


Electrochemical and Spectroscopic Characterization of Oxidized Intermediate Forms of Vitamin E

Abstract
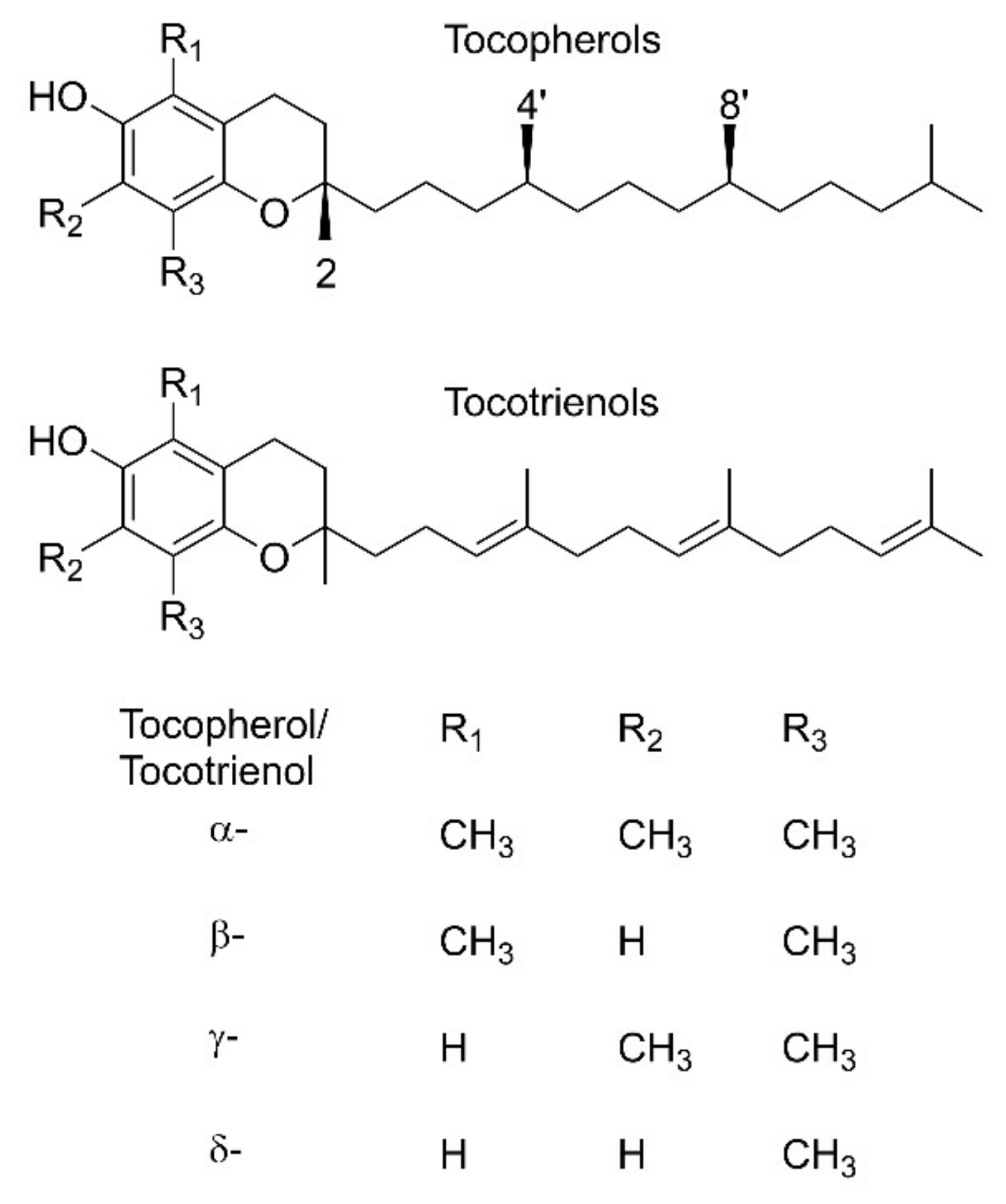
1. Introduction
2. Preparation of Oxidized Compounds through Chemical, Photochemical, and Electrochemical Processes
2.1. Hydrogen Atom Transfer (HAT) Reactions
2.2. Electrochemical Oxidation
2.2.1. Aqueous Conditions
2.2.2. Aprotic Conditions
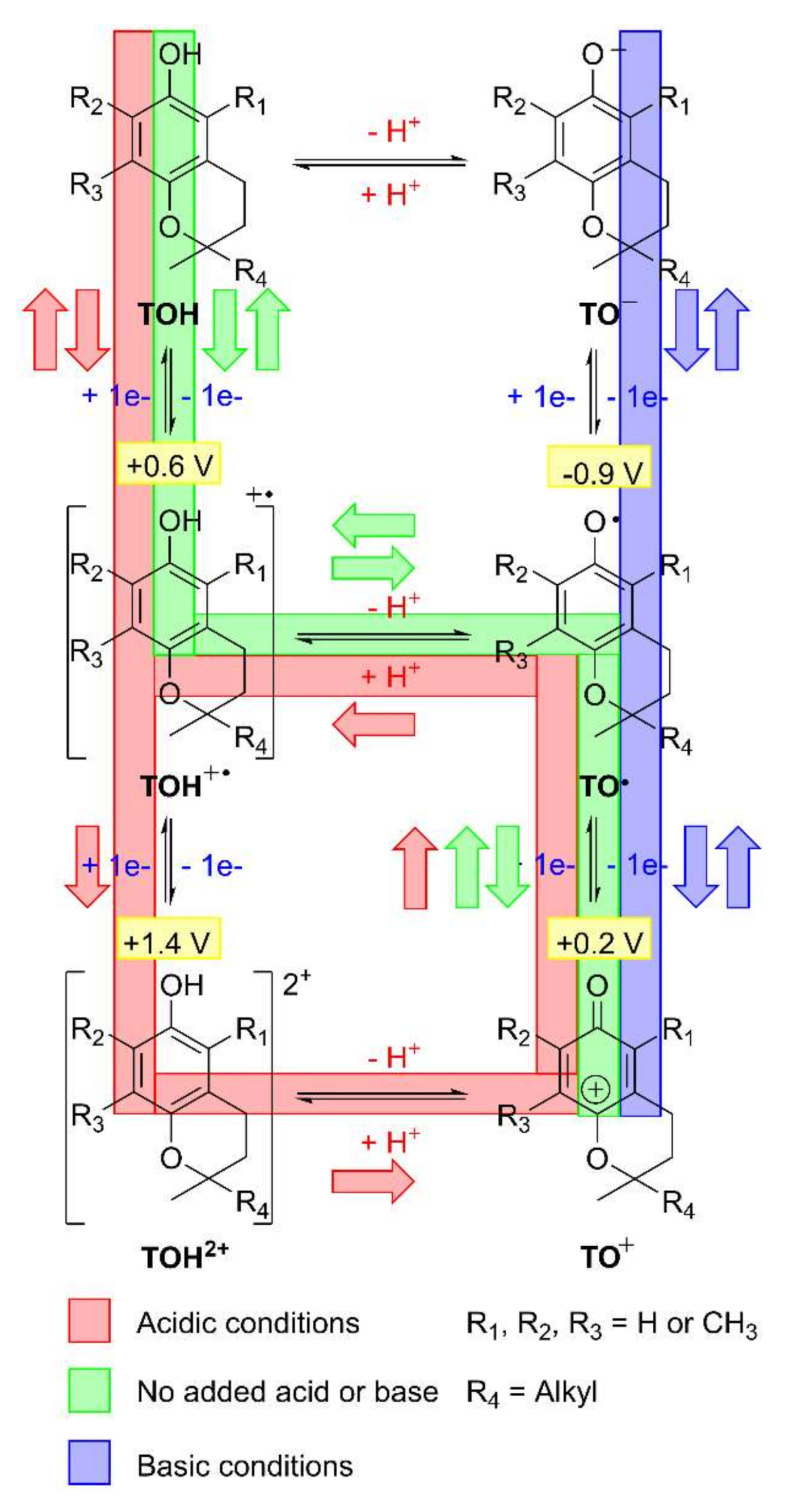
2.2.3. Square-Scheme Mechanism and pH Effects
2.2.4. Kinetic Measurements and Effects of Methyl-Substitution from Different Tocopherols/Tocotrienols
2.2.5. Difference between ECE and Disproportionation Mechanisms
2.2.6. Diamagnetic Cation
2.2.7. Quinone Methide
2.2.8. Hydrolysis Products
3. Spectroscopic Characterization of Each Class of Compound
3.1. Tocopherols and Tocotrienols (1)
3.2. Phenolate Anions (2)
3.3. Phenol Cation Radicals (3)
3.4. Phenoxyl Radicals (4)
3.5. Dications (5)
3.6. Diamagnetic Cations (Phenoxeniums) (6)
3.7. Hemiketals and Hemiketal Anions (8,9)
3.8. Quinones and Quinone Anions (10,11)
3.9. Quinone Methides (12)
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.M. The Pioneer History of Vitamin E. Vitam. Horm. 1962, 20, 379–387. [Google Scholar]
- Evans, H.M.; Emerson, O.H.; Emerson, G.A. The isolation from wheat germ oil of an alcohol, α-tocopherol, having the properties of vitamin E. J. Biol. Chem. 1936, 113, 319–332. [Google Scholar] [CrossRef]
- Fernholz, E. On the Constitution of α-Tocopherol. J. Am. Chem. Soc. 1938, 60, 700–705. [Google Scholar] [CrossRef]
- Karrer, P.; Fritzsche, H.; Ringier, B.H.; Salomon, H. Synthese des α-Tocopherols. Helv. Chim. Acta 1938, 21, 820–825. [Google Scholar] [CrossRef]
- Emerson, O.H. The structure of beta and gamma tocopherols. J. Am. Chem. Soc. 1938, 60, 1741–1742. [Google Scholar] [CrossRef]
- Niki, E.; Traber, M.G. A History of Vitamin E. Ann. Nutr. Metab. 2012, 61, 207–212. [Google Scholar] [CrossRef]
- Wang, X.Y.; Quinn, P.J. Vitamin E and its function in membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef]
- Olcott, H.S.; Mattill, H.A. The unsaponifiable lipids of lettuce: III. Antioxidant. J. Biol. Chem. 1931, 93, 65–70. [Google Scholar] [CrossRef]
- Mattill, H.A. Antioxidants. Annu. Rev. Biochem. 1947, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; Shmarakov, I.O.; Traber, M.G. Vitamin A and Vitamin E: Will the Real Antioxidant Please Stand Up? Annu. Rev. Nutr. 2021, 41, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Vitamin in Search of a Disease. J. Am. Med. Assoc. 1967, 201, 195–196. [Google Scholar] [CrossRef]
- Melton, L. The Antioxidant Myth. New Sci. 2006, 2563, 40–43. [Google Scholar]
- Brigelius-Flohé, R.; Davies, K.J.A. Is vitamin E an antioxidant, a regulator of signal transduction and gene expression, or a ‘junk’ food? Comments on the two accompanying papers: “Molecular mechanism of α-tocopherol action” by A. Azzi and “Vitamin E, antioxidant and nothing more” by M. Traber and J. Atkinson. Free Rad. Biol. Med. 2007, 43, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Rad. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef]
- Niki, E. Vitamin E function. Free Rad. Bio. Med. 2007, 43, 1466–1467. [Google Scholar] [CrossRef]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Rad. Biol. Med. 2007, 43, 16–21. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Vitamin E: Application of the Principles of Physical Organic Chemistry to the Exploration of its Structure and Function. Acc. Chem. Res. 1986, 19, 194–201. [Google Scholar] [CrossRef]
- Niki, E.; Noguchi, N. Dynamics of antioxidant action of vitamin E. Acc. Chem. Res. 2004, 37, 45–51. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of Cell-Proliferation by α-Tocopherol—Role of Protein-Kinase-C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar] [CrossRef]
- Tasinato, A.; Boscoboinik, D.; Bartoli, G.M.; Maroni, P.; Azzi, A. d-α-Tocopherol Inhibition of Vascular Smooth Muscle Cell Proliferation Occurs at Physiological Concentrations, Correlates with Protein Kinase C Inhibition, and is Independent of Its Antioxidant Properties. Proc. Natl. Acad. Sci. USA 1995, 92, 12190–12194. [Google Scholar] [CrossRef] [PubMed]
- Rimbach, G.; Minihane, A.M.; Majewicz, J.; Fischer, A.; Pallauf, J.; Virgli, F.; Weinberg, P.D. Regulation of cell signalling by vitamin E. Proc. Nutr. Soc. 2002, 61, 415–425. [Google Scholar] [CrossRef]
- Hosomi, A.; Arita, M.; Sato, Y.; Kiyose, C.; Ueda, T.; Igarashi, O.; Arai, H.; Inoue, K. Affinity For α-Tocopherol Transfer Protein as a Determinant of the Biological Activities of Vitamin E Analogs. FEBS Lett. 1997, 409, 105–108. [Google Scholar] [CrossRef]
- Min, K.C.; Kovall, R.A.; Hendrickson, W.A. Crystal Structure of Human α-Tocopherol Transfer Protein Bound to Its Ligand: Implications For Ataxia with Vitamin E Deficiency. Proc. Natl. Acad. Sci. USA 2003, 100, 14713–14718. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Burton, G.W.; Hughes, L.; Ingold, K.U.; Hidaka, H.; Malloy, M.; Kane, J.; Hyams, J.; Kayden, H.J. Discrimination between forms of vitamin E by humans with and without genetic abnormalities of lipoprotein metabolism. J. Lipid Res. 1992, 33, 1171–1182. [Google Scholar] [CrossRef]
- Pryor, W.A.; Cornicelli, J.A.; Devall, L.J.; Tait, B.; Trivedi, B.K.; Witiak, D.T.; Wu, M. A Rapid Screening Test to Determine the Antioxidant Potencies of Natural and Synthetic Antioxidants. J. Org. Chem. 1993, 58, 3521–3532. [Google Scholar] [CrossRef]
- Meagher, E.A.; Barry, O.P.; Lawson, J.A.; Rokach, J.; FitzGerald, G.A. Effects of Vitamin E on Lipid Peroxidation in Healthy Persons. J. Am. Med. Assoc. 2001, 285, 1178–1182. [Google Scholar] [CrossRef]
- The National Health Service (NHS) of the United Kingdom Recommends 3 or 4 mg per Day for Females and Males, respectively. Available online: https://www.nhs.uk/conditions/vitamins-and-minerals/vitamin-e (accessed on 14 August 2022).
- The National Institute of Health (NIH) of the USA Recommends 15 mg per Day for Adults. Available online: https://ods.od.nih.gov/factsheets/vitamine-consumer (accessed on 14 August 2022).
- Packer, J.E.; Slater, T.F.; Willson, R.L. Direct Observation of a Free Radical Interaction Between Vitamin E and Vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef]
- Atkinson, J.; Epand, R.F.; Epand, R.M. Tocopherols and tocotrienols in membranes: A critical review. Free Rad. Biol. Med. 2008, 44, 739–764. [Google Scholar] [CrossRef]
- Marquardt, D.; Williams, J.A.; Kucěrka, N.; Atkinson, J.; Wassall, S.R.; Katsaras, J.; Harroun, T.A. Tocopherol Activity Correlates with Its Location in a Membrane: A New Perspective on the Antioxidant Vitamin E. J. Am. Chem. Soc. 2013, 135, 7523–7533. [Google Scholar] [CrossRef]
- Atkinson, J.; Marquardt, D.; DiPasquale, M.; Harroun, T. From fat to bilayers: Understanding where and how vitamin E works. Free Rad. Biol. Med. 2021, 176, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ingold, K.U.; Burton, G.W.; Foster, D.O.; Hughes, L. Is methyl-branching in α-tocopherol’s “tail” important for its in vivo activity? Rat curative myopathy bioassay measurements of the vitamin E activity of three 2RS-n-alkyl-2,5,7,8-tetramethyl-6-hydroxychromans. Free Rad. Biol. Med. 1990, 9, 205–210. [Google Scholar] [CrossRef]
- Ranard, K.M.; Erdman, J.W., Jr. Effects of dietary RRR α-tocopherol vs all-racemic α-tocopherol on health outcomes. Nutr. Rev. 2018, 76, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Traber, M.G.; Acuff, R.V.; Walters, D.N.; Kayden, H.; Hughes, L.; Ingold, K.U. Human plasma and tissue α-tocopherol concentrations in response to supplementation with deuterated natural and synthetic vitamin E. Am. J. Clin. Nutr. 1998, 67, 669–684. [Google Scholar] [CrossRef]
- Smith, L.I.; Ungnade, H.E.; Hoehn, H.H.; Wawzonek, S. The Chemistry of Vitamin E. VI. The Addition of Dienes to Phenols and Hydroquinones. J. Org. Chem. 1939, 4, 311–317. [Google Scholar] [CrossRef]
- Schmölz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 7, 14–43. [Google Scholar] [CrossRef] [PubMed]
- Suarna, C.; Craig, D.C.; Cross, K.J.; Southwell-Keely, P.T. Oxidations of Vitamin E (α-Tocopherol) and Its Model Compound 2,2,5,7,8-Pentamethyl-6-hydroxychroman. A New Dimer. J. Org. Chem. 1988, 53, 1281–1284. [Google Scholar] [CrossRef]
- Valgimigli, L.; Banks, J.T.; Ingold, K.U.; Lusztyk, J. Kinetic Solvent Effects on Hydroxylic Hydrogen Atom Abstractions Are Independent of the Nature of the Abstracting Radical. Two Extreme Tests Using Vitamin E and Phenol. J. Am. Chem. Soc. 1995, 117, 9966–9971. [Google Scholar] [CrossRef]
- Bowry, V.W.; Ingold, K.U. Extraordinary Kinetic Behavior of the α-Tocopheroxyl (Vitamin E) Radical. J. Org. Chem. 1995, 60, 5456–5467. [Google Scholar] [CrossRef]
- Rosenau, T.; Kloser, E.; Gille, L.; Mazzini, F.; Netscher, T. Vitamin E Chemistry. Studies into Initial Oxidation Intermediates of α-Tocopherol: Disproving the Involvement of 5a-C-Centered “Chromanol Methide” Radicals. J. Org. Chem. 2007, 72, 3268–3281. [Google Scholar] [CrossRef]
- Lars, J.; Nilsson, G.; Doyle Davis, G., Jr.; Folkers, K. The Oxidative Dimerization of α, β, γ and δ-Tocopherols. Acta Chem. Scand. 1968, 22, 207–218. [Google Scholar] [CrossRef]
- Torres, L.M.; Gil, A.F.; Galicia, L.; González, I. Understanding the Difference between Inner- and Outer-Sphere Mechanisms: An Electrochemical Experiment. J. Chem. Educ. 1996, 73, 808–810. [Google Scholar] [CrossRef]
- Giacomelli, C.; Giacomelli, F.C.; Alves, L.O.; Timbola, A.K.; Spinelli, A. Electrochemistry of Vitamin E Hydro-Alcoholic Solutions. J. Braz. Chem. Soc. 2004, 15, 748–755. [Google Scholar] [CrossRef]
- Malyszko, J.; Karbarz, M. Electrochemical oxidation of trolox and α-tocopherol in acetic acid: A comparative study. J. Electroanal. Chem. 2006, 595, 136–144. [Google Scholar] [CrossRef]
- Okugaki, T.; Kasuno, M.; Maeda, K.; Kihara, S. Redox reactions of vitamin E in 1,2-dichloroethane with oxidants in water at the water/1,2-dichloroethane interface. J. Electroanal. Chem. 2010, 639, 67–76. [Google Scholar] [CrossRef]
- Wain, A.J.; Wadhawan, J.D.; France, R.R.; Compton, R.G. Biphasic redox chemistry of α-tocopherol: Evidence for electrochemically induced hydrolysis and dimerization on the surface of and within femtolitre droplets immobilized onto graphite electrodes. Phys. Chem. Chem. Phys. 2004, 6, 836–842. [Google Scholar] [CrossRef]
- Yao, W.W.; Peng, H.M.; Webster, R.D. Electrochemistry of α-Tocopherol (Vitamin E) and α-Tocopherol Quinone Films Deposited on Electrode Surfaces in the Presence and Absence of Lipid Bilayers. J. Phys. Chem. C 2009, 113, 21805–21814. [Google Scholar] [CrossRef]
- Yao, W.W.; Lau, C.; Hui, Y.; Poh, H.L.; Webster, R.D. Electrode-Supported Biomembrane for Examining Electron-Transfer and Ion-Transfer Reactions of Encapsulated Low Molecular Weight Biological Molecules. J. Phys. Chem. C 2011, 115, 2100–2113. [Google Scholar] [CrossRef]
- Parker, V.D. Anodic Alkyl Transfer from Hydroquinone Ethers. II. Anodic Oxidation of an α-Tocopherol Model Compound. J. Am. Chem. Soc. 1969, 91, 5380–5381. [Google Scholar] [CrossRef]
- Svanholm, U.; Bechgaard, K.; Parker, V.D. Electrochemistry in Media of Intermediate Acidity. VIII. Reversible Oxidation Products of the α-Tocopherol Model Compound. Cation Radical, Cation, and Dication. J. Am. Chem. Soc. 1974, 96, 2409–2413. [Google Scholar] [CrossRef]
- Marcus, M.F.; Hawley, M.D. Electrochemical Studies of The Redox Behavior of α-Tocopherol. Biochim. Biophys. Acta 1970, 201, 163–173. [Google Scholar] [CrossRef]
- Williams, L.L.; Webster, R.D. Electrochemically Controlled Chemically Reversible Transformation of α-Tocopherol (Vitamin E) into its Phenoxonium Cation. J. Am. Chem. Soc. 2004, 126, 12441–12450. [Google Scholar] [CrossRef]
- Lee, S.B.; Lin, C.Y.; Gill, P.M.W.; Webster, R.D. Transformation of α-Tocopherol (Vitamin E) and Related Chromanol Model Compounds into Their Phenoxonium Ions by Chemical Oxidation with the Nitrosonium Cation. J. Org. Chem. 2005, 70, 10466–10473. [Google Scholar] [CrossRef]
- Wilson, G.J.; Lin, C.Y.; Webster, R.D. Significant Differences in the Electrochemical Behavior of the α-, β-, γ- and δ-Tocopherols (Vitamin E). J. Phys. Chem. B 2006, 110, 11540–11548. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Willis, A.C.; Webster, R.D. Synthesis of the Phenoxonium Cation of an α-Tocopherol Model Compound Crystallized with Non-Nucleophilic [B(C6F5)4]− and (CB11H6Br6)− Anions. J. Am. Chem. Soc. 2006, 128, 9332–9333. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.D. New Insights into the Oxidative Electrochemistry of Vitamin E. Acc. Chem. Res. 2007, 40, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.M.; Webster, R.D. Investigation into Phenoxonium Cations Produced during the Electrochemical Oxidation of Chroman-6-ol and Dihydrobenzofuran-5-ol Substituted Compounds. J. Org. Chem. 2008, 73, 2169–2175. [Google Scholar] [CrossRef]
- Yao, W.W.; Peng, H.M.; Webster, R.D.; Gill, P.M.W. Variable Scan Rate Cyclic Voltammetry and Theoretical Studies on Tocopherol (Vitamin E) Model Compounds. J. Phys. Chem. B 2008, 112, 6847–6855. [Google Scholar] [CrossRef][Green Version]
- Peng, H.M.; Choules, B.F.; Yao, W.W.; Zhang, Z.; Webster, R.D.; Gill, P.M.W. Long-Lived Radical Cations as Model Compounds for the Reactive One-Electron Oxidation Product of Vitamin E. J. Phys. Chem. B 2008, 112, 10367–10374. [Google Scholar] [CrossRef]
- Naqvi, K.R.; Li, H.; Melø, T.B.; Zhang, Y.; Webster, R.D. Spectroscopic Characterization of Neutral and Cation Radicals of α-Tocopherol and Related Molecules: A Satisfactory Denouement. J. Phys. Chem. A 2010, 114, 10795–10802. [Google Scholar] [CrossRef]
- Chen, S.S.; Peng, H.M.; Webster, R.D. Infrared and UV-vis spectra of phenoxonium cations produced during the oxidation of phenols with structures similar to vitamin E. Electrochim. Acta 2010, 55, 8863–8869. [Google Scholar] [CrossRef]
- Tan, Y.S.; Webster, R.D. Electron Transfer Reactions between the Diamagnetic Cation of α-Tocopherol (Vitamin E) and β-Carotene. J. Phys. Chem. B 2011, 115, 4244–4250. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Chen, S.S.; Hong, W.M.; Kan, J.M.; Kwek, E.S.E.; Lim, S.Y.; Lim, Z.H.; Tessensohn, M.E.; Zhang, Y.-L.; Webster, R.D. The role of low levels of water in the electrochemical oxidation of α-tocopherol (vitamin E) and other phenols in acetonitrile. Phys. Chem. Chem. Phys. 2011, 13, 12745–12754. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.D. Voltammetry of the Liposoluble Vitamins (A, D, E and K) in Organic Solvents. Chem. Rec. 2012, 12, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Novianti, M.; Tessensohn, M.E.; Hirao, H.; Webster, R.D. Optimizing the Lifetimes of Phenoxonium Cations Derived from Vitamin E via Structural Modifications. Org. Biomol. Chem. 2015, 13, 11732–11739. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.D. Electrochemical Modeling of Biological Processes. In Organic Electrochemistry, 5th ed.; Chapter 40; Hammerich, O., Speiser, B., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 1543–1567. ISBN 978-1-4200-8401-6. [Google Scholar]
- Webster, R.D. In situ electrochemical-ATR-FTIR spectroscopic studies on solution phase 2,4,6-tri-substituted phenoxyl radicals. Electrochem. Commun. 2003, 5, 6–11. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.H.; Jimenez, P.J.; Kelly, M.J. Reversible Dimerization of Phenoxyl Radicals Formed by Anodic Oxidation of Phenolates. J. Electroanal. Chem. 1984, 163, 145–157. [Google Scholar] [CrossRef]
- Hapiot, P.; Pinson, J. Multiple reaction pathways for the oxidation of 2,6-diphenylphenolates. J. Electroanal. Chem. 1993, 362, 257–265. [Google Scholar] [CrossRef]
- Francke, R.; Quell, T.; Wiebe, A.; Waldvogel, S.R. Oxygen Containing Compounds. In Organic Electrochemistry, 5th ed.; Chapter 26; Hammerich, O., Speiser, B., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 981–1034. ISBN 978-1-4200-8401-6. [Google Scholar]
- Webster, R.D. Voltammetric studies on the α-tocopherol anion and α-tocopheroxyl (Vitamin E) radical in acetonitrile. Electrochem. Commun. 1999, 1, 581–584. [Google Scholar] [CrossRef]
- Chan, Y.Y.; Yue, Y.; Li, Y.; Webster, R.D. Electrochemical/Chemical Oxidation of Bisphenol A in a Four-Electron/Two-Proton Process in Aprotic Organic Solvents. Electrochim. Acta 2013, 112, 287–294. [Google Scholar] [CrossRef]
- Chan, Y.-Y.; Webster, R.D. Electrochemical Oxidation of the Phenolic Benzotriazoles UV-234 and UV-327 in Organic Solvents. ChemElectroChem 2019, 6, 4297–4306. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Electrochemical and Homogeneous Proton-Coupled Electron Transfers: Concerted Pathways in the One-Electron Oxidation of a Phenol Coupled with an Intramolecular Amine-Driven Proton Transfer. J. Am. Chem. Soc. 2006, 128, 4552–4553. [Google Scholar] [CrossRef]
- Mukai, K.; Uemoto, Y.; Fukuhara, M.; Nagaoka, S.-I.; Ishizu, K. ENDOR Study of the Cation Radicals of Vitamin E Derivatives. Relation between Antioxidant Activity and Molecular Structure. Bull. Chem. Soc. Jpn. 1992, 65, 2016–2020. [Google Scholar] [CrossRef]
- Mukai, K.; Ohbayashi, S.; Nagaoka, S.-I.; Ozawa, T.; Azuma, N. X-Ray Crystallographic Studies of Vitamin E Derivatives. Relationship between Antioxidant Activity and Molecular Structure. Bull. Chem. Soc. Jpn. 1993, 66, 3808–3810. [Google Scholar] [CrossRef]
- Shi, R.R.S.; Tessensohn, M.E.; Lauw, S.J.L.; Foo, N.A.B.Y.; Webster, R.D. Tuning the reduction potential of quinones by controlling the effects of hydrogen bonding, protonation and proton-coupled electron transfer reactions. Chem. Commun. 2019, 55, 2277–2280. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Webster, R.D. Absorption of Water into Organic Solvents used for Electrochemistry under Conventional Operating Conditions. Anal. Chem. 2011, 83, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Linschitz, H. Hydrogen-Bonding and Protonation Effects in Electrochemistry of Quinones in Aprotic Solvents. J. Am. Chem. Soc. 1997, 119, 6384–6391. [Google Scholar] [CrossRef]
- Quan, M.; Sanchez, D.; Wasylkiw, M.F.; Smith, D.K. Voltammetry of Quinones in Unbuffered Aqueous Solution: Reassessing the Roles of Proton Transfer and Hydrogen Bonding in the Aqueous Electrochemistry of Quinones. J. Am. Chem. Soc. 2007, 129, 12847–12856. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Chng, E.L.K.; Chng, C.Y.L.; Poh, H.L.; Webster, R.D. Hydrogen Bonding Interactions between Water and the One- and Two-Electron Reduced forms of Vitamin K1: Applying Quinone Electrochemistry to Determine the Moisture Content of Non-Aqueous Solvents. J. Am. Chem. Soc. 2009, 131, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Chng, E.L.K.; Chua, L.P.-L.; Liu, W.Z.; Webster, R.D. Voltammetric Method for Determining the Trace Moisture Content of Organic Solvents Based on Hydrogen-Bonding Interactions with Quinones. Anal. Chem. 2010, 82, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- Tessensohn, M.E.; Hirao, H.; Webster, R.D. Electrochemical Properties of Phenols and Quinones in Organic Solvents are Strongly Influenced by Hydrogen-Bonding with Water. J. Phys. Chem. C 2013, 117, 1081–1090. [Google Scholar] [CrossRef]
- Tessensohn, M.E.; Webster, R.D. Using voltammetry to measure hydrogen-bonding interactions in non-aqueous solvents: A mini-review. Electrochem. Commun. 2016, 62, 38–43. [Google Scholar] [CrossRef]
- Tessensohn, M.E.; Webster, R.D. Voltammetric applications of hydrogen bonding and proton-coupled electron-transfer reactions of organic molecules. Curr. Opin. Electrochem. 2019, 15, 27–33. [Google Scholar] [CrossRef]
- Amatore, C.; Savéant, J.M. ECE and disproportionation: Part V. Stationary state general solution application to linear sweep voltammetry. J. Electroanal. Chem. 1977, 85, 27–46. [Google Scholar] [CrossRef]
- Costentin, C. Electrochemical Approach to the Mechanistic Study of Proton-Coupled Electron Transfer. Chem. Rev. 2008, 108, 2145–2179. [Google Scholar] [CrossRef] [PubMed]
- Speiser, B.; Rieker, A. Electrochemical Oxidations. Part 1. Phenoxy cations in the Anodic Oxidation of Phenolic Compounds. J. Chem. Res. 1977, 9, 314–315. [Google Scholar]
- Speiser, B.; Rieker, A. Electrochemical oxidations: Part IV. Electrochemical investigations into the behaviour of 2,6-di-tert-butyl-4-(4-dimethylaminophenyl)-phenol part 1. Phenol and the species derived from it: Phenoxy radical, phenolate anion and phenoxenium cation. J. Electroanal. Chem. 1979, 102, 373–395. [Google Scholar] [CrossRef]
- Speiser, B.; Rieker, A. Electrochemical oxidations: Part V. Electrochemical investigations into the behaviour of 2,6-di-tert-butyl-4-(4-dimethylamino-phenyl)-phenol Part 2: Anodic oxidation in the presence of 2,6-dimethylpyridine and the mechanism of the formation of the phenoxenium ion. J. Electroanal. Chem. 1980, 110, 231–246. [Google Scholar] [CrossRef]
- Kim, E.K.; Kochi, J.K. Oxidative Aromatic Nitration with Charge-Transfer Complexes of Arenes and Nitrosonium salts. J. Org. Chem. 1989, 54, 1692–1702. [Google Scholar] [CrossRef]
- Novak, M.; Glover, S.A. Generation and Trapping of the 4-Biphenylyloxenium Ion by Water and Azide: Comparisons with the 4-Biphenylylnitrenium Ion. J. Am. Chem. Soc. 2004, 126, 7748–7749. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Wang, J.; Platz, M.S.; Novak, M. Direct Detection of a Transient Oxenium Ion in Water Generated by Laser Flash Photolysis. J. Am. Chem. Soc. 2007, 129, 14566–14567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Jin, K.J.; Leopold, S.H.; Wang, J.; Peng, H.-L.; Platz, M.S.; Xue, J.; Phillips, D.L.; Glover, S.A.; Novak, M. Characterization of Reactive Intermediates Generated During Photolysis of 4-Acetoxy-4-aryl-2,5-cyclohexadienones: Oxenium Ions and Aryloxy Radicals. J. Am. Chem. Soc. 2008, 130, 16021–16030. [Google Scholar] [CrossRef] [PubMed]
- Hanway, P.J.; Winter, A.H. Phenyloxenium Ions: More Like Phenylnitrenium Ions than Isoelectronic Phenylnitrenes? J. Am. Chem. Soc. 2011, 133, 5086–5093. [Google Scholar] [CrossRef][Green Version]
- Hanway, P.J.; Xue, J.; Bhattacharjee, U.; Milot, M.J.; Ruixue, Z.; Phillips, D.L.; Winter, A.H. Direct Detection and Reactivity of the Short-Lived Phenyloxenium Ion. J. Am. Chem. Soc. 2013, 135, 9078–9082. [Google Scholar] [CrossRef]
- Rosenau, T.; Potthast, A.; Elder, T.; Kosma, P. Stabilization and First Direct Spectroscopic Evidence of the o-Quinone Methide Derived from Vitamin E. Org. Lett. 2002, 4, 4285–4288. [Google Scholar] [CrossRef]
- Bohmdorfer, S.; Rosenau, T. Oxidation with a “Stopover”—Stable Zwitterions as Intermediates in the Oxidation of α-Tocopherol (Vitamin E) Model Compounds to their Corresponding ortho-Quinone Methides. ChemistryOpen 2021, 10, 421–429. [Google Scholar] [CrossRef]
- Patel, A.; Netscher, T.; Gille, L.; Mereiter, K.; Rosenau, T. Novel tocopheryl compounds XXV: Synthesis and comparison of the para-quinones of all four homologous tocopherol model compounds and their 3,4-dehydro derivatives. Tetrahedron 2007, 63, 5312–5318. [Google Scholar] [CrossRef]
- Dürckheimer, W.; Cohen, L.A. Mechanisms of α-tocopherol oxidation: Synthesis of the highly labile 9-hydroxy-α-tocopherone. Biochem. Biophys. Res. Commun. 1962, 9, 262–265. [Google Scholar] [CrossRef]
- Borchardt, R.T.; Cohen, L.A. Stereopopulation control. IV. Facilitation of intramolecular conjugate addition of the hydroxyl group. J. Am. Chem. Soc. 1973, 95, 8308–8313. [Google Scholar] [CrossRef]
- Nilsson, A.; Palmquist, U.; Pettersson, T.; Ronlán, A. Anodic oxidation of phenolic compounds. Part 5. Anodic methoxylation of phenols. A simple synthesis of quinones, quinone acetals, and 4-methyl-α-methoxycyclohexa-2,5-dienones. J. Chem. Soc. Perkin Trans. 1 1978, 7, 696–707. [Google Scholar] [CrossRef]
- Omura, K. Iodine oxidation of alpha-tocopherol and its model compound in alkaline methanol: Unexpected isomerization of the product quinone monoketals. J. Org. Chem. 1989, 54, 1987–1990. [Google Scholar] [CrossRef]
- Pelter, A.; Elgendy, S.M.A. Phenolic oxidations with phenyliodonium diacetate. J. Chem. Soc. Perkin Trans. 1 1993, 16, 1891–1896. [Google Scholar] [CrossRef]
- Baker, J.K.; Myers, C.W. One-dimensional and two-dimensional 1H- and 13C-nuclear magnetic resonance (NMR) analysis of vitamin E raw materials or analytical reference standards. Pharm. Res. 1991, 8, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Lodge, J.K. Mass spectrometry approaches for vitamin E research. Biochem. Soc. Trans. 2008, 36, 1066–1070. [Google Scholar] [CrossRef]
- Tahir, H.E.; Xiaobo, Z.; Jianbo, X.; Mahunu, G.K.; Jiyong, S.; Xu, J.-L.; Sun, D.-W. Recent Progress in Rapid Analyses of Vitamins, Phenolic, and Volatile Compounds in Foods Using Vibrational Spectroscopy Combined with Chemometrics: A Review. Food Anal. Methods 2019, 12, 2361–2382. [Google Scholar] [CrossRef]
- Nagy, K.; Courtet-Compondu, M.-C.; Holst, B.; Kussmann, M. Comprehensive analysis of vitamin E constituents in human plasma by liquid chromatography-mass spectrometry. Anal. Chem. 2007, 79, 7087–7096. [Google Scholar] [CrossRef]
- Wiley Online Databases. Available online: https://sciencesolutions.wiley.com/ (accessed on 19 August 2022).
- Bernal, J.; Mendiola, J.A.; Ibáñez, E.; Cifuentes, A. Advanced analysis of nutraceuticals. J. Pharm. Biomed. Anal. 2011, 55, 758–774. [Google Scholar] [CrossRef]
- Nakanishi, I.; Miyazaki, K.; Shimada, T.; Iizuka, Y.; Inami, K.; Mochizuki, M.; Urano, S.; Okuda, H.; Ozawa, T.; Fukuzumi, S.; et al. Kinetic study of the electron-transfer oxidation of the phenolate anion of a vitamin E model by molecular oxygen generating superoxide anion in an aprotic medium. Org. Biomol. Chem. 2003, 1, 4085–4088. [Google Scholar] [CrossRef]
- Lauw, S.J.L.; Yeo, J.Y.H.; Zhong, C.; Webster, R.D. Comparing the Relative Reactivities of Food and Vitamin Molecules Towards Electrochemically Generated Superoxide. ChemElectroChem 2017, 4, 1190–1198. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R.; Kuwta, K.; Usui, S.; Uni, B. Electrochemical and Mechanistic Study of Reactivities of α, β, γ, and δ-Tocopherol toward Electrogenerated Superoxide in N,N-Dimethylformamide through Proton-Coupled Electron Transfer. Antioxidants 2022, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, A.G.; Moreno, R.B.; Navaratnam, S.; Wilkinson, J.A.; Bisby, R.H. Generation of superoxide and singlet oxygen from α-tocopherolquinone and analogues. Free Radic. Res. 2007, 41, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Bonin, J.; Robert, M. Photoinduced Proton-Coupled Electron Transfers in Biorelevant Phenolic Systems. Photochem. Photobiol. 2011, 87, 1190–1203. [Google Scholar] [CrossRef]
- Yusof, M.S.B.M.; Song, H.; Debnath, T.; Lowe, B.; Yang, M.; Loh, Z.-H. Ultrafast proton transfer of the aqueous phenol radical cation. Phys. Chem. Chem. Phys. 2022, 24, 12236–12248. [Google Scholar] [CrossRef]
- Mukai, K.; Tsuzuki, N.; Ishizu, K.; Ouchi, S.; Fukuzawa, K. Electron Spin Resonance and Electron Nuclear Double Resonance Studies of Cation Radicals Derived From Tocopherol Model Compounds. Chem. Phys. Lipids 1984, 35, 199–208. [Google Scholar] [CrossRef]
- Lehtovuori, P.; Joela, H. Radical Cations of Vitamin E. Phys. Chem. Chem. Phys. 2002, 4, 1928–1933. [Google Scholar] [CrossRef]
- Edge, R.; Land, E.J.; McGarvey, D.; Mulroy, L.; Truscott, T.G. Relative One-Electron Reduction Potentials of Carotenoid Radical Cations and the Interactions of Carotenoids with the Vitamin E Radical Cation. J. Am. Chem. Soc. 1998, 120, 4087–4090. [Google Scholar] [CrossRef]
- Parker, A.W.; Bisby, R.H. Time-Resolved Resonance Raman Spectroscopy of α-Tocopheroxyl and Related Radicals in Solvent, Micellar and Membrane Systems. J. Chem. Soc. Faraday Trans. 1993, 89, 2873–2878. [Google Scholar] [CrossRef]
- Thomas, M.J.; Bielski, B.H.J. Oxidation and Reaction of Trolox c, a Tocopherol Analogue, in Aqueous Solution. A Pulse-Radiolysis Study. J. Am. Chem. Soc. 1989, 111, 3315–3319. [Google Scholar] [CrossRef]
- Mukai, K.; Tsuzuki, N.; Ishizu, K.; Ouchi, S.; Fukuzawa, K. Electron Nuclear Double Resonance Studies of Radicals Produced by the PbO2 Oxidation of α-Tocopherol and Its Model Compound in Solution. Chem. Phys. Lipids 1981, 29, 129–135. [Google Scholar] [CrossRef]
- Matsuo, M.; Matsumoto, S.; Ozawa, T. Electron Spin Resonance Spectra and Hyperfine Coupling Constants of the α-Tocopheroxyl and 2,2,5,7,8-Pentamethylchroman-6-oxyl Radicals Derived from Vitamin E and its Model and Deuterated Model Compounds. Org. Magn. Reson. 1983, 21, 261–264. [Google Scholar] [CrossRef]
- Marcus, M.F.; Hawley, M.D. Electrochemical Studies of The Redox Behavior of α-Tocopherylquinone and a Related Model Quinone. Biochim. Biophys. Acta 1970, 222, 163–173. [Google Scholar] [CrossRef]
- Lim, Z.H.; Chng, E.L.K.; Hui, Y.; Webster, R.D. The Hydrogen-Bonded Dianion of Vitamin K1 Produced in Aqueous-Organic Solutions Exists in Equilibrium with its Hydrogen-Bonded Semiquinone Anion Radical. J. Phys. Chem. B 2013, 117, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Danylchuk, D.I.; Jouard, P.-H.; Klymchenko, A.S. Targeted Solvatochromic Fluorescent Probes for Imaging Lipid Order in Organelles under Oxidative and Mechanical Stress. J. Am. Chem. Soc. 2021, 143, 912–924. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, W.; Li, X.; Ma, H. Recent advances in fluorescent probes for lipid droplets. Chem. Commun. 2022, 58, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Sakaya, A.; Durantini, A.M.; Gidi, Y.; Šverko, T.; Wieczny, V.; McCain, J.; Cosa, G. Fluorescence-Amplified Detection of Redox Turnovers in Supported Lipid Bilayers Illuminates Redox Processes of α-Tocopherol. ACS Appl. Mater. Interfaces 2022, 14, 13872–13882. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solvent | λmax/nm | Literature |
|---|---|---|---|
| α–TOH | hexane | 298 | [62] |
| CH3CN | 294 | [54] | |
| (CH3)α–TOH | CH3CN | 294 | [55] |
| (CH3)α–TO− | CH3CN | 325 | [114] |
| (CH3)α–TOH+• | CH2Cl2/CF3COOH | 461 | [62] |
| CH2Cl2/CF3COOH | 308,465 | [56] | |
| α–TOH+• | CH3CN/CF3SO3H | 301, 464 | [54] |
| α–TO• | chlorobenzene | 424 | [41] |
| (CH3)α–TO• | CH2Cl2 | 425 | [62] |
| α–TO+ | CH3CN | 298, 425 | [54] |
| (CH3)α–ΤO+ | CH3CN | 298, 425 | [55] |
| (CH3)α–TOQ(OH) | CH3OH | 239 | [104] |
| (CH3)α–TOQ | CH3CN | 258, 266 | [63] |
| (COOH)α–TOQ−• | H2O | 440 | [117] |
| Compound b | UV-Vis and Pulse Radiolysis | Infrared and Raman | EPR | NMR | X-ray Crystallography |
|---|---|---|---|---|---|
| Literature Numbers from Reference Section | |||||
| TO− (2) | [114] | ||||
| TOH+• (3) | [52,54,56,62,122] | [56,123] | [52,54,56,120,121] | ||
| TO• (4) | [30,41,62,124] | [123] | [41,69,125,126] | ||
| TO+ (6) | [54,55,56] | [54,55,56,63] | [55] | [57] | |
| TOQ(OH) (8) | [104] | [63,104] | [104] | ||
| TOQ (10) | [63,104] | [63,104] | [104] | ||
| TOQ−•(11) | [117] | ||||
| QM (12) | [100,101] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, R.D. Electrochemical and Spectroscopic Characterization of Oxidized Intermediate Forms of Vitamin E. Molecules 2022, 27, 6194. https://doi.org/10.3390/molecules27196194
Webster RD. Electrochemical and Spectroscopic Characterization of Oxidized Intermediate Forms of Vitamin E. Molecules. 2022; 27(19):6194. https://doi.org/10.3390/molecules27196194
Chicago/Turabian StyleWebster, Richard D. 2022. "Electrochemical and Spectroscopic Characterization of Oxidized Intermediate Forms of Vitamin E" Molecules 27, no. 19: 6194. https://doi.org/10.3390/molecules27196194
APA StyleWebster, R. D. (2022). Electrochemical and Spectroscopic Characterization of Oxidized Intermediate Forms of Vitamin E. Molecules, 27(19), 6194. https://doi.org/10.3390/molecules27196194