


Determination of Local Anesthetic Drugs in Human Plasma Using Magnetic Solid-Phase Extraction Coupled with High-Performance Liquid Chromatography


Abstract
1. Introduction
2. Results and Discussion
2.1. Preparation and characterization of Mag-CCNT-TEPA
2.2. Optimization of Extraction Conditions
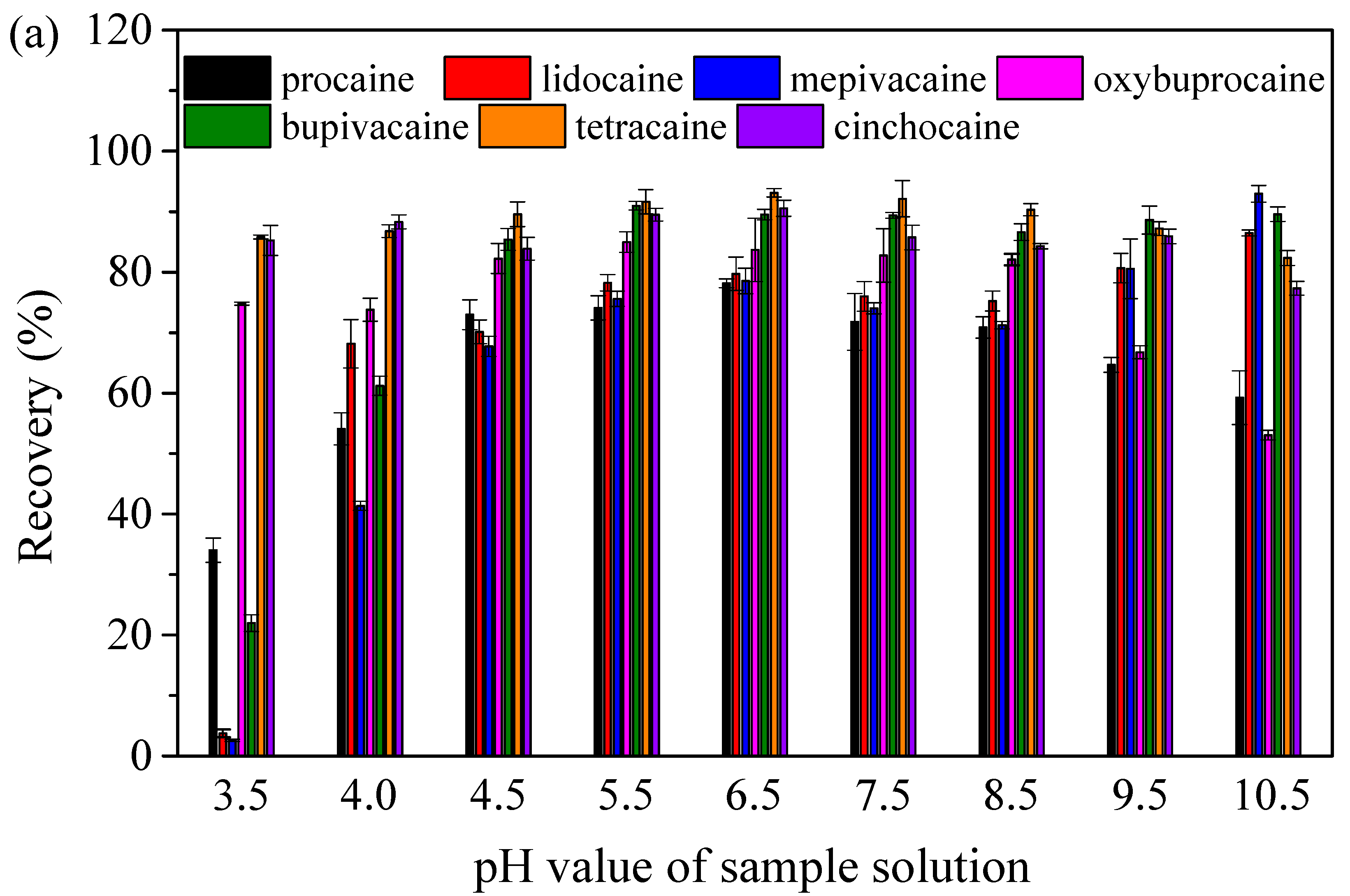
2.2.1. The Effect of the pH of Sample Solutions
2.2.2. Effect of the Amount of Adsorbents
2.2.3. Effect of the Extraction Time
2.2.4. Effects of the Elution Conditions
2.3. Regeneration and Reusability of Adsorbent
2.4. Adsorbent Type
2.5. Method Validation
2.6. Analysis of Real Samples
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Equipment
3.3. HPLC Analysis
3.4. Preparation of Mag-CCNT-TEPA
3.5. Standard Preparation
3.6. Application of Mag-CCNT-TEPA for the Extraction of Local Anesthetic Drugs from Human Plasma
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
Sample Availability
References
- Lombardo-Agui, M.; Cruces-Blanco, C.; Garcia-Campana, A.M. Capillary zone electrophoresis with diode-array detection for analysis of local anaesthetics and opium alkaloids in urine samples. J. Chromatogr. B. 2009, 877, 833–836. [Google Scholar] [CrossRef]
- Weissberg, A.; Drug, E.; Prihed, H.; Madmon, M.; Shamai Yamin, T. Structural elucidation of amino amide-type local anesthetic drugs and their main metabolites in urine by LC-MS after derivatization and its application for differentiation between positional isomers of prilocaine. J. Mass Spectrom. 2020, 55, e4654. [Google Scholar] [CrossRef]
- Busardo, F.P.; Tritapepe, L.; Montana, A.; Indorato, F.; Zaami, S.; Romano, G. A fatal accidental subarachnoid injection of lidocaine and levobupivacaine during a lumbar paravertebral block. Forensic Sci. Int. 2015, 256, 17–20. [Google Scholar] [CrossRef]
- Collins, S.; Neubrander, J.; Vorst, Z.; Sheffield, B. Lipid Emulsion in Treatment of Local Anesthetic Toxicity. J. Perianesth. Nurs. 2015, 30, 308–320. [Google Scholar] [CrossRef]
- Care, W.; Larabi, I.A.; Langrand, J.; Medernach, C.; Alvarez, J.C.; Villa, A. Poisoning associated with inappropriate use of a eutectic mixture of lidocaine and prilocaine before laser-assisted hair removal: About 3 cases. Int. J. Legal Med. 2019, 133, 843–846. [Google Scholar] [CrossRef]
- ter Weijden, E.; van den Broek, M.P.H.; Ververs, F.F.T. Easy and fast LC-MS/MS determination of lidocaine and MEGX in plasma for therapeutic drug monitoring in neonates with seizures. J. Chromatogr. B 2012, 881–882, 111–114. [Google Scholar] [CrossRef]
- Xie, C.; Li, Q.; Han, G.; Liu, H.; Yang, J.; Li, J. Stable isotope dilution assay for the accurate determination of tricaine in fish samples by HPLC-MS-MS. Biomed. Chromatogr. 2019, 33, e4512. [Google Scholar] [CrossRef]
- Daryanavard, S.M.; Jeppsson-Dadoun, A.; Andersson, L.I.; Hashemi, M.; Colmsjo, A.; Abdel-Rehim, M. Molecularly imprinted polymer in microextraction by packed sorbent for the simultaneous determination of local anesthetics: Lidocaine, ropivacaine, mepivacaine and bupivacaine in plasma and urine samples. Biomed. Chromatogr. 2013, 27, 1481–1488. [Google Scholar] [CrossRef]
- Fiorentin, T.R.; Fogarty, M.; Limberger, R.P.; Logan, B.K. Determination of cutting agents in seized cocaine samples using GC-MS, GC-TMS and LC-MS/MS. Forensic Sci. Int. 2019, 295, 199–206. [Google Scholar] [CrossRef]
- Tonooka, K.; Naruki, N.; Honma, K.; Agei, K.; Okutsu, M.; Hosono, T.; Kunisue, Y.; Terada, M.; Tomobe, K.; Shinozuka, T. Sensitive liquid chromatography/tandem mass spectrometry method for the simultaneous determination of nine local anesthetic drugs. Forensic Sci. Int. 2016, 265, 182–185. [Google Scholar] [CrossRef]
- Grigoriev, A.; Nikitina, A.; Yaroshenko, I.; Sidorova, A. Development of a HPLC-MS/MS method for the simultaneous determination of nifedipine and lidocaine in human plasma. J. Pharm. Biomed. Anal. 2016, 131, 13–19. [Google Scholar] [CrossRef]
- Oliveira Souza, M.C.; Marques, M.P.; Duarte, G.; Lanchote, V.L. Analysis of bupivacaine enantiomers in plasma as total and unbound concentrations using LC-MS/MS: Application in a pharmacokinetic study of a parturient with placental transfer. J. Pharm. Biomed. Anal. 2019, 164, 268–275. [Google Scholar] [CrossRef]
- Bertol, E.; Argo, A.; Capretti, C.; Ciolini, A.; Umani Ronchi, F.; Zerbo, S.; Mari, F.; Vaiano, F. A novel LC-MS/MS analytical method for detection of articaine and mepivacaine in blood and its application to a preliminary pharmacokinetic study. J. Pharm. Biomed. Anal. 2020, 187, 113335. [Google Scholar] [CrossRef]
- Karimiyan, H.; Uheida, A.; Hadjmohammadi, M.; Moein, M.M.; Abdel-Rehim, M. Polyacrylonitrile/graphene oxide nanofibers for packed sorbent microextraction of drugs and their metabolites from human plasma samples. Talanta 2019, 201, 474–479. [Google Scholar] [CrossRef]
- Iadaresta, F.; Crescenzi, C.; Amini, A.; Colmsjo, A.; Koyi, H.; Abdel-Rehim, M. Application of graphitic sorbent for online microextraction of drugs in human plasma samples. J. Chromatogr. A 2015, 1422, 34–42. [Google Scholar] [CrossRef]
- Socas-Rodriguez, B.; Herrera-Herrera, A.V.; Asensio-Ramos, M.; Hernandez-Borges, J. Recent applications of carbon nanotube sorbents in analytical chemistry. J. Chromatogr. A 2014, 1357, 110–146. [Google Scholar] [CrossRef]
- Liang, R.; Hu, Y.; Li, G. Photochemical synthesis of magnetic covalent organic framework/carbon nanotube composite and its enrichment of heterocyclic aromatic amines in food samples. J. Chromatogr. A 2020, 1618, 460867. [Google Scholar] [CrossRef]
- Mashkoor, F.; Nasar, A.; Inamuddin. Carbon nanotube-based adsorbents for the removal of dyes from waters: A review. Environ. Chem. Lett. 2020, 18, 605–629. [Google Scholar] [CrossRef]
- Jacobs, C.B.; Peairs, M.J.; Venton, B.J. Review: Carbon nanotube based electrochemical sensors for biomolecules. Anal. Chim. Acta. 2010, 662, 105–127. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Gonzalez-Curbelo, M.A.; Hernandez-Borges, J.; Rodriguez-Delgado, M.A. Carbon nanotubes applications in separation science: A review. Anal. Chim. Acta. 2012, 734, 1–30. [Google Scholar] [CrossRef]
- Vasconcelos, I.; Fernandes, C. Magnetic solid phase extraction for determination of drugs in biological matrices. TrAC Trends Anal. Chem. 2017, 89, 41–52. [Google Scholar] [CrossRef]
- Ghorbani, M.; Aghamohammadhasan, M.; Shams, A.; Tajfirooz, F.; Pourhassan, R.; Bana Khosravi, S.R.; Karimi, E.; Jampour, A. Ultrasonic assisted magnetic dispersive solid phase microextraction for preconcentration of two nonsteroidal anti-inflammatory drugs in real water, biological and milk samples employing an experimental design. Microchem. J. 2019, 145, 1026–1035. [Google Scholar] [CrossRef]
- Escamilla-Lara, K.A.; Heredia, A.C.; Pena-Alvarez, A.; Ibarra, I.S.; Barrado, E.; Rodriguez, J.A. Magnetic Solid-Phase Extraction Based on Poly 4-Vinyl Pyridine for HPLC-FLD Analysis of Naproxen in Urine Samples. Molecules 2020, 25, 2924. [Google Scholar] [CrossRef]
- Wang, Y.; Ou, Y.; Xie, S.; Chen, D.; Wang, X.; Pan, Y.; Wang, Y.; Huang, L.; Cheng, G.; Qu, W.; et al. Magnetic Graphene Solid-Phase Extraction for the Determination of 47 Kinds of Non-steroidal Anti-inflammatory Drug Residues in Animal Food with Liquid Chromatography Tandem Mass Spectrometry. Food Anal. Methods 2019, 12, 1346–1368. [Google Scholar] [CrossRef]
- Baciu, T.; Borrull, F.; Neususs, C.; Aguilar, C.; Calull, M. Capillary electrophoresis combined in-line with solid-phase extraction using magnetic particles as new adsorbents for the determination of drugs of abuse in human urine. Electrophoresis 2016, 37, 1232–1244. [Google Scholar] [CrossRef]
- Kolaei, M.; Dashtian, K.; Rafiee, Z.; Ghaedi, M. Ultrasonic-assisted magnetic solid phase extraction of morphine in urine samples by new imprinted polymer-supported on MWCNT-Fe3O4-NPs: Central composite design optimization. Ultrason. Sonochem. 2016, 33, 240–248. [Google Scholar] [CrossRef]
- Guo, S.; Li, D.; Zhang, L.; Li, J.; Wang, E. Monodisperse mesoporous superparamagnetic single-crystal magnetite nanoparticles for drug delivery. Biomaterials 2009, 30, 1881–1889. [Google Scholar] [CrossRef]
- Ohshima, T.; Takayasu, T. Simultaneous determination of local anesthetics including ester-type anesthetics in human plasma and urine by gas chromatography–mass spectrometry with solid-phase extraction. J. Chromatogr. B 1999, 726, 185–194. [Google Scholar] [CrossRef]
- Al Nebaihi, H.M.; Primrose, M.; Green, J.S.; Brocks, D.R. A high-performance liquid chromatography assay method for the determination of lidocaine in human serum. Pharmaceutics 2017, 9, 52. [Google Scholar] [CrossRef]
- Qin, W.W.; Jiao, Z.; Zhong, M.K.; Shi, X.J.; Zhang, J.; Li, Z.D.; Cui, X.Y. Simultaneous determination of procaine, lidocaine, ropivacaine, tetracaine and bupivacaine in human plasma by high-performance liquid chromatography. J. Chromatogr. B 2010, 878, 1185–1189. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Linear Range (mg/L) | Linear Equation | Correlation Coefficient (r2) | Spiked Level (mg/L) | Average Recovery (%) | RSD (%) | Batch–Batch ReProducibility RSD% | LOD (mg/L) | LOQ (mg/L) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||||||
| procaine | 0.02–5.00 | Y = 2.5147X − 0.0657 | 0.9993 | 0.1 1.0 4.0 | 82.0 103 100 | 2.0 2.3 1.6 | 3.0 7.2 4.6 | 3.9 | 0.004 | 0.014 |
| lidocaine | 0.02–5.00 | Y = 2.7307X − 0.0057 | 0.9995 | 0.1 1.0 4.0 | 91.6 93.3 98.9 | 2.0 5.7 2.8 | 4.0 3.8 5.4 | 4.2 | 0.007 | 0.025 |
| mepivacaine | 0.02–5.00 | Y = 2.5830X + 0.2243 | 0.9980 | 0.1 1.0 4.0 | 97.7 87.7 101 | 7.1 5.6 4.0 | 1.7 7.5 4.7 | 4.0 | 0.007 | 0.023 |
| oxybuprocaine | 0.04–5.00 | Y = 2.1061X − 0.0648 | 0.9998 | 0.1 1.0 4.0 | 98.7 99.6 94.7 | 7.7 1.5 1.5 | 6.0 2.5 2.3 | 1.5 | 0.008 | 0.028 |
| bupivacaine | 0.02–5.00 | Y = 2.7547X + 0.0792 | 0.9998 | 0.1 1.0 4.0 | 89.4 93.7 99.3 | 4.7 6.3 5.1 | 8.3 4.4 2.3 | 3.5 | 0.006 | 0.022 |
| tetracaine | 0.02–5.00 | Y = 3.9807X + 0.0555 | 0.9998 | 0.1 1.0 4.0 | 82.8 90.0 94.7 | 1.7 1.9 1.7 | 1.5 1.6 1.8 | 1.9 | 0.004 | 0.015 |
| cinchocaine | 0.02–5.00 | Y = 7.3979X − 0.0917 | 0.9999 | 0.1 1.0 4.0 | 91.1 108 108 | 1.8 2.5 4.3 | 2.6 2.3 2.6 | 3.3 | 0.003 | 0.011 |
| Analysis | Preparation Method | Samples | Analytes | Correlation Coefficient (r2) | LOQ (µg/L) | Recovery (%) | Ref. |
|---|---|---|---|---|---|---|---|
| LC-MS/MS | SPE | Human serum | Nine local anesthetic drugs | >0.9858 | 10 | 81.4–144 | [10] |
| HPLC-MS/MS | LLE | Human plasma | Lidocaine | >0.9996 | 1.0 | 95.2–104 | [11] |
| LC-MS/MS | LLE | Human plasma | Bupivacaine | >0.998 | 0.25 | / | [12] |
| LC-MS/MS | LLE | Blood | Two local anesthetic drugs | >0.9991 | 0.1–0.8 | 85.4–87.5 | [13] |
| LC-MS/MS | MEPS | Human plasma | Two local anesthetic drugs | >0.995 | 0.44–0.47 | 91.0–118 | [14] |
| GC-MS | SPE | Plasma and urine | Seven local anesthetic drugs | >0.983 | 50–100 | 73.0–95.0 | [28] |
| HPLC-UV | LLE | Human serum | Lidocaine | 0.9995 | 43 | 80.4–93.9 | [29] |
| HPLC-UV | LLE | Human plasma | Five local anesthetic drugs | >0.9980 | 50 | 91.7–106 | [30] |
| HPLC-UV | MSPE | Human plasma | Seven local anesthetic drugs | >0.9980 | 11–28 | 82.0–108 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, S.-Y.; Shi, F.; Zhao, Y.-G.; Wang, H.-W. Determination of Local Anesthetic Drugs in Human Plasma Using Magnetic Solid-Phase Extraction Coupled with High-Performance Liquid Chromatography. Molecules 2022, 27, 5509. https://doi.org/10.3390/molecules27175509
Liang S-Y, Shi F, Zhao Y-G, Wang H-W. Determination of Local Anesthetic Drugs in Human Plasma Using Magnetic Solid-Phase Extraction Coupled with High-Performance Liquid Chromatography. Molecules. 2022; 27(17):5509. https://doi.org/10.3390/molecules27175509
Chicago/Turabian StyleLiang, Shan-Yan, Fang Shi, Yong-Gang Zhao, and Hong-Wei Wang. 2022. "Determination of Local Anesthetic Drugs in Human Plasma Using Magnetic Solid-Phase Extraction Coupled with High-Performance Liquid Chromatography" Molecules 27, no. 17: 5509. https://doi.org/10.3390/molecules27175509
APA StyleLiang, S.-Y., Shi, F., Zhao, Y.-G., & Wang, H.-W. (2022). Determination of Local Anesthetic Drugs in Human Plasma Using Magnetic Solid-Phase Extraction Coupled with High-Performance Liquid Chromatography. Molecules, 27(17), 5509. https://doi.org/10.3390/molecules27175509