
Gallic Acid Alkyl Esters: Trypanocidal and Leishmanicidal Activity, and Target Identification via Modeling Studies

 and
and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Gallic Acid Alkyl Esters
2.2. Biological Activity of Gallic Acid Alkyl Esters
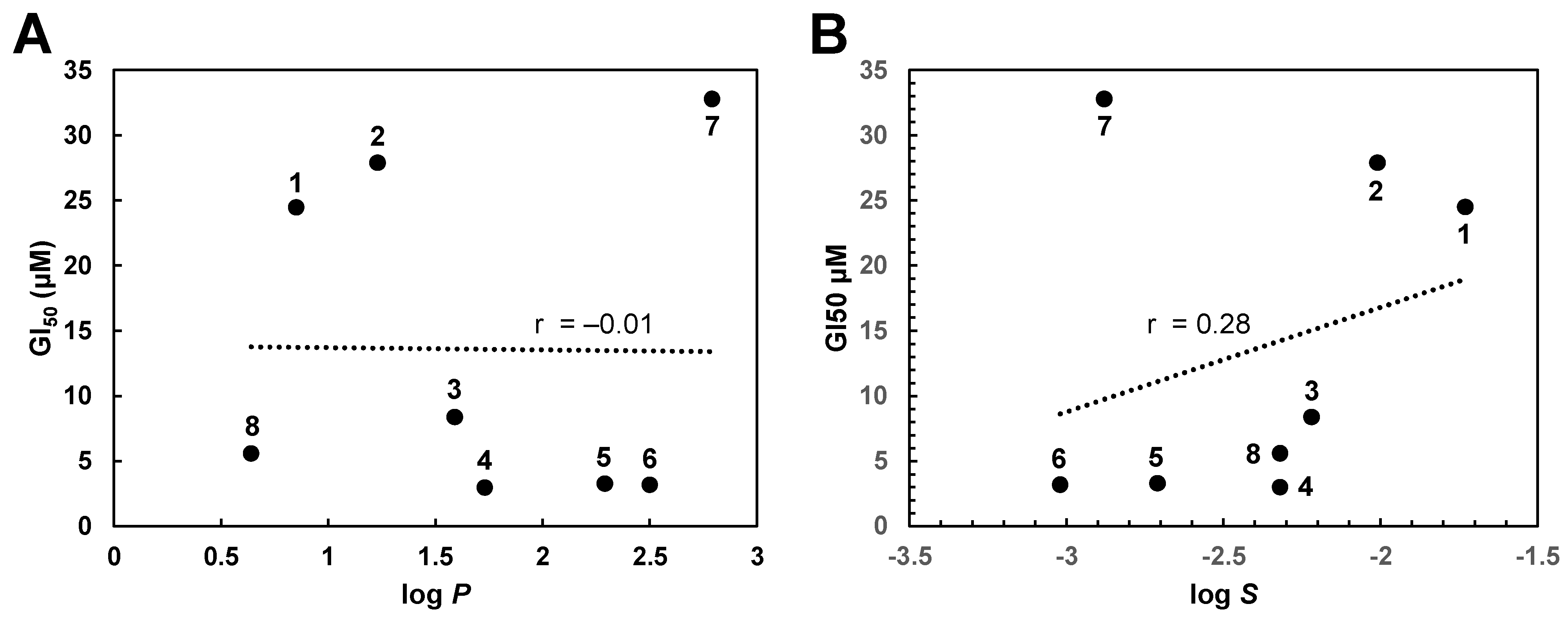
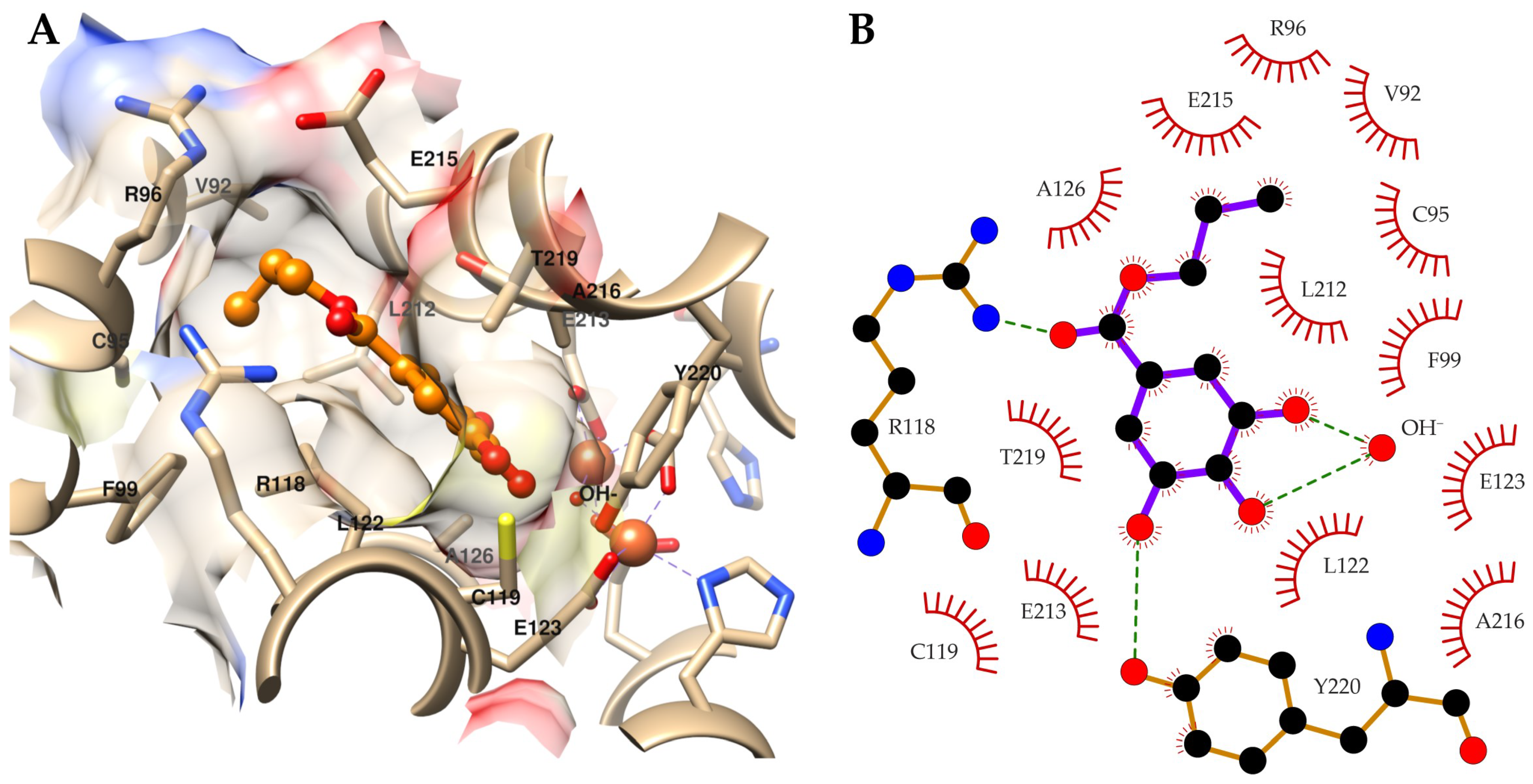
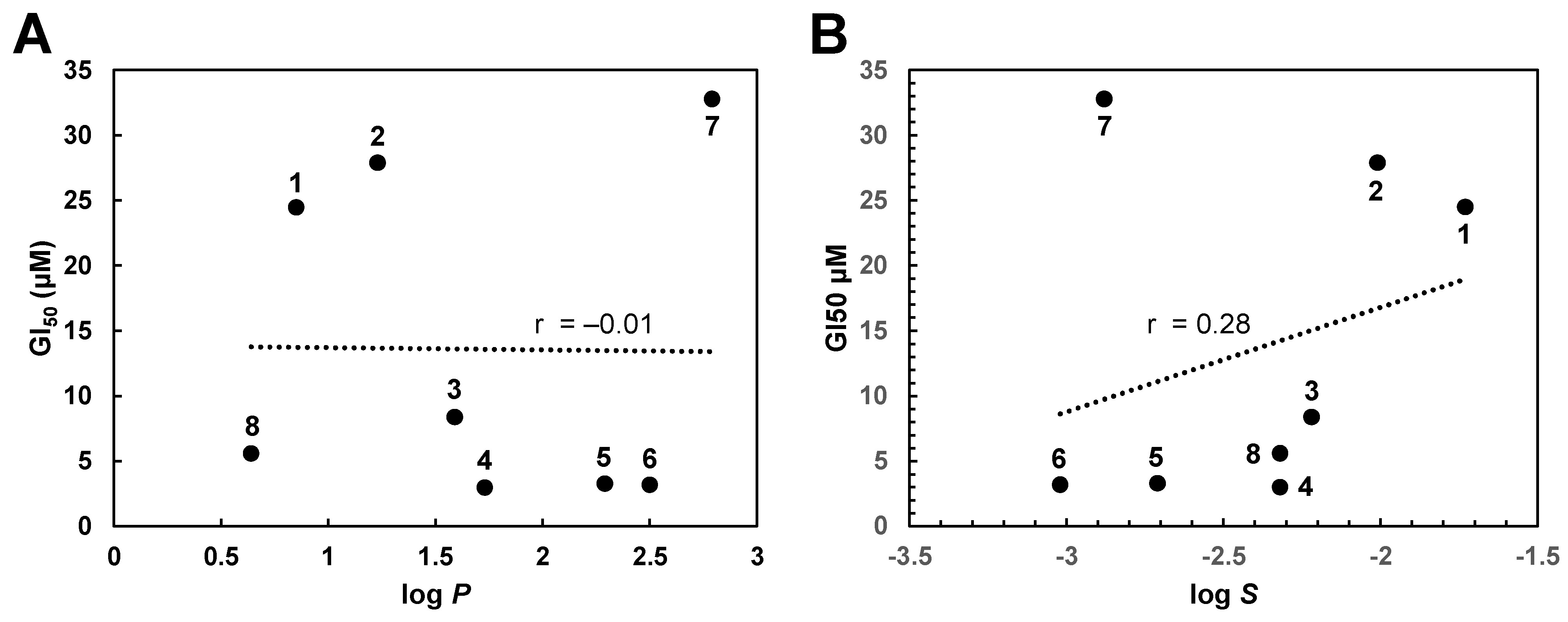
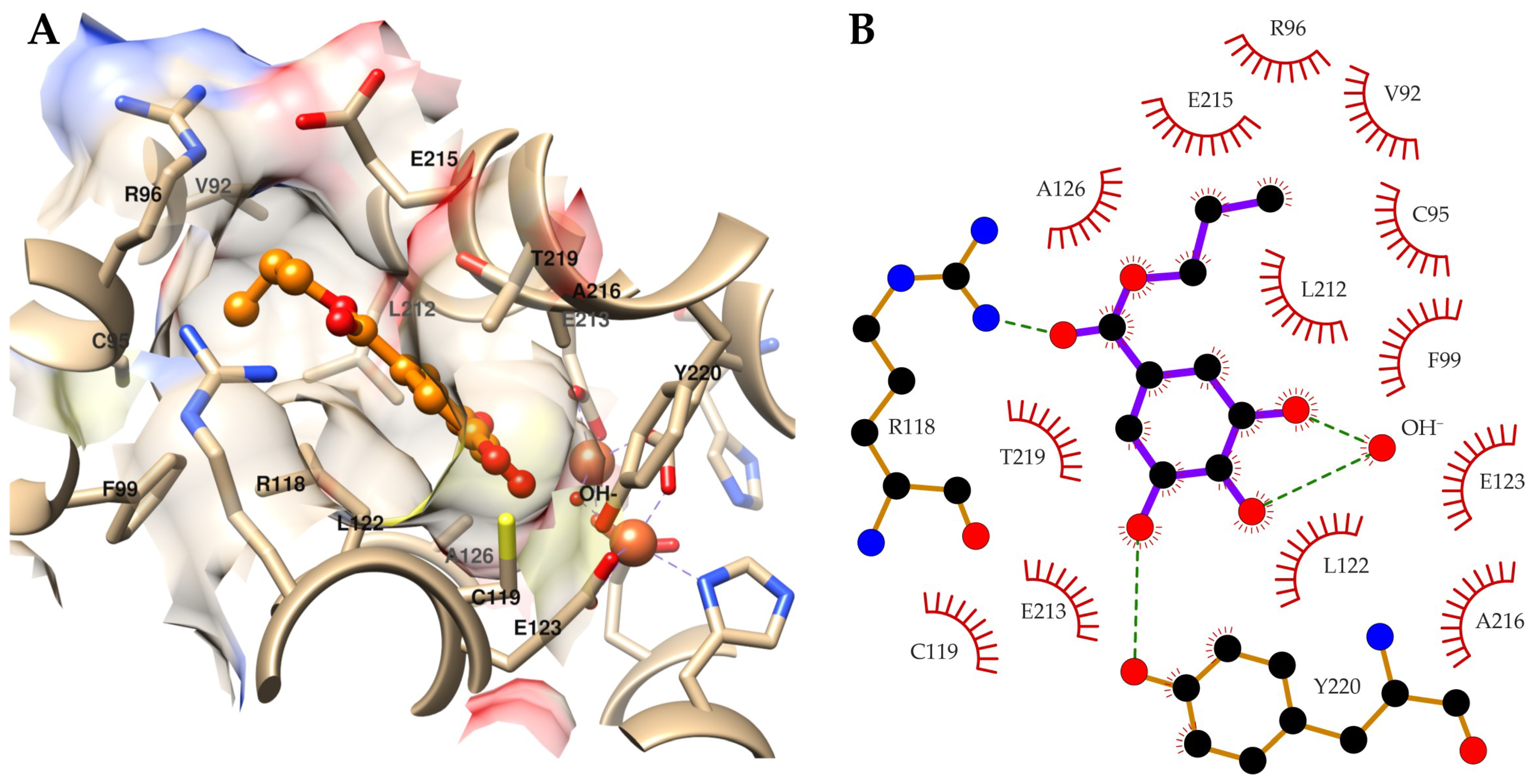
2.3. Target Identification via Molecular Modeling Studies
2.4. ADMET and Druglikeness Properties of n-Propyl Gallate (Compound 4)
2.5. Effect of Gallic Acid Alkyl Esters on the Motility of Trypanosomes
2.6. Conclusions
3. Materials and Methods
3.1. Chemistry
3.2. In Vitro Toxicity Assays
3.3. Motility Assay
3.4. Modeling Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Steverding, D. The history of African trypanosomiasis. Parasit. Vectors 2008, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The history of Chagas disease. Parasit. Vectors 2014, 7, 317. [Google Scholar] [CrossRef]
- Steverding, D. The history of leishmaniasis. Parasit. Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, C.R.; Steverding, D.; Ferreira, R.S.; de Oliveira, R.B.; O’Donoghue, A.J.; Monti, L.; Ballatore, C.; Bachovchin, K.A.; Ferrins, L.; Pollastri, M.P.; et al. Drug discovery and development for kinetoplastid diseases. In Burger’s Medicinal Chemistry, Drug Discovery and Development, 8th ed.; Abraham, J.D., Myers, M., Eds.; John Wiley & Sons: New York, NY, USA, 2021; Volume 7, pp. 255–334. [Google Scholar]
- Okello, I.; Mafie, E.; Eastwood, G.; Nzalawahe, J.; Mboera, L.E.G. African animal trypanosomiasis: A systematic review on prevalence, risk factors and drug resistance in sub-Saharan Africa. J. Med. Entomol. 2022, 59, 1099–1143. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Cheuka, P.M.; Mayoka, G.; Mutai, P.; Chibale, K. The role of natural products in drug discovery and development against neglected tropical diseases. Molecules 2016, 22, 58. [Google Scholar] [CrossRef]
- Cueva, C.; Moreno-Arribas, M.V.; Martín-Álverez, P.J.; Bills, G.; Vincete, M.F.; Basilio, A.; López Rivas, C.; Requena, T.; Rodríguez, J.M.; Bartolomé, B. Antimicrobial activity of phenolic acids against commensal, probiotic and pathogenic bacteria. Res. Microbiol. 2010, 161, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D.; da Nóbrega, F.R.; Rushworth, S.A.; de Sousa, D.P. Trypanocidal and cysteine protease inhibitory activity of isopentyl caffeate is not linked in Trypanosoma brucei. Parasitol. Res. 2016, 115, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Amisigo, C.M.; Antwi, C.A.; Adjimani, J.P.; Gwira, T.M. In vitro anti-trypanosomal effects of selected phenolic acids on Trypanosoma brucei. PLoS ONE 2019, 14, e0216078. [Google Scholar] [CrossRef]
- Otoguro, K.; Iwatsuki, M.; Ishiyama, A.; Namatame, M.; Nishihara-Tsukashima, A.; Kiyohara, H.; Hashimoto, T.; Asakawa, Y.; Omura, S.; Yamada, H. In vitro antitrypanosomal activity of some phenolic compounds from propolis and lactones from Fijian Kawa (Piper methysticum). J. Nat. Med. 2012, 66, 558–561. [Google Scholar] [CrossRef]
- Burke, T.R., Jr.; Fesen, M.R.; Mazumder, A.; Wang, J.; Carothers, A.M.; Grunberger, D.; Driscoll, J.; Kohn, K.; Pommier, Y. Hydroxylated aromatic inhibitors of HIV-1 integrase. J. Med. Chem. 1995, 38, 4171–4178. [Google Scholar]
- Savi, L.A.; Leal, P.C.; Vieira, T.O.; Rosso, R.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B.; Barardi, C.R.M.; Simões, C.M.O. Evaluation of anti-herpetic and antioxidant activities, and cytotoxic and genotoxic effects of synthetic alkyl-esters of gallic acid. Arzneimittelforschung 2005, 55, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-H.; Li, N.-G.; Tang, Y.-P.; Shi, Q.-P.; Zhang, W.; Zhang, P.-X.; Li, W.; Dong, Z.-X.; Duan, J.-A. Microwave-assisted esterification of gallic acid. Asian J. Chem. 2015, 27, 1351–1354. [Google Scholar]
- Mpousis, S.; Thysiadis, S.; Avramidis, N.; Katsamakas, S.; Efthimiopoulos, S.; Sarli, V. Synthesis and evaluation of gallocyanine dyes as potential agents for the treatment of Alzheimer’s disease and related neurodegenerative tauopathies. Eur. J. Med. Chem. 2016, 108, 28–38. [Google Scholar] [PubMed]
- Hirun, N.; Dokmaisrijan, S.; Tantishaiyakul, V. Experimental FTIR and theoretical studies of gallic acid–acetonitrile clusters. Spectrochim. Acta Mol. Biomol. Spectrosc. 2012, 86, 93–100. [Google Scholar]
- Kubo, I.; Fujita, K.-I.; Nihei, K.-I. Anti-Salmonella activity of alkyl gallates. J. Agric Food Chem. 2002, 50, 6692–6696. [Google Scholar] [CrossRef] [PubMed]
- Kubo, I.; Xiao, P.; Fujita, K.I. Anti-MRSA activity of alkyl gallates. Bioorg. Med. Chem. Lett. 2002, 12, 113–116. [Google Scholar]
- Kubo, I.; Fujita, K.-I.; Nihei, K.-I.; Masuoka, N. Non-antibiotic antibacterial activity of dodecyl gallate. Bioorg. Med. Chem. 2003, 11, 573–580. [Google Scholar]
- Silva, I.C.; Regasini, L.O.; Petrônio, M.S.; Silva, D.H.S.; Bolzani, V.S.; Belasque, J., Jr.; Sacramento, L.V.S.; Ferreira, H. Antibacterial activity of alkyl gallates against Xanthomonas citri subsp. Citri. J. Bacteriol. 2013, 195, 85–94. [Google Scholar] [CrossRef]
- Nihai, K.-i.; Nihe, i.A.; Kubo, I. Rational design of antimicrobial agents: Antifungal activity of alk(en)yl dihydroxybenzoates and dihydroxyphenyl alkanoates. Bioorg. Med. Chem. Lett. 2003, 13, 3993–3996. [Google Scholar]
- Serrano, A.; Palacios, C.; Roy, G.; Cespón, C.; Villar, M.L.; Nocito, M.; González-Porqué, P. Derivatives of gallic acid induce apoptosis in tumoral cell lines and inhibit lymphocyte proliferation. Arch. Biochem. Biophys. 1998, 350, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Interactive logP Calculator. Available online: https://www.molinspiration.com/services/logp.html (accessed on 3 June 2022).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.P.; Yepes, L.M.; Pérez-Castillo, Y.; Robledo, S.M.; de Sousa, D.P. Alkyl and aryl derivatives based on p-coumaric acid modification and inhibitory action against Leishmania braziliensis and Plasmodium falciparum. Molecules 2020, 25, 3178. [Google Scholar] [CrossRef]
- Ott, R.; Chibale, K.; Anderson, S.; Chipeleme, A.; Chaudhuri, M.; Guerrah, A.; Colowick, N.; Hill, G.S. New inhibitors of the trypanosome alternative oxidase inhibit Trypanosoma brucei brucei growth and respiration. Acta Trop. 2006, 100, 172–184. [Google Scholar] [CrossRef]
- Meco-Navas, A.; Ebiloma, G.U.; Martín-Domínguez, A.; Martínez-Benayas, I.; Cueto-Díaz, E.J.; Alhejely, A.S.; Balogun, E.O.; Saito, M.; Matsui, M.; Arai, N.; et al. SAR of 4-alkoxybenzoic acid inhibitors of the trypanosome alternative Oxidase. ACS Med. Chem. Lett. 2018, 9, 923–928. [Google Scholar] [CrossRef]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acid Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- Mazurek, J. Evaluation of ranking similarity in ordinal ranking problems. Acta Acad. Karviniensia 2011, 11, 119–128. [Google Scholar] [CrossRef]
- Shiba, T.; Kido, Y.; Sakamoto, K.; Inaoka, D.K.; Tsuge, C.; Tatsumi, R.; Takahashi, G.; Balogun, E.O.; Nara, T.; Aoki, T.; et al. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 4580–4585. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank. Available online: https://www.rcsb.org (accessed on 19 July 2022).
- Luo, Q.; Zhao, L.; Hu, J.; Jin, H.; Liu, Z.; Zhang, L. The scoring bias in reverse docking and the score normalization strategy to improve success rate of target fishing. PLoS ONE 2017, 12, e0171433. [Google Scholar] [CrossRef] [PubMed]
- Lapillo, M.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Giordano, A.; Poli, G. Extensive reliability evaluation of docking-based target-fishing strategies. Int. J. Mol. Sci. 2019, 20, 1023. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.C.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Balogun, E.O.; Inaoka, D.K.; Shiba, T.; Tsuge, C.; May, B.; Sato, T.; Kido, Y.; Nara, T.; Aoki, T.; Honma, T.; et al. Discovery of trypanocidal coumarins with dual inhibition of both the glycerol kinase and alternative oxidase of Trypanosoma brucei brucei. FASEB J. 2019, 33, 13002–13013. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven year itch: Pan-assay interference compounds (PAINS) in 2017–Utility and limitations. ACS Chem. Biol. 2018, 13, 36–244. [Google Scholar] [CrossRef]
- Michels, P.A.M.; Villafraz, O.; Pineda, E.; Alencar, M.B.; Cáceres, A.J.; Silber, A.M.; Bringaud, F. Carbohydrate metabolism in trypanosomatids: New insights revealing novel complexity, diversity and species-unique features. Exp. Parasitol. 2021, 224, 108102. [Google Scholar] [CrossRef]
- Pineda, E.; Thonnus, M.; Mazet, M.; Mourier, A.; Cahoreau, E.; Kulyk, H.; Dupuy, J.-W.; Biran, M.; Masante, C.; Allmann, S.; et al. Glycerol supports growth of the Trypanosoma brucei bloodstream forms in the absence of glucose: Analysis of metabolic adaptations on glycerol-rich conditions. PLoS Pathog. 2018, 14, e1007412. [Google Scholar] [CrossRef]
- Opperdoes, F.R.; Borst, P.; Fonck, K. The potential use of inhibitors of glycerol-3-phosphate oxidase for chemotherapy for African trypanosomiasis. FEBS Lett. 1976, 62, 169–172. [Google Scholar] [CrossRef]
- Nwaka, S.; Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006, 5, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Andrighetti-Fröhner, C.R.; Kolling, D.J.; Leal, P.C.; Cirne-Santos, C.C.; Yunes, R.A.; Nunes, R.J.; Trybala, E.; Bergström, T.; Frugulhetti, I.C.P.P.; et al. Anti-HSV-1 and anti-HIV-1 activity of gallic acid and pentyl gallate. Mem. Inst. Oswaldo Cruz 2008, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. What is the current value of MM/PBSA and MM/GBSA methods in drug discovery? Expert Opin. Drug Discov. 2021, 16, 1233–1237. [Google Scholar] [CrossRef]
- Khatkar, A.; Nanda, A.; Kumar, P.; Narasimhan, B. Synthesis, antimicrobial evaluation and QSAR studies of gallic acid derivatives. Arab. J. Chem. 2017, 10, S2870–S2880. [Google Scholar] [CrossRef] [Green Version]
- Hirumi, H.; Hirumi, K.; Doyle, J.J.; Cross, G.A.M. In vitro cloning of animal-infective bloodstream forms of Trypanosoma brucei. Parasitology 1980, 80, 371–382. [Google Scholar] [CrossRef]
- Ivens, A.C.; Blackwell, J.M. Unravelling the Leishmania genome. Curr. Opin. Genet. Dev. 1996, 6, 704–710. [Google Scholar] [CrossRef]
- Collins, S.J.; Gallo, R.C.; Gallagher, R.E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 1977, 270, 347–349. [Google Scholar] [CrossRef]
- Merschjohann, K.; Sporer, F.; Steverding, D.; Wink, M. In vitro effect of alkaloids on bloodstream forms of Trypanosoma brucei and T. congolense. Planta Med. 2001, 67, 623–627. [Google Scholar] [CrossRef]
- Mikus, J.; Steverding, D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue®. Parasitol. Int. 2000, 48, 265–269. [Google Scholar] [CrossRef]
- Huber, W.; Koella, J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific. Available online: http://www.eyesopen.com (accessed on 18 July 2022).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022 Reference Manual; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Ferreira, A.R.; Alves, D.d.N.; de Castro, R.D.; Perez-Castillo, Y.; de Sousa, D.P. Synthesis of coumarin and homoisoflavonoid derivatives and analogs: The search for new antifungal agents. Pharmaceuticals 2022, 15, 712. [Google Scholar] [CrossRef] [PubMed]
- Amber Parameter Database. Available online: http://amber.manchester.ac.uk/index.html (accessed on 18 July 2022).
- Li, P.; Merz, K.M., Jr. MCPB.py: A python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. Split the charge difference in two! A rule of thumb for adding proper amounts of ions in MD simulations. J. Chem. Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Alkyl Chain | T. brucei | HL-60 | ||
|---|---|---|---|---|---|
| MIC (μM) | GI50 (μM) | MIC (μM) | GI50 (μM) | ||
| 1 | methyl | 100 | 24.5 ± 4.5 | >100 | >100 |
| 2 | ethyl | 100 | 27.9 ± 2.4 | >100 | >100 |
| 3 | isopropyl | 100 | 8.4 ± 2.2 | >100 | 87.9 ± 8.2 |
| 4 | n-propyl | 10 | 3.0 ± 0.1 | >100 | 82.0 ± 8.2 |
| 5 | n-butyl | 10 | 3.3 ± 0.1 | >100 | 74.8 ± 4.5 |
| 6 | isopentyl | 10 | 3.2 ± 0.1 | >100 | 90.4 ± 21.6 |
| 7 | n-pentyl | 100 | 32.8 ± 0.9 | >100 | >100 |
| 8 | 2-methoxylethyl | 100 | 5.6 ± 1.3 | >100 | 99.2 ± 9.4 |
| Suramin | – | 1 | 0.04 ± 0.0 | >100 | >100 |
| Compound | Alkyl Chain | MIC (μM) | Growth Inhibition (% at 100 μM) |
|---|---|---|---|
| 1 | methyl | >100 | 2 |
| 2 | ethyl | >100 | 0 |
| 3 | isopropyl | >100 | 34 |
| 4 | n-propyl | 100 | 61 (50.4 μM) 1 |
| 5 | n-butyl | 100 | 58 (62.4 μM) 1 |
| 6 | isopentyl | 100 | 45 |
| 7 | n-pentyl | >100 | 0 |
| 8 | 2-methoxylethyl | 100 | 47 |
| Amphotericin B | – | 0.1 | 100 2 (0.04 μM) 1 |
| Compound | Alkyl Chain | MIC Ratio 1 | GI50 Ratio 1 |
|---|---|---|---|
| 1 | methyl | >1 | >4.1 |
| 2 | ethyl | >1 | >3.6 |
| 3 | isopropyl | >1 | 10.5 |
| 4 | n-propyl | >10 | 27.3 |
| 5 | n-butyl | >10 | 22.7 |
| 6 | isopentyl | >10 | 28.3 |
| 7 | n-pentyl | >1 | >3.0 |
| 8 | 2-methoxylethyl | >1 | 17.7 |
| Suramin | – | >100 | >2500 |
| Target | Pose | GOLDScore | CHEMScore | MM-PBSA Binding Energy (kcal/mol) 1 |
|---|---|---|---|---|
| G6PD substrate binding site | 1 | 11.65 | 3.42 | 4.02 |
| 2 | 18.08 | 2.81 | −3.98 | |
| 3 | 41.00 | 2.81 | 0.49 | |
| G6PD cofactor binding site | 1 | 48.14 | 15.45 | −7.25 |
| 2 | 38.90 | 11.85 | 0.71 | |
| 3 | 34.68 | 10.65 | −5.71 | |
| PKA1 | 1 | 45.45 | 14.07 | −8.09 |
| 2 | 40.13 | 8.65 | −7.93 | |
| 3 | 38.34 | 8.64 | −4.64 | |
| FT | 1 | 31.57 | 12.23 | −2.48 |
| 2 | 34.38 | 12.11 | −6.61 | |
| 3 | 33.83 | 11.51 | −1.95 | |
| IleRL | 1 | 22.53 | 7.97 | −4.58 |
| 2 | 20.68 | 6.58 | −1.76 | |
| 3 | 21.65 | 6.56 | −0.14 | |
| TAO with hydroxide anion | 1 | 32.65 | 16.33 | −12.45 |
| 2 | 52.42 | 14.07 | −10.24 | |
| 3 | 55.03 | 13.92 | −9.38 | |
| TAO without hydroxide anion | 1 | 61.06 | 21.49 | −8.02 |
| 2 | 55.84 | 19.80 | −2.58 | |
| 3 | 54.15 | 18.20 | −5.76 |
| Parameter | Compound 4 | Suramin |
|---|---|---|
| Physiochemical properties | ||
| Molecular weight (g/mol) | 212.2 | 1297.28 |
| Rotatable bonds | 4 | 22 |
| H-bond acceptors | 5 | 23 |
| H-bond donors | 3 | 12 |
| Fraction Csp3 | 0.3 | 0.04 |
| TSPA (A2) | 86.99 | 534.03 |
| Lipophilicity (Log Po/w) | ||
| iLOGP | 1.92 | −2.33 |
| XLOBP3 | 1.8 | 1.54 |
| MLOGP | 0.8 | 3.51 |
| Consensus | 1.38 | 2.64 |
| Absorption | ||
| Water solubility (Log S) | −2.32 | −7.78 |
| Gastrointestinal absorption (%) | 93.13 | 0 |
| Skin permeability (Log KP) | −2.819 | −2.735 |
| Distribution | ||
| Blood–brain permeability (Log BB) | −1.115 | −4.044 |
| CNS permeability (Log PS) | −3.362 | −4.943 |
| VDSS (human, Log(L/kg)) | 0.351 | −0.02 |
| Metabolism | ||
| CYP1A2 inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2C19 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| Excretion | ||
| Total clearance (Log(mL/min/kg)) | 0.443 | −4.246 |
| Renal OCT2 substrate | No | Yes |
| Toxicity | ||
| AMES toxicity | No | No |
| Max. tolerated dose (human, Log(mg/kg/day)) | −0.27 | 0.438 |
| hERG I inhibitor | No | No |
| hERG II inhibitor | No | No |
| Oral rat acute toxicity (LD50, mol/kg) | 1.993 | 2.482 |
| Oral rat chronic toxicity (LOAEL, Log(mg/kg_bw/day)) | 2.399 | 7.095 |
| Hepatotoxicity | No | No |
| Skin Sensitization | No | No |
| Compound (200 μM) | Motility after 5 min 1 | |
|---|---|---|
| 55 mM Glucose | 55 mM Glycerol | |
| – | + + + | + + |
| 1 | + + + | – |
| 2 | + + + | – |
| 3 | + + + | – |
| 4 | + + + | – |
| 5 | + + + | – |
| 6 | + + + | – |
| 7 | + + + | – |
| 8 | + + + | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steverding, D.; do Nascimento, L.G.; Perez-Castillo, Y.; de Sousa, D.P. Gallic Acid Alkyl Esters: Trypanocidal and Leishmanicidal Activity, and Target Identification via Modeling Studies. Molecules 2022, 27, 5876. https://doi.org/10.3390/molecules27185876
Steverding D, do Nascimento LG, Perez-Castillo Y, de Sousa DP. Gallic Acid Alkyl Esters: Trypanocidal and Leishmanicidal Activity, and Target Identification via Modeling Studies. Molecules. 2022; 27(18):5876. https://doi.org/10.3390/molecules27185876
Chicago/Turabian StyleSteverding, Dietmar, Lázaro Gomes do Nascimento, Yunierkis Perez-Castillo, and Damião Pergentino de Sousa. 2022. "Gallic Acid Alkyl Esters: Trypanocidal and Leishmanicidal Activity, and Target Identification via Modeling Studies" Molecules 27, no. 18: 5876. https://doi.org/10.3390/molecules27185876
APA StyleSteverding, D., do Nascimento, L. G., Perez-Castillo, Y., & de Sousa, D. P. (2022). Gallic Acid Alkyl Esters: Trypanocidal and Leishmanicidal Activity, and Target Identification via Modeling Studies. Molecules, 27(18), 5876. https://doi.org/10.3390/molecules27185876