
PyPLIF HIPPOS and Receptor Ensemble Docking Increase the Prediction Accuracy of the Structure-Based Virtual Screening Protocol Targeting Acetylcholinesterase
 ,
,  ,
,
Abstract
:1. Introduction
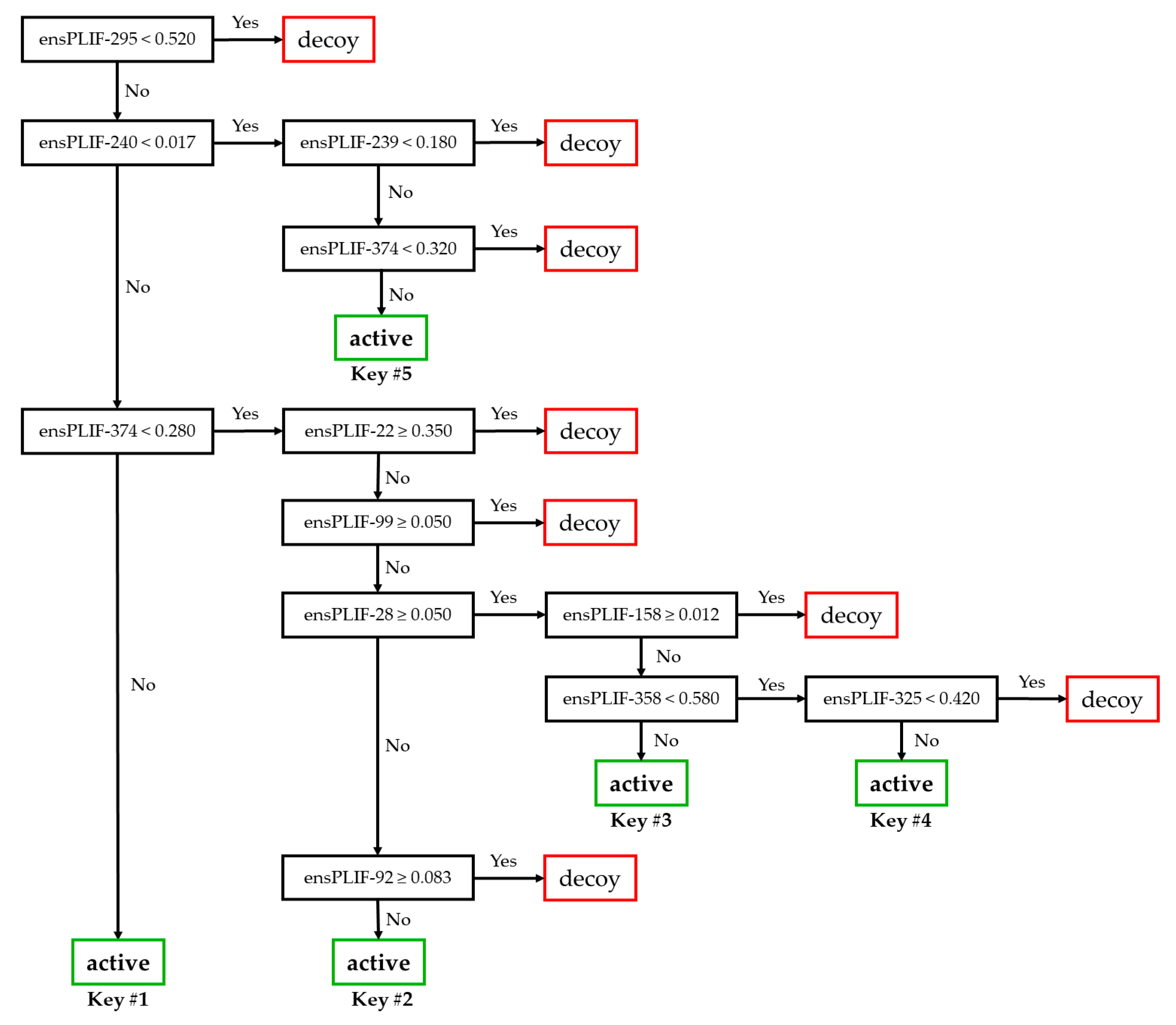
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Ligand Preparation
4.2.2. Replication Optimization
4.2.3. Proteins Preparation
4.2.4. Automated Molecular Docking Simulations Using AutoDock Vina
4.2.5. Ensemble Docking Scores and ensplif Calculations
4.2.6. Analysis Using RPART in R
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.T.; Shah, R.C.; Bennett, D.A. Alzheimer’s Disease: Unique markers for diagnosis & new treatment modalities. Indian J. Med. Res. 2015, 142, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Kukreja, H.; Chugh, R.; Silakari, O.; Singh, D. Acetylcholinesterase inhibitors as Alzheimer therapy: From nerve toxins to neuroprotection. Eur. J. Med. Chem. 2013, 70, 165–188. [Google Scholar] [CrossRef]
- Bryson, H.M.; Benfield, P. Donepezil. Drugs Aging 1997, 10, 234–239. [Google Scholar] [CrossRef]
- Knapp, M.; King, D.; Romeo, R.; Adams, J.; Baldwin, A.; Ballard, C.; Banerjee, S.; Barber, R.; Bentham, P.; Brown, R.G.; et al. Cost-effectiveness of donepezil and memantine in moderate to severe Alzheimer’s disease (the DOMINO-AD Trial). Int. J. Geriatr. Psychiatry 2017, 32, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Heydorn, W.E. Donepezil (E2020): A New Acetylcholinesterase Inhibitor. Review of Its Pharmacology, Pharmacokinetics, and Utility in the Treatment of Alzheimer’s Disease. Expert Opin. Investig. Drugs 1997, 6, 1527–1535. [Google Scholar] [CrossRef]
- Riswanto, F.D.O.; Hariono, M.; Yuliani, S.H.; Istyastono, E.P. Computer-aided design of chalcone derivatives as lead compounds targeting acetylcholinesterase. Indonesian J. Pharm. 2017, 28, 100–111. [Google Scholar] [CrossRef]
- Riswanto, F.D.O.; Rawa, M.S.A.; Murugaiyah, V.; Salin, N.H.; Istyastono, E.P.; Hariono, M.; Wahab, H.A. Anti-cholinesterase activity of chalcone derivatives: Synthesis, in vitro assay and molecular docking study. Med. Chem. 2021, 17, 442–452. [Google Scholar] [CrossRef]
- Prasasty, V.D.; Istyastono, E.P. Data of small peptides in SMILES and three-dimensional formats for virtual screening campaigns. Data Brief 2019, 27, 104607. [Google Scholar] [CrossRef]
- Prasasty, V.; Radifar, M.; Istyastono, E. Natural peptides in drug discovery targeting acetylcholinesterase. Molecules 2018, 23, 2344. [Google Scholar] [CrossRef] [Green Version]
- Prasasty, V.D.; Istyastono, E.P. Structure-based design and molecular dynamics simulations of pentapeptide AEYTR as a potential acetylcholinesterase inhibitor. Indones. J. Chem. 2020, 20, 953–959. [Google Scholar] [CrossRef]
- Istyastono, E.P.; Prasasty, V.D. Computer-aided discovery of pentapeptide AEYTR as a potent acetylcholinesterase inhibitor. Indones. J. Chem. 2021, 21, 243–350. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Shen, C.; Hu, X.; Gao, J.; Li, D.; Cao, D.; Hou, T. Combined strategies in structure-based virtual screening. Phys. Chem. Chem. Phys. 2020, 22, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Daoud, I.; Melkemi, N.; Salah, T.; Ghalem, S. Combined QSAR, molecular docking and molecular dynamics study on new acetylcholinesterase and butyrylcholinesterase inhibitors. Comput. Biol. Chem. 2018, 74, 304–326. [Google Scholar] [CrossRef]
- Liu, H.; Men, X.; Gao, X.; Liu, L.; Fan, H.; Xia, X.; Wang, Q. Discovery of potent and selective acetylcholinesterase (AChE) inhibitors: Acacetin 7-O-methyl ether mannich base derivatives synthesised from easy access natural product naringin. Nat. Prod. Res. 2018, 32, 743–747. [Google Scholar] [CrossRef]
- van der Westhuizen, C.J.; Stander, A.; Riley, D.L.; Panayides, J.L. Discovery of novel acetylcholinesterase inhibitors by virtual screening, in vitro screening, and molecular dynamics simulations. J. Chem. Inf. Model. 2022, 62, 1550–1572. [Google Scholar] [CrossRef]
- Atanasova, M.; Dimitrov, I.; Ivanov, S.; Georgiev, B.; Berkov, S.; Zheleva-Dimitrova, D.; Doytchinova, I. virtual screening and hit selection of natural compounds as acetylcholinesterase inhibitors. Molecules 2022, 27, 3139. [Google Scholar] [CrossRef]
- Koshland, D.E. The key–lock theory and the induced fit theory. Angew. Chem. Int. Ed. Engl. 1994, 33, 2375–2378. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015, 11, 1–28. [Google Scholar] [CrossRef]
- Ogrizek, M.; Turk, S.; Lešnik, S.; Sosič, I.; Hodošček, M.; Mirković, B.; Kos, J.; Janežič, D.; Gobec, S.; Konc, J. Molecular dynamics to enhance structure-based virtual screening on cathepsin B. J. Comput. Aided Mol. Des. 2015, 29, 707–712. [Google Scholar] [CrossRef]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Methods Mol. Biol.; Humana Press Inc.: New York, NY, USA, 2019; Volume 2053, pp. 13–34. [Google Scholar]
- Mohammadi, S.; Narimani, Z.; Ashouri, M.; Firouzi, R.; Karimi-Jafari, M.H. Ensemble learning from ensemble docking: Revisiting the optimum ensemble size problem. Sci. Rep. 2022, 12, 1–15. [Google Scholar] [CrossRef]
- Radifar, M.; Yuniarti, N.; Istyastono, E.P. PyPLIF: Python-based protein-ligand interaction fingerprinting. Bioinformation 2013, 9, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Istyastono, E.P. Employing recursive partition and regression tree method to increase the quality of structure-based virtual screening in the estrogen receptor alpha ligands identification. Asian J. Pharm. Clin. Res. 2015, 8, 21–24. [Google Scholar]
- Istyastono, E.P.; Yuniarti, N.; Hariono, M.; Yuliani, S.H.; Riswanto, F.D.O. Binary quantitative structure-activity relationship analysis in retrospective structure based virtual screening campaigns targeting estrogen receptor alpha. Asian J. Pharm. Clin. Res. 2017, 10, 206–211. [Google Scholar] [CrossRef]
- Istyastono, E.P.; Radifar, M.; Yuniarti, N.; Prasasty, V.D.; Mungkasi, S. PyPLIF HIPPOS: A molecular interaction fingerprinting tool for docking results of AutoDock Vina and PLANTS. J. Chem. Inf. Model. 2020, 60, 3697–3702. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Dvir, H.; Wong, D.M.; Harel, M.; Barril, X.; Orozco, M.; Luque, F.J.; Munoz-Torrero, D.; Camps, P.; Rosenberry, T.L.; Silman, I.; et al. 3D Structure of torpedo californica acetylcholinesterase complexed with huprine X at 2.1 Å resolution: Kinetic and molecular dynamic correlates. Biochemistry 2002, 41, 2970–2981. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Stein, R.M.; Yang, Y.; Balius, T.E.; O’Meara, M.J.; Lyu, J.; Young, J.; Tang, K.; Shoichet, B.K.; Irwin, J.J. Property-unmatched decoys in docking benchmarks. J. Chem. Inf. Model. 2021, 61, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Istyastono, E.P.; Yuniarti, N.; Prasasty, V.D.; Mungkasi, S. PyPLIF HIPPOS-assisted prediction of molecular determinants of ligand binding to receptors. Molecules 2021, 26, 2452. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein–ligand docking. Proc. IEEE Swarm Intell. Symp. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- ten Brink, T.; Exner, T.E. Influence of protonation, tautomeric, and stereoisomeric states on protein-ligand docking results. J. Chem. Inf. Model. 2009, 49, 1535–1546. [Google Scholar] [CrossRef]
- Therneau, T.; Atkinson, B.; Ripley, B. RPART: Recursive Partitioning and Regression Trees. R Package Version 4.1-9. 2015. Available online: https://CRAN.R-project.org/package=rpart (accessed on 28 September 2019).
- R Core Team. R: A Language and Environment for Statistical Computing. Vienna. 2019. Available online: http://www.r-project.org (accessed on 28 September 2019).
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Cappel, D.; Dixon, S.L.; Sherman, W.; Duan, J. Exploring conformational search protocols for ligand-based virtual screening and 3-D QSAR modeling. J. Comput. Aided Mol. Des. 2015, 29, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Waite, G.P. Nonlinear moment-tensor inversion of repetitive long-periods events recorded at Pacaya Volcano, Guatemala. Front. Earth Sci. 2018, 6, 1–16. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Variable No. | Descriptor | Corresponding Residue | Corresponding Interaction Type 1 |
|---|---|---|---|
| V22 | ensPLIF-22 | Asp72 | hydrophobic |
| V28 | ensPLIF-28 | Asp72 | ionic (residue as the anion) |
| V92 | ensPLIF-92 | Asn85 | hydrophobic |
| V99 | ensPLIF-99 | Pro86 | hydrophobic |
| V158 | ensPLIF-158 | Ser122 | H-bond (residue as the donor) |
| V239 | ensPLIF-239 | Trp279 | hydrophobic |
| V240 | ensPLIF-240 | Trp279 | aromatic (face-to-face) |
| V295 | ensPLIF-295 | Phe330 | hydrophobic |
| V325 | ensPLIF-325 | Tyr334 | aromatic (edge-to-face) |
| V358 | ensPLIF-358 | His440 | hydrophobic |
| V374 | ensPLIF-374 | Tyr442 | aromatic (edge-to-face) |
| SBVS Protocol | Confusion Matrix | Statistical Significance | |||||
|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | EF | F1 | BA | |
| Mysinger et al. [30] 1 | 91 | 362 | 25988 | 262 | 20.1 | 0.225 | 0.595 |
| Riswanto et al. [7] | 124 | 329 | 26226 | 24 | 299.393 | 0.413 | 0.636 |
| van der Westhuizen et al. [16] 1 | 127 | 326 | 25988 | 262 | 28 | 0.301 | 0.635 |
| SBVS developed in this article | 214 | 239 | 25886 | 364 | 34.068 | 0.415 | 0.729 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istyastono, E.P.; Riswanto, F.D.O.; Yuniarti, N.; Prasasty, V.D.; Mungkasi, S. PyPLIF HIPPOS and Receptor Ensemble Docking Increase the Prediction Accuracy of the Structure-Based Virtual Screening Protocol Targeting Acetylcholinesterase. Molecules 2022, 27, 5661. https://doi.org/10.3390/molecules27175661
Istyastono EP, Riswanto FDO, Yuniarti N, Prasasty VD, Mungkasi S. PyPLIF HIPPOS and Receptor Ensemble Docking Increase the Prediction Accuracy of the Structure-Based Virtual Screening Protocol Targeting Acetylcholinesterase. Molecules. 2022; 27(17):5661. https://doi.org/10.3390/molecules27175661
Chicago/Turabian StyleIstyastono, Enade P., Florentinus Dika Octa Riswanto, Nunung Yuniarti, Vivitri D. Prasasty, and Sudi Mungkasi. 2022. "PyPLIF HIPPOS and Receptor Ensemble Docking Increase the Prediction Accuracy of the Structure-Based Virtual Screening Protocol Targeting Acetylcholinesterase" Molecules 27, no. 17: 5661. https://doi.org/10.3390/molecules27175661
APA StyleIstyastono, E. P., Riswanto, F. D. O., Yuniarti, N., Prasasty, V. D., & Mungkasi, S. (2022). PyPLIF HIPPOS and Receptor Ensemble Docking Increase the Prediction Accuracy of the Structure-Based Virtual Screening Protocol Targeting Acetylcholinesterase. Molecules, 27(17), 5661. https://doi.org/10.3390/molecules27175661