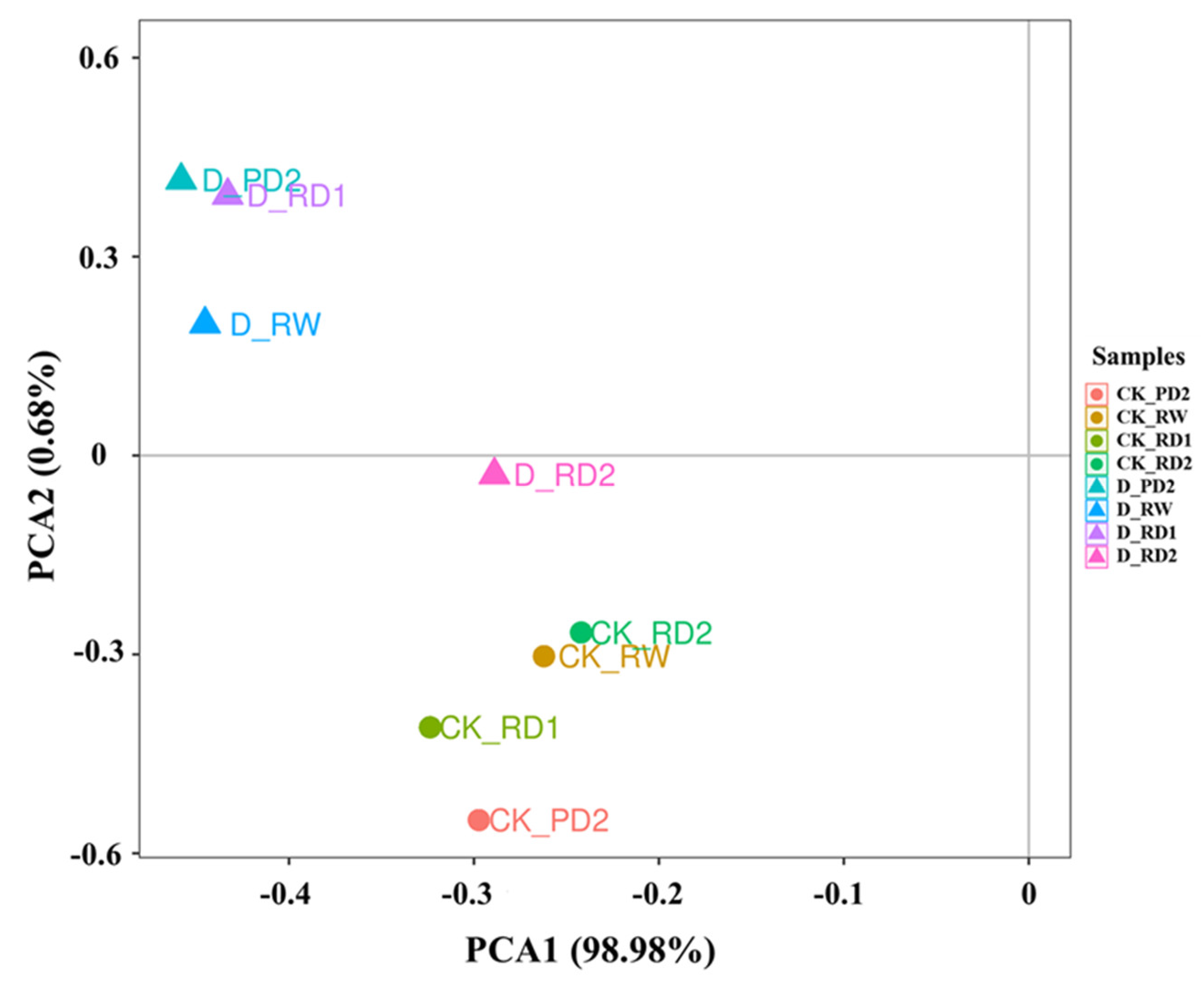
2.3. Principal Component Analysis (PCA) of Responses to Drought Stress
2.4. Sequencing Data and Annotation Analysis of B. chinense under Drought Stress
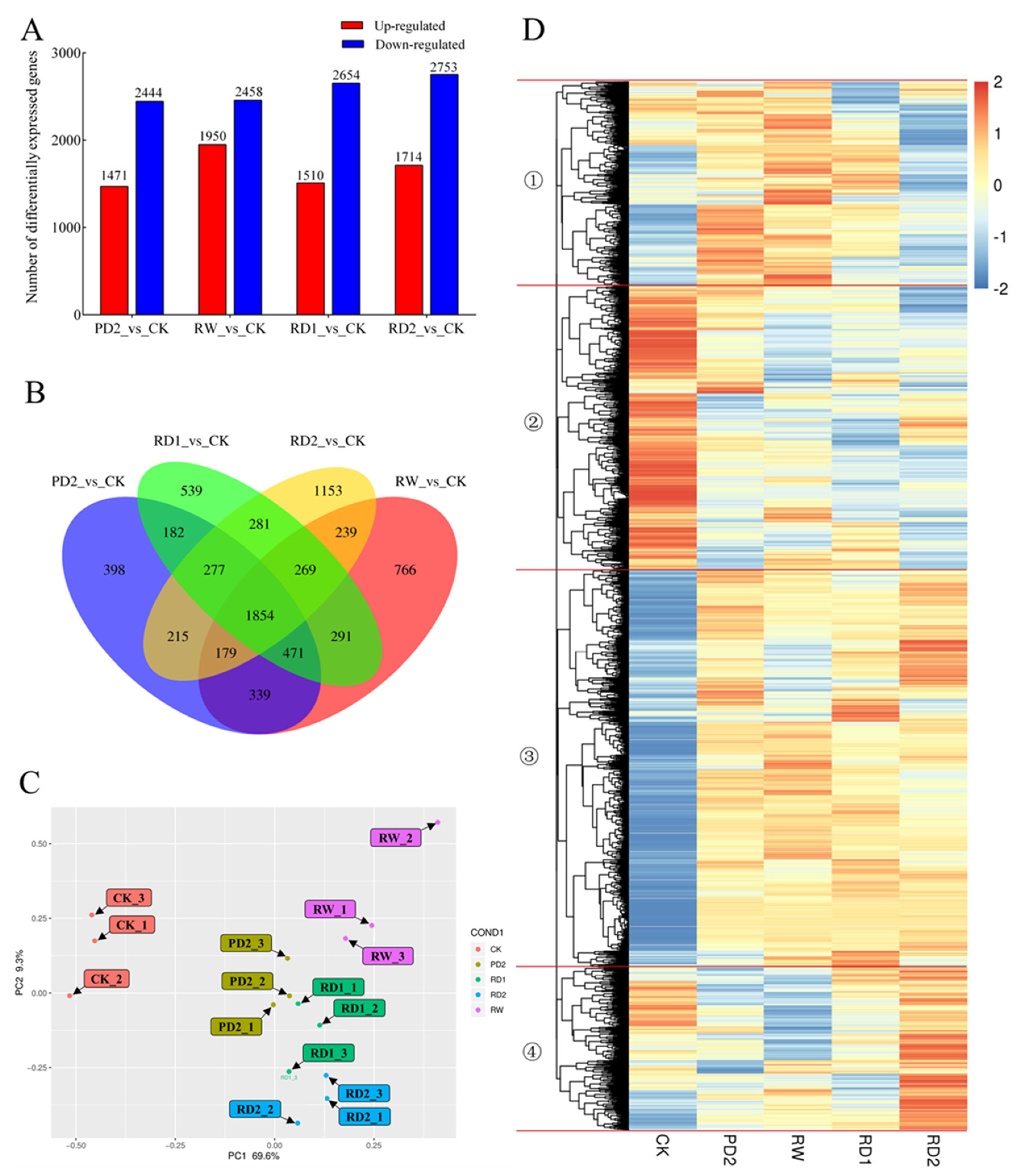
2.5. Analysis of DEGs among Different Samples of B. chinense under Drought Stress
2.6. GO and KEGG Functional Annotation and Classification of B. chinense under Drought Stress
2.7. KEGG Enrichment of MAPK Signaling Pathway—Plant and Plant Hormone Signal Transduction
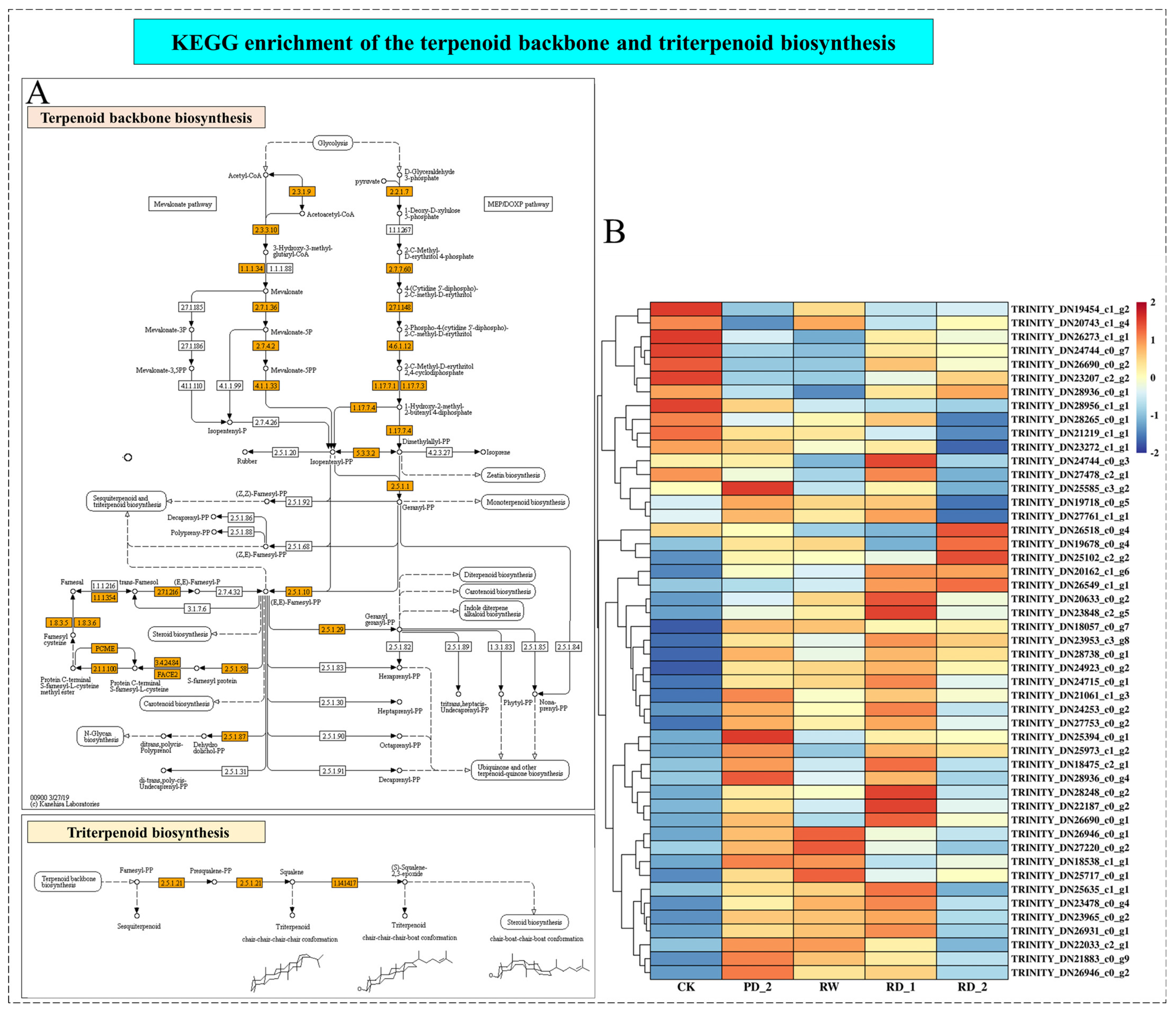
2.8. Analysis of Terpenoid Backbone and Triterpenoid Biosynthesis of B. chinense under Drought Stress
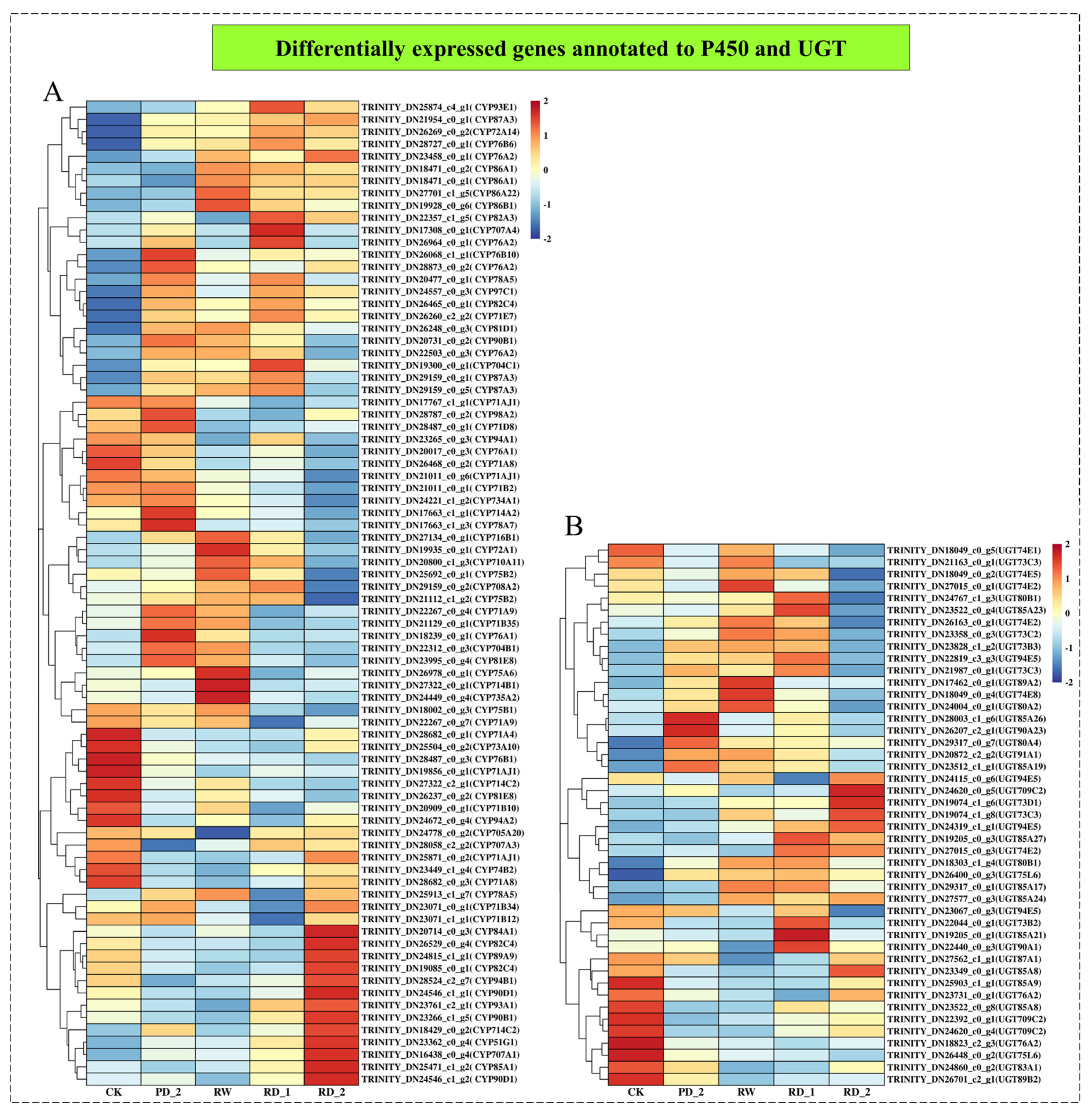
2.9. Analysis of Differentially Expressed Genes Encoding P450 and UGT
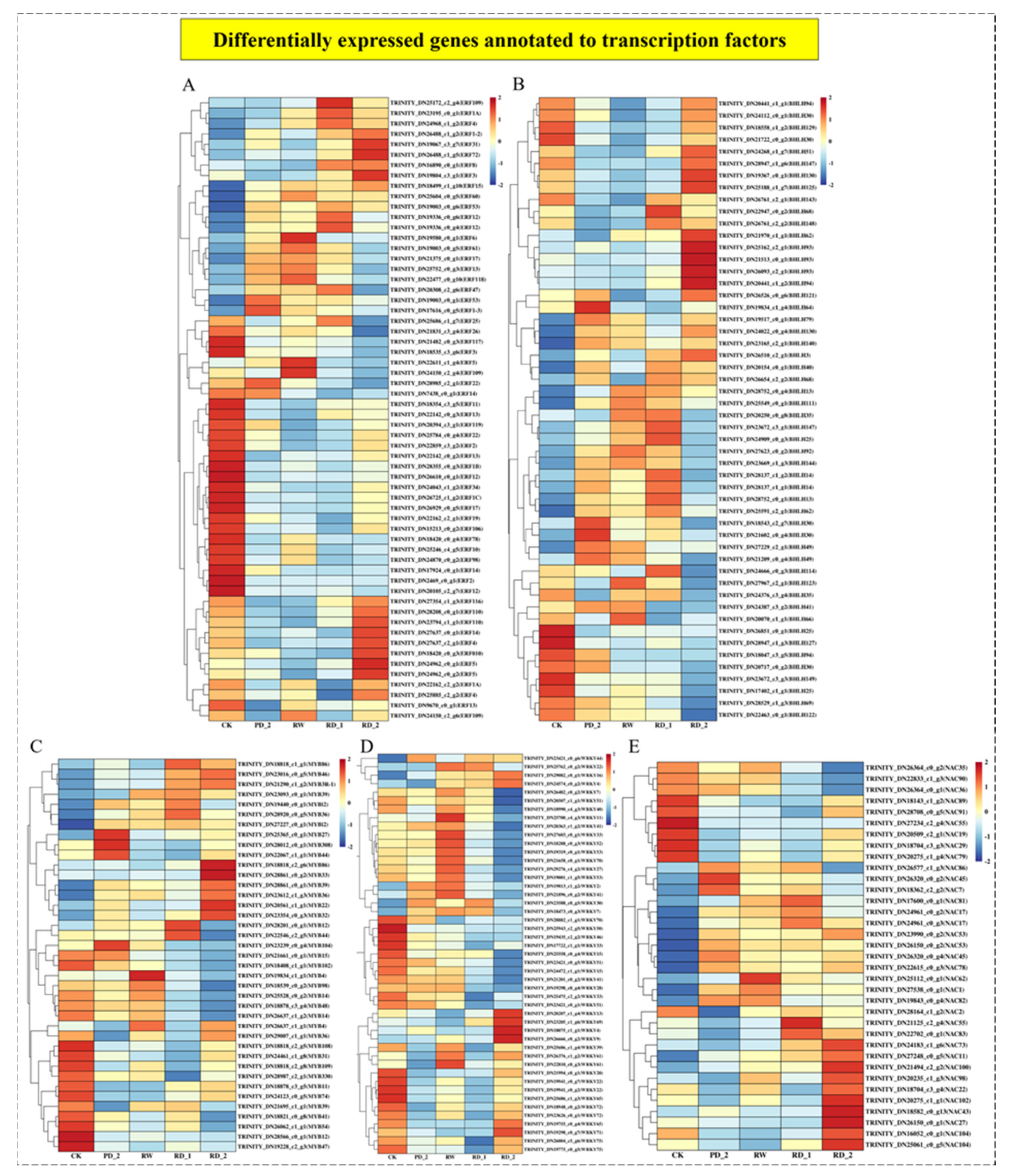
2.10. Analysis of Differentially Expressed Genes of Transcription Factors
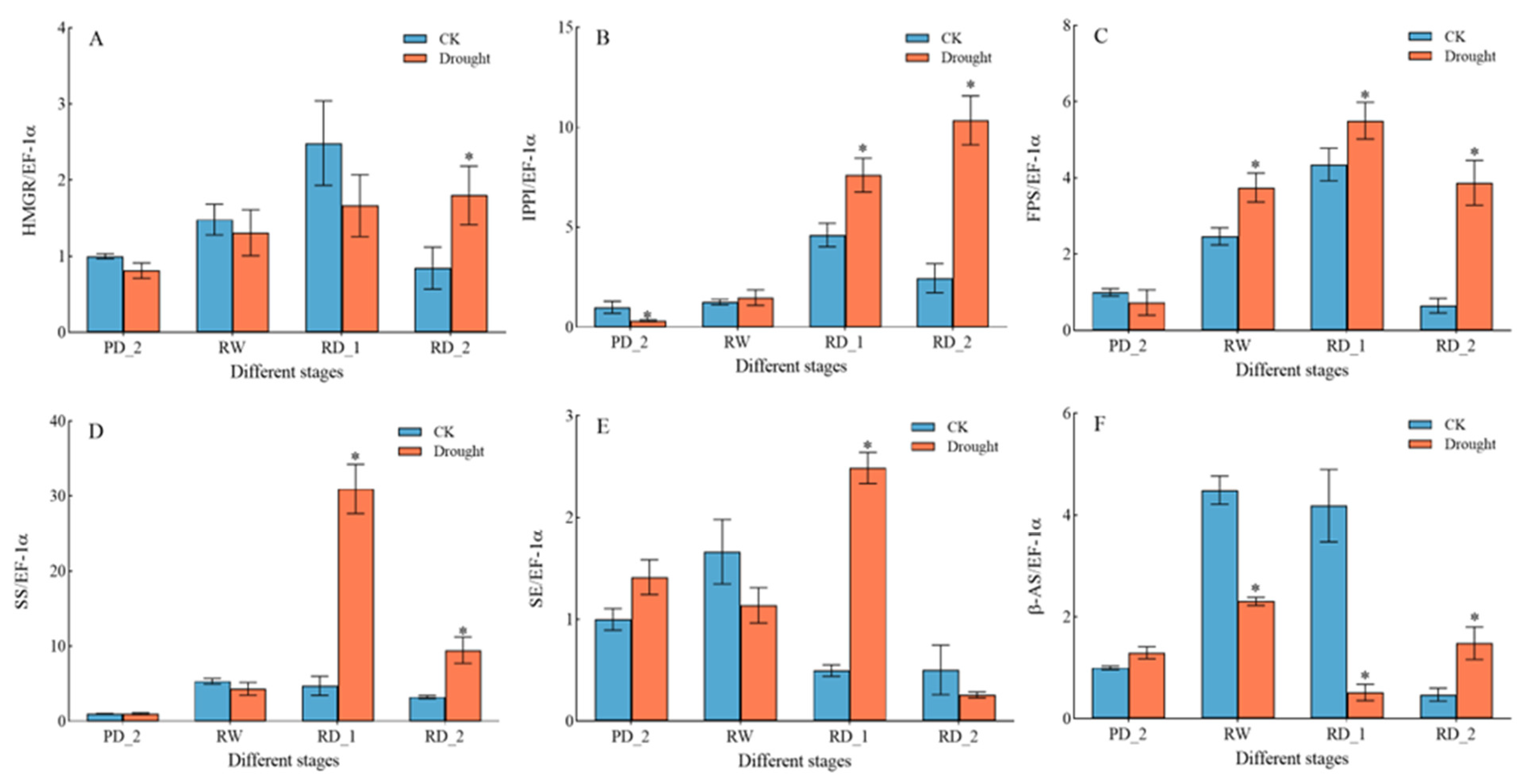
2.11. Expression of Key Genes Encoding Saikosaponin Biosynthesis Enzymes in Response to Drought Stress
2.12. Screening of Transcription Factors Involved in Saikosaponin Synthesis Regulation
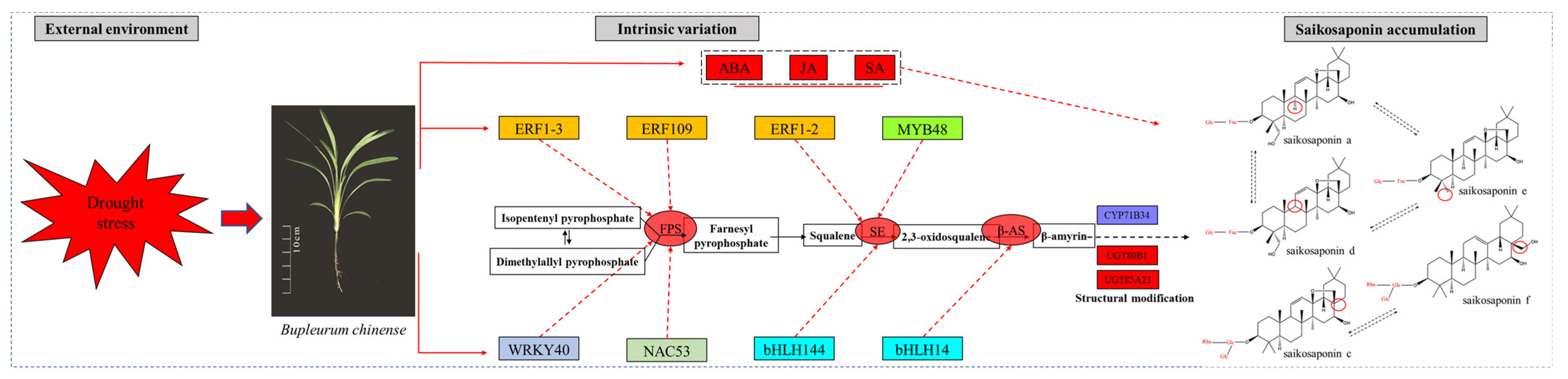
2.13. Preliminary Construction of the Saikosaponin Synthesis Regulation Network
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Experimental Treatment
5.2. Determination of Saikosaponin Content in Roots
5.3. Determination of Endogenous Hormone Content
5.4. Identification and Functional Annotation of Differentially Expressed Genes
5.4.1. Total RNA Extraction and Sequencing
5.4.2. De Novo Assembly, Unigene Annotation, and Functional Classification
5.4.3. Differentially Expressed Gene (DEG) Analysis
5.5. Determination of Key Genes in the Saikosaponin Synthesis Pathway
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shahrajabian, M.H.; Sun, W.; Cheng, Q. Traditional Chinese medicine and agriculture; organic life and sustainability for future. GSC Biol. Pharm. Sci. 2019, 7, 91–95. [Google Scholar]
- Tian, R.-T.; Xie, P.-S.; Liu, H.-P. Evaluation of traditional Chinese herbal medicine: Chaihu (Bupleuri Radix) by both high-performance liquid chromatographic and high-performance thin-layer chromatographic fingerprint and chemometric analysis. J. Chromatogr. 2009, 1216, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liang, Z.-T.; Yi, T.; Ma, Y.; Zhao, Z.-Z.; Guo, B.-L.; Zhang, J.-Y.; Chen, H.-B. Comparison of chemical profiles between the root and aerial parts from three Bupleurum species based on a UHPLC-QTOF-MS metabolomics approach. BMC Complement. Altern. Med. 2017, 17, 305. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.-L.; Zhang, A.-H.; Wang, X.-J. Traditional Chinese medicine for COVID-19 treatment. Pharmacol. Res. 2020, 155, 104743. [Google Scholar] [CrossRef]
- Mohiuddin, A. Environmental impact of secondary metabolism on medicinal plants. J. Glob. Biosci. 2019, 8, 5890–5899. [Google Scholar]
- Liu, Z.; Yang, L.; Zhang, Y.; Han, M.; Lin, H.; Yang, L. Effects of soil factors on saikosaponin content of Bupleurum chinense in different habitats. Chin. Tradit. Herb. Drugs 2020, 51, 5328–5336. [Google Scholar]
- Huang, L.; Xiao, P.; Guo, L.; Gao, W. Molecular pharmacognosy. Sci. China Life Sci. 2010, 53, 643–652. [Google Scholar] [CrossRef]
- Wentao, Q.; Jianchao, L.; Chen, W.; Rong, L.; Changli, L. Analysis of Seed Characteristics and Plant Growth Status of Bupleurum chinense From Different Geographical Provenances. J. Agric. Sci. Technol. 2020, 22, 68. [Google Scholar]
- Li, D.; Yue, D.; Liu, D.; Liu, X.; Song, S. Chemical constituents from Bupleurum chinese and their chemotaxonomic significance. Biochem. Syst. Ecol. 2019, 86, 103929. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Mishra, P.; Mishra, A.K. Effect of climate change on plant secondary metabolism: An ecological perspective. In Evolutionary Diversity as a Source for Anticancer Molecules; Elsevier: Amsterdam, The Netherlands, 2021; pp. 47–76. [Google Scholar]
- Sui, C.; Han, W.-J.; Zhu, C.-R.; Wei, J.-H. Recent Progress in Saikosaponin Biosynthesis in Bupleurum. Curr. Pharm. Biotechnol. 2021, 22, 329–340. [Google Scholar] [CrossRef]
- Ramawat, K.; Dass, S.; Mathur, M. The chemical diversity of bioactive molecules and therapeutic potential of medicinal plants. In Herbal Drugs: Ethnomedicine to Modern Medicine; Springer: Berlin/Heidelberg, Germany, 2009; pp. 7–32. [Google Scholar]
- Parra, A.; Rivas, F.; Garcia-Granados, A.; Martinez, A. Microbial transformation of triterpenoids. Mini Rev. Org. Chem. 2009, 6, 307–320. [Google Scholar] [CrossRef]
- Seki, H.; Tamura, K.; Muranaka, T. P450s and UGTs: Key players in the structural diversity of triterpenoid saponins. Plant Cell Physiol. 2015, 56, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Chiou, C.-Y.; Chiou, S.-J. Putative genes involved in saikosaponin biosynthesis in Bupleurum species. Int. J. Mol. Sci. 2013, 14, 12806–12826. [Google Scholar] [CrossRef] [PubMed]
- Szakiel, A.; Pączkowski, C.; Henry, M. Influence of environmental abiotic factors on the content of saponins in plants. Phytochem. Rev. 2011, 10, 471–491. [Google Scholar] [CrossRef]
- Yang, L.-L.; Yang, L.; Lan, Y.-M.; Zhao, Y.; Han, M.; Yang, L.-M. Exogenous abscisic acid reduces saikosaponin accumulation by inhibiting saikosaponin synthesis pathway gene expression under drought stress in Bupleurum chinense DC. Ind. Crops Prod. 2020, 154, 112686. [Google Scholar] [CrossRef]
- Tripathi, S.; Jadaun, J.S.; Chandra, M.; Sangwan, N.S. Medicinal plant transcriptomes: The new gateways for accelerated understanding of plant secondary metabolism. Plant Genet. Resour. 2016, 14, 256–269. [Google Scholar] [CrossRef]
- Zhao, N.; Wang, G.; Norris, A.; Chen, X.; Chen, F. Studying plant secondary metabolism in the age of genomics. Crit. Rev. Plant Sci. 2013, 32, 369–382. [Google Scholar] [CrossRef]
- Li, Y.; Kong, D.; Fu, Y.; Sussman, M.R.; Wu, H. The effect of developmental and environmental factors on secondary metabolites in medicinal plants. Plant Physiol. Biochem. 2020, 148, 80–89. [Google Scholar] [CrossRef]
- Yang, D.; Du, X.; Yang, Z.; Liang, Z.; Guo, Z.; Liu, Y. Transcriptomics, proteomics, and metabolomics to reveal mechanisms underlying plant secondary metabolism. Eng. Life Sci. 2014, 14, 456–466. [Google Scholar] [CrossRef]
- Gandhi, S.G.; Mahajan, V.; Bedi, Y.S. Changing trends in biotechnology of secondary metabolism in medicinal and aromatic plants. Planta 2015, 241, 303–317. [Google Scholar] [CrossRef]
- Sui, C.; Zhang, J.; Wei, J.; Chen, S.; Li, Y.; Xu, J.; Jin, Y.; Xie, C.; Gao, Z.; Chen, H. Transcriptome analysis of Bupleurum chinense focusing on genes involved in the biosynthesis of saikosaponins. BMC Genom. 2011, 12, 539. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Chen, M.; Xu, J.; Wei, J.; Jin, Y.; Xu, Y.; Sun, J.; Gao, K.; Yang, C.; Zhang, Z. Comparison of root transcriptomes and expressions of genes involved in main medicinal secondary metabolites from Bupleurum chinense and Bupleurum scorzonerifolium, the two Chinese official Radix bupleuri source species. Physiol. Plant. 2015, 153, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Reiter, R.J.; Chan, Z. Phytomelatonin: A universal abiotic stress regulator. J. Exp. Bot. 2018, 69, 963–974. [Google Scholar] [CrossRef]
- Yang, C.Q.; Fang, X.; Wu, X.M.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Transcriptional regulation of plant secondary metabolism F. J. Integr. Plant Biol. 2012, 54, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Pant, P.; Pandey, S.; Dall’Acqua, S. The influence of environmental conditions on secondary metabolites in medicinal plants: A literature review. Chem. Biodivers. 2021, 18, e2100345. [Google Scholar] [CrossRef]
- Rungsung, W.; Ratha, K.K.; Dutta, S.; Dixit, A.K.; Hazra, J. Secondary metabolites of plants in drugs discovery. World J. Pharm. Res. 2015, 4, 604–613. [Google Scholar]
- Li, W.-F.; Jiang, J.-G.; Chen, J. Chinese medicine and its modernization demands. Arch. Med. Res. 2008, 39, 246–251. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Y.; Zhang, Q.; Cheng, L.; Han, M.; Ren, Y.; Yang, L. Effects of drought-re-watering-drought on the photosynthesis physiology and secondary metabolite production of Bupleurum chinense DC. Plant Cell Rep. 2019, 38, 1181–1197. [Google Scholar] [CrossRef]
- Cheng, L.; Han, M.; Yang, L.-M.; Li, Y.; Sun, Z.; Zhang, T. Changes in the physiological characteristics and baicalin biosynthesis metabolism of Scutellaria baicalensis Georgi under drought stress. Ind. Crops Prod. 2018, 122, 473–482. [Google Scholar] [CrossRef]
- Moore, B.D.; Andrew, R.L.; Külheim, C.; Foley, W.J. Explaining intraspecific diversity in plant secondary metabolites in an ecological context. New Phytol. 2014, 201, 733–750. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Y.; Yang, T.; Zhang, L.; Xu, S.; Xue, L.; An, L. Diverse signals converge at MAPK cascades in plant. Plant Physiol. Biochem. 2006, 44, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Luan, S. ABA signal transduction at the crossroad of biotic and abiotic stress responses. Plant Cell Environ. 2012, 35, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jun, L.; Juan, W.; Wen-Yuan, G. Advances in biosynthesis of triterpenoid saponins in medicinal plants. Chin. J. Nat. Med. 2020, 18, 417–424. [Google Scholar]
- Jiang, H.; Yang, L.; Hou, A.; Zhang, J.; Wang, S.; Man, W.; Zheng, S.; Yu, H.; Wang, X.; Yang, B. Botany, traditional uses, phytochemistry, analytical methods, processing, pharmacology and pharmacokinetics of Bupleuri Radix: A systematic review. Biomed. Pharmacother. 2020, 131, 110679. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Dong, S.; Zhang, S.; Wei, X.; Xie, Q.; Ding, Q.; Xia, R.; Zhang, X. Chromosome-level reference genome assembly provides insights into aroma biosynthesis in passion fruit (Passiflora edulis). Mol. Ecol. Resour. 2021, 21, 955–968. [Google Scholar] [CrossRef]
- Mishra, M.K.; Singh, G.; Tiwari, S.; Singh, R.; Kumari, N.; Misra, P. Characterization of Arabidopsis sterol glycosyltransferase TTG15/UGT80B1 role during freeze and heat stress. Plant Signal. Behav. 2015, 10, e1075682. [Google Scholar] [CrossRef] [Green Version]
- Nagatoshi, M.; Terasaka, K.; Nagatsu, A.; Mizukami, H. Iridoid-specific glucosyltransferase from Gardenia jasminoides. J. Biol. Chem. 2011, 286, 32866–32874. [Google Scholar] [CrossRef]
- Yang, L.-L.; Yang, L.; Yang, X.; Zhang, T.; Lan, Y.-M.; Zhao, Y.; Han, M.; Yang, L.-M. Drought stress induces biosynthesis of flavonoids in leaves and saikosaponins in roots of Bupleurum chinense DC. Phytochemistry 2020, 177, 112434. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Raw Bases | Valid Reads | Valid Bases | Valid (%) | Q20% | Q30% | GC% |
|---|---|---|---|---|---|---|---|---|
| CK_1 | 54,026,434 | 7.62 G | 53,451,132 | 7.45 G | 98.94 | 98.26 | 94.77 | 43.95 |
| CK_2 | 56,586,800 | 7.98 G | 55,938,322 | 7.82 G | 98.85 | 98.46 | 94.99 | 43.92 |
| CK_3 | 53,820,460 | 7.59 G | 53,229,136 | 7.44 G | 98.90 | 98.44 | 94.91 | 43.20 |
| PD2_1 | 41,709,950 | 6.26 G | 39,579,492 | 5.90 G | 94.89 | 98.72 | 95.48 | 42.87 |
| PD2_2 | 42,729,440 | 6.41 G | 42,223,028 | 6.31 G | 98.81 | 98.91 | 95.82 | 42.47 |
| PD2_3 | 44,270,858 | 6.64 G | 43,101,032 | 6.43 G | 97.36 | 98.84 | 95.73 | 42.42 |
| RW_1 | 43,486,690 | 6.52 G | 42,148,790 | 6.29 G | 96.92 | 98.81 | 95.63 | 42.27 |
| RW_2 | 49,878,582 | 7.48 G | 49,477,892 | 7.37 G | 99.20 | 98.81 | 95.72 | 42.05 |
| RW_3 | 50,165,512 | 7.52 G | 49,848,972 | 7.44 G | 99.37 | 98.90 | 95.87 | 42.34 |
| RD1_1 | 45,543,552 | 6.83 G | 45,263,368 | 6.75 G | 99.38 | 98.90 | 95.91 | 42.37 |
| RD1_2 | 46,609,874 | 6.99 G | 46,239,616 | 6.89 G | 99.21 | 98.83 | 95.77 | 42.38 |
| RD1_3 | 52,905,820 | 7.94 G | 51,958,506 | 7.74 G | 98.21 | 98.73 | 95.47 | 42.62 |
| RD2_1 | 49,930,204 | 7.49 G | 49,394,108 | 7.37 G | 98.93 | 98.84 | 95.68 | 42.30 |
| RD2_2 | 57,051,090 | 8.56 G | 56,531,180 | 8.44 G | 99.09 | 98.83 | 95.67 | 42.43 |
| RD2_3 | 55,426,212 | 8.31 G | 54,754,658 | 8.17 G | 98.79 | 98.80 | 95.64 | 42.47 |
| Databases | Number of Unigenes Annotated | Ratio (%) |
|---|---|---|
| NR | 22,320 | 66.87 |
| GO | 19,905 | 59.63 |
| eggNOG | 21,448 | 64.26 |
| KEGG | 15,227 | 45.62 |
| Pfam | 17,667 | 52.93 |
| Swiss-Prot | 16,554 | 49.59 |
| All | 33,379 | 100.00 |
| Hormone | Parent Ion (m∙s−1) | Daughter Ion (m∙s−1) | Retention Time (min) |
|---|---|---|---|
| ABA | 262.9 | 153.1 | 6.09 |
| SA | 137.0 | 93.0 | 5.81 |
| JA | 209.0 | 58.9 | 6.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Qiao, L.; Su, X.; Ji, B.; Dong, C. Drought Stress Stimulates the Terpenoid Backbone and Triterpenoid Biosynthesis Pathway to Promote the Synthesis of Saikosaponin in Bupleurum chinense DC. Roots. Molecules 2022, 27, 5470. https://doi.org/10.3390/molecules27175470
Yang L, Qiao L, Su X, Ji B, Dong C. Drought Stress Stimulates the Terpenoid Backbone and Triterpenoid Biosynthesis Pathway to Promote the Synthesis of Saikosaponin in Bupleurum chinense DC. Roots. Molecules. 2022; 27(17):5470. https://doi.org/10.3390/molecules27175470
Chicago/Turabian StyleYang, Linlin, Lu Qiao, Xiuhong Su, Baoyu Ji, and Chengming Dong. 2022. "Drought Stress Stimulates the Terpenoid Backbone and Triterpenoid Biosynthesis Pathway to Promote the Synthesis of Saikosaponin in Bupleurum chinense DC. Roots" Molecules 27, no. 17: 5470. https://doi.org/10.3390/molecules27175470
APA StyleYang, L., Qiao, L., Su, X., Ji, B., & Dong, C. (2022). Drought Stress Stimulates the Terpenoid Backbone and Triterpenoid Biosynthesis Pathway to Promote the Synthesis of Saikosaponin in Bupleurum chinense DC. Roots. Molecules, 27(17), 5470. https://doi.org/10.3390/molecules27175470