
Multicomponent Molecular Systems Based on Porphyrins, 1,3,5-Triazine and Carboranes: Synthesis and Characterization
 , , ,
, , ,
Abstract
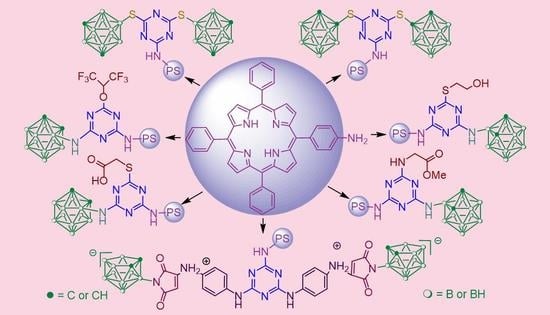
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
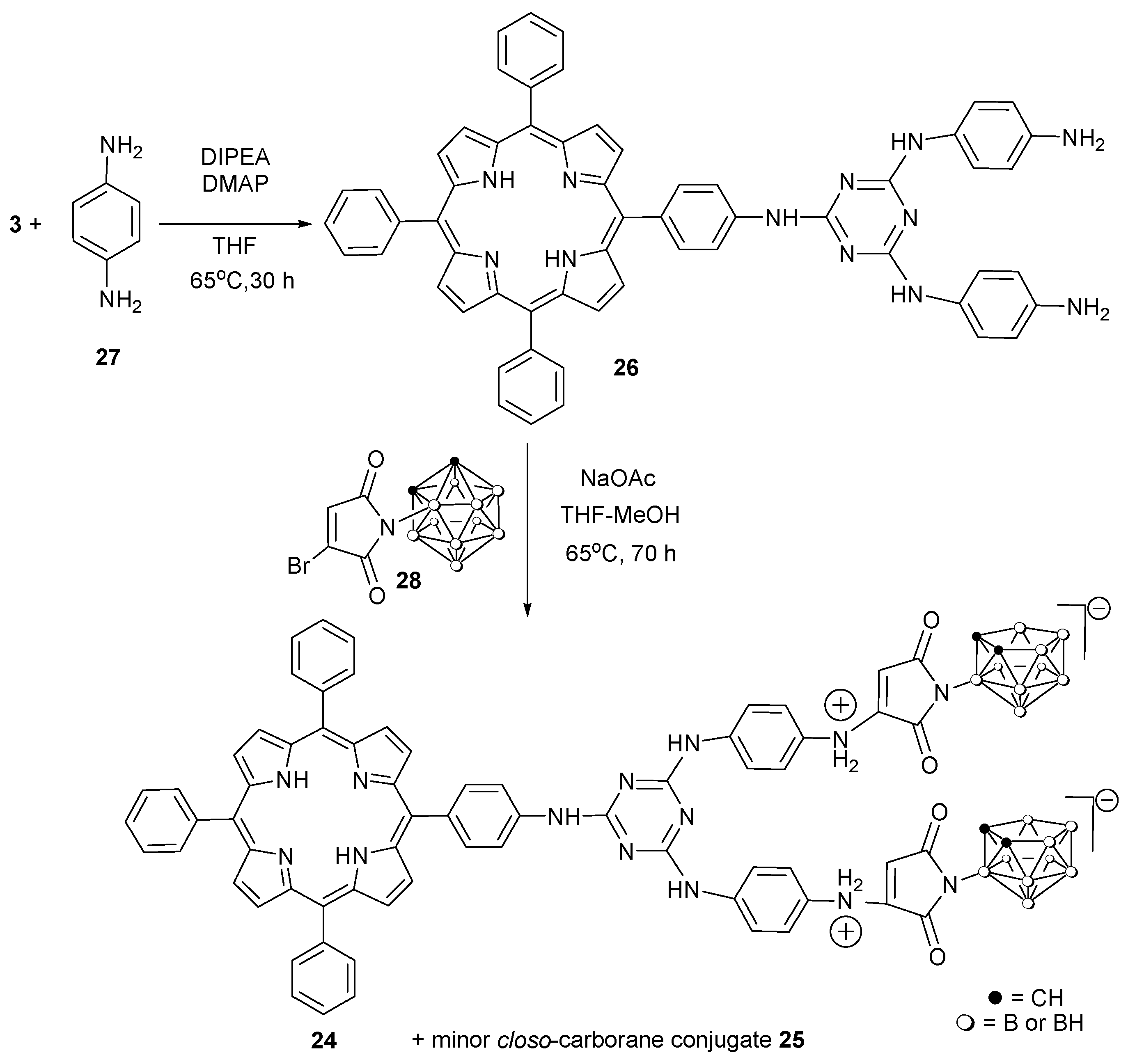
2.1. Synthesis
2.2. Spectroscopic Data
2.3. UV–Visible Absorption and Fluorescence Spectra
2.4. Cytotoxicity
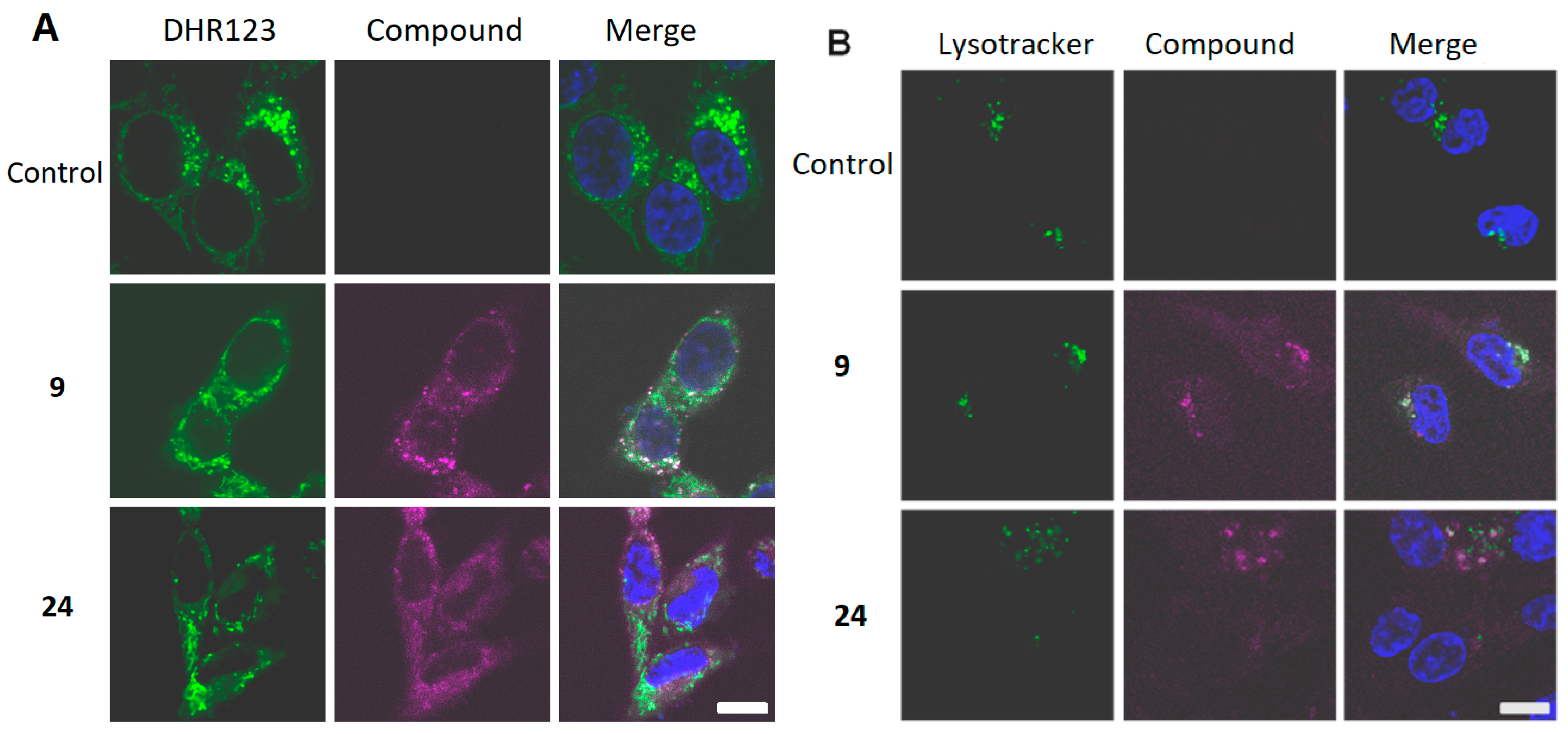
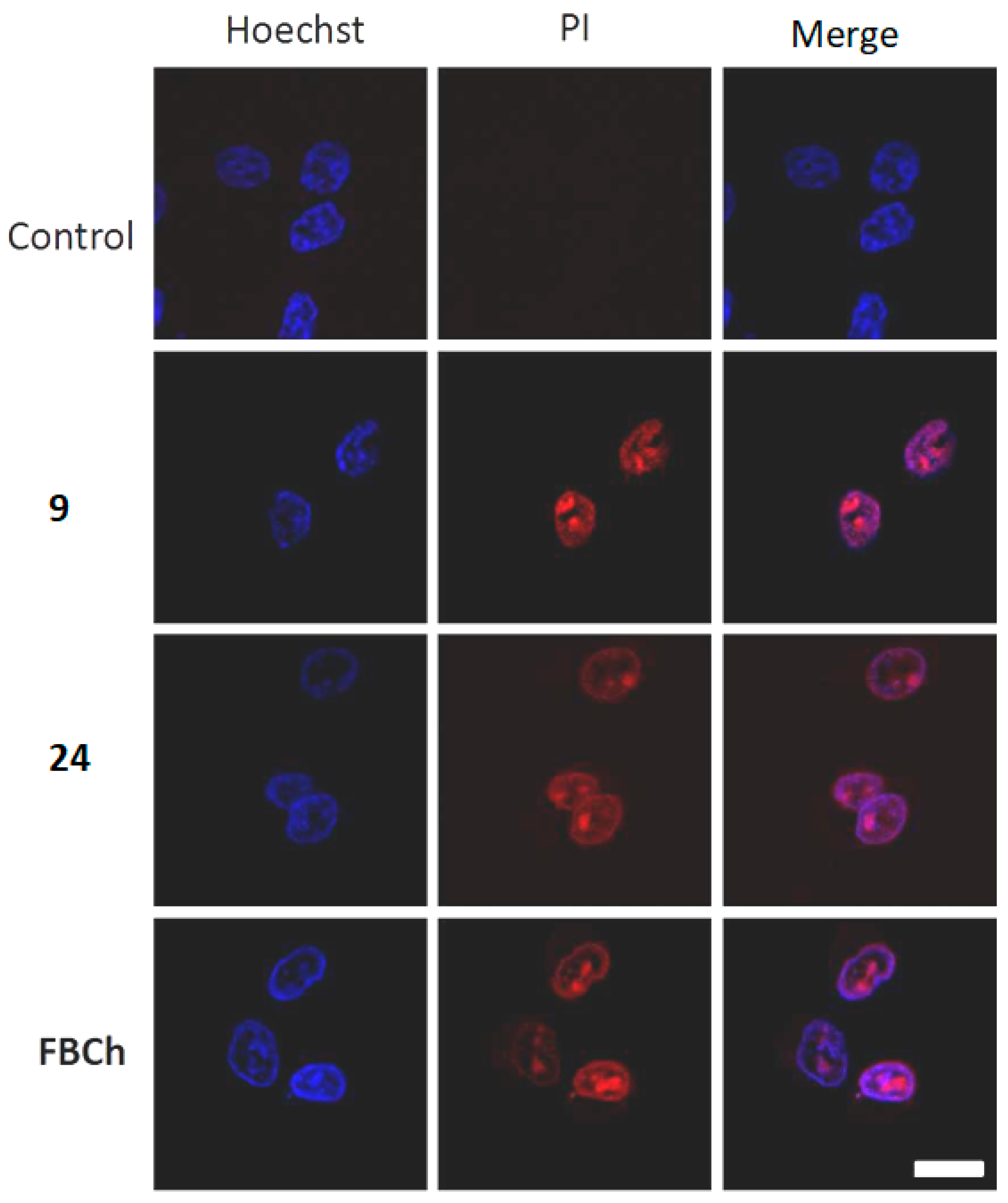
2.5. Intracellular Localization and Photoinduced Cell Death
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Analysis of Compounds
3.3. Fluorescence Spectroscopy
3.4. Cell Culture
3.5. Cytotoxicity Studies
3.6. Confocal Laser Scanning Fluorescence Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. Handbook of Porphyrin Science; World Scientific Publishing: Singapore, 2012; pp. 10–12. [Google Scholar]
- Mukhopadhyay, R.D.; Kim, Y.; Koo, J.; Kim, K. Porphyrin boxes. Acc. Chem. Res. 2018, 51, 2730–2738. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Zhua, W.; Xie, Y. Fluorescent and colorimetric ion probes based on conjugated oligopyrroles. Chem. Soc. Rev. 2015, 44, 1101–1112. [Google Scholar] [CrossRef]
- Ding, Y.; Zhu, W.H.; Xie, Y. Development of ion chemosensors based on porphyrin analogues. Chem. Rev. 2017, 117, 2203–2256. [Google Scholar] [CrossRef]
- Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Silva, A.M.N.; Medforth, C.J.; Freire, C. Iron(III) fluorinated porphyrins: Greener chemistry from synthesis to oxidative catalysis reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef]
- Li, L.L.; Diau, E.W.G. Porphyrin-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 291–304. [Google Scholar] [CrossRef]
- Urbani, M.; Grätzel, M.; Nazeeruddin, M.K.; Torres, T. Meso-substituted porphyrins for dye-sensitized solar cells. Chem. Rev. 2014, 114, 12330–12396. [Google Scholar] [CrossRef] [PubMed]
- Lukšiene, Ž. New approach to inactivation of harmful and pathogenic microorganisms by photosensitization. Food Technol. Biotechnol. 2005, 43, 411–418. [Google Scholar]
- Almeida-Marrero, V.; van de Winckel, E.; Anaya-Plaza, E.; Torres, T.; de la Escosura, A. Porphyrinoid biohybrid materials as an emerging toolbox for biomedical light management. Chem. Soc. Rev. 2018, 47, 7369–7400. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, T.; Bai, R.; Xie, Y. Chemical approaches for the enhancement of porphyrin skeleton-based photodynamic therapy. J. Enzym. Inhib. Med. Chem. 2020, 34, 1080–1099. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- dos Santos, A.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.S.; Labriola, L. Photodynamic therapy in cancer treatment—An update review. J. Cancer Metastasis Treat. 2019, 5, 25–44. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, C.Y.; Gao, J.; Wang, Z. Recent advances in photodynamic therapy for cancer and infectious diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, 1560. [Google Scholar] [CrossRef]
- Marko, A.J.; Patel, N.J.; Joshi, P.; Missert, J.R.; Pandey, R.K. Multifunctional agents for cancer-imaging and photodynamic therapy: Monomers vs. polyacrylamide-based nanoplatforms. In Handbook of Photodynamic Therapy—Updates on Recent Applications of Porphyrin-Based Compounds; Pandey, R.K., Kessel, D., Dougherty, T.J., Eds.; World Scientific Publishing: Hackensack, NJ, USA, 2016; pp. 3–43. [Google Scholar]
- Fayter, D.; Corbett, M.; Heirs, M.; Fox, D.; Eastwood, A. A systematic review of photodynamic therapy in the treatment of pre-cancerous skin conditions, Barrett’s oesophagus and cancers of the biliary tract, brain, head and neck, lung, oesophagus and skin. Health Technol. Assess. 2010, 14, 1–288. [Google Scholar] [CrossRef]
- Meng, S.; Xu, Z.; Hong, G.; Zhao, L.; Zhao, Z.; Guo, J.; Ji, H.; Liu, T. Synthesis, characterization and in vitro photodynamic antimicrobial activity of basic amino acid-porphyrin conjugates. Eur. J. Med. Chem. 2015, 92, 35–48. [Google Scholar] [CrossRef]
- Cieplik, F.; Deng, D.; Crielaard, W.; Buchalla, W.; Hellwig, E.; Al-Ahmad, A.; Maisch, T. Antimicrobial photodynamic therapy—What we know and what we don’t. Crit. Rev. Microbiol. 2018, 44, 571–589. [Google Scholar] [CrossRef]
- Mahmoudi, H.; Bahador, A.; Pourhajibagher, M.; Alikhani, M.Y. Antimicrobial photodynamic therapy: An effective alternative approach to control bacterial infections. J. Lasers Med. Sci. 2018, 9, 154–160. [Google Scholar] [CrossRef]
- Zou, Q.; Abbas, M.; Zhao, L.; Li, S.; Shen, G.; Yan, X. Biological photothermal nanodots based on self-assembly of peptide-porphyrin conjugates for antitumor therapy. J. Am. Chem. Soc. 2017, 139, 1921–1927. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, X.; Wang, X.; Guan, X.; Zhang, W.; Ma, J. Recent advances in selective photothermal therapy of tumor. J. Nanobiotechnol. 2021, 19, 335. [Google Scholar] [CrossRef]
- Wan, G.Y.; Liu, Y.; Chen, B.W.; Liu, Y.Y.; Wang, Y.S.; Zhang, N. Recent advances of sonodynamic therapy in cancer treatment. Cancer Biol. Med. 2016, 13, 335–337. [Google Scholar] [CrossRef] [Green Version]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Josefsen, L.B.; Boyle, R.W. Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics 2012, 2, 916–966. [Google Scholar] [CrossRef]
- Luo, D.; Carter, K.A.; Miranda, D.; Lovell, J.F. Chemophototherapy: An emerging treatment option for solid tumors. Adv. Sci. 2017, 4, 1600106. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cunha, Â.; Almeida, A. Photodynamic inactivation of mammalian viruses and bacteriophages. Viruses 2012, 4, 1034–1074. [Google Scholar] [CrossRef]
- Kiesslich, T.; Gollmer, A.; Maisch, T.; Berneburg, M.; Plaetzer, K. A comprehensive tutorial on in vitro characterization of new photosensitizers for photodynamic antitumor therapy and photodynamic inactivation of microorganisms. Biomed. Res. Int. 2013, 2013, 840417. [Google Scholar] [CrossRef]
- Costa, L.; Tomé, J.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Cunha, A.; Faustino, M.A.F.; Almeida, A. Susceptibility of non-enveloped DNA- and RNA-type viruses to photodynamic inactivation. Photochem. Photobiol. Sci. 2012, 11, 1520–1523. [Google Scholar] [CrossRef]
- Ol’shevskaya, V.A.; Zaitsev, A.V.; Makarenkov, A.V.; Kononova, E.G.; Markova, A.A.; Kostyukov, A.A.; Egorov, A.E.; Klimovich, M.A.; Koroleva, O.A.; Kuzmin, V.A. Synthesis of boronated meso-arylporphyrins via copper-catalyzed 1,3-dipolar cycloaddition reaction and their binding ability towards albumin and low density lipoproteins. J. Organomet. Chem. 2020, 916, 121248. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. 1,3,5-Triazines: A promising scaffold for anticancer drugs development. Eur. J. Med. Chem. 2017, 142, 523–549. [Google Scholar] [CrossRef]
- Moreno, L.M.; Quiroga, J.; Abonia, R.; Ramírez-Prada, J.; Insuasty, B. Synthesis of new 1,3,5-triazine-based 2-pyrazolines as potential anticancer agents. Molecules 2018, 23, 1956. [Google Scholar] [CrossRef]
- Pathak, M.; Ojha, H.; Tiwari, A.K.; Sharma, D.; Saini, M.; Kakkar, R. Design, synthesis and biological evaluation of antimalarial activity of new derivatives of 2,4,6-s-triazine. Chem. Cent. J. 2017, 11, 132. [Google Scholar] [CrossRef] [Green Version]
- Jensen, N.P.; Ager, A.L.; Bliss, R.A.; Canfield, C.J.; Kotecka, B.M.; Rieckmann, K.H.; Terpinski, J.; Jacobus, D.P. Phenoxypropoxybiguanides, prodrugs of DHFR-inhibiting diaminotriazine antimalarials. J. Med. Chem. 2001, 44, 3925–3931. [Google Scholar] [CrossRef]
- Agarwal, A.; Srivastava, K.; Puri, S.K.; Chauhan, P.M.S. Antimalarial activity and synthesis of new trisubstituted pyrimidines. Biorg. Med. Chem. Lett. 2005, 15, 531–533. [Google Scholar] [CrossRef]
- Srinivas, K.; Srinivas, U.; Rao, V.J.; Bhanuprakash, K.; Kishore, K.H.; Murty, U.S.N. Synthesis and antibacterial activity of 2,4,6-tri substituted s-triazines. Biorg. Med. Chem. Lett. 2005, 15, 1121–1123. [Google Scholar] [CrossRef]
- McKay, G.A.; Reddy, R.; Arhin, F.; Belley, A.; Lehoux, D.; Moeck, G.; Sarmiento, I.; Parr, T.R.; Gros, P.; Pelletier, J.; et al. Triaminotriazine DNA helicase inhibitors with antibacterial activity. Biorg. Med. Chem. Lett. 2006, 16, 1286–1290. [Google Scholar] [CrossRef]
- Ghaib, A.; Ménager, S.; Vérité, P.; Lafont, O. Synthesis of variously 9,9-dialkylated octahydropyrimido [3,4-a]-s-triazines with potential antifungal activity. Farmaco 2002, 57, 109–116. [Google Scholar] [CrossRef]
- Sharma, M.; Chauhan, K.; Chauhan, S.S.; Kumar, A.; Singh, S.V.; Saxena, J.K.; Agarwal, P.; Srivastava, K.; Kumar, S.R.; Puri, S.K.; et al. Synthesis of hybrid 4-anilinoquinoline triazines as potent antimalarial agents, their in silico modeling and bioevaluation as Plasmodium falciparumtran sketolase and β-hematin inhibitors. Med. Chem. Commun. 2012, 3, 71–79. [Google Scholar] [CrossRef]
- Mibu, N.; Yokomizo, K.; Sano, M.; Kawaguchi, Y.; Morimoto, K.; Shimomura, S.; Sato, R.; Hiraga, N.; Matsunaga, A.; Zhou, J.R.; et al. Preparation and antiviral activity of some new C3- and CS-symmetrical tri-substituted triazine derivatives having benzylamine substituents. Chem. Pharm. Bull. 2018, 66, 830–838. [Google Scholar] [CrossRef]
- Maarouf, A.R.; Farahat, A.A.; Selim, K.B.; Eisa, H.M. Synthesis and antiviral activity of benzimidazolyl- and triazolyl-1,3,5-triazines. Med. Chem. Res. 2012, 21, 703–710. [Google Scholar] [CrossRef]
- Liu, H.; Long, S.; Rakesh, K.P.; Zha, G.F. Structure-activity relationships (SAR) of triazine derivatives: Promising antimicrobial agents. Eur. J. Med. Chem. 2020, 185, 111804. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.A.M.; Lourenco, N.; Rosatella, A.A. Synthesis of 2,4,6-tri-substituted-1,3,5-triazines. Molecules 2006, 11, 81–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ol’shevskaya, V.A.; Alpatova, V.M.; Radchenko, A.S.; Ramonova, A.A.; Petrova, A.S.; Tatarskiy, V.V.; Zaitsev, A.V.; Kononova, E.G.; Ikonnikov, N.S.; Kostyukov, A.A.; et al. β-Maleimide substituted meso-arylporphyrins: Synthesis, transformations, physico-chemical and antitumor properties. Dye Pigment. 2019, 171, 107760. [Google Scholar] [CrossRef]
- Ol’shevskaya, V.A.; Nikitina, R.G.; Zaitsev, A.V.; Luzgina, V.N.; Kononova, E.G.; Morozova, T.G.; Drozhzhina, V.V.; Ivanov, O.G.; Kaplan, M.A.; Kalinin, V.N.; et al. Boronated protohaemins: Synthesis and in vivo antitumour efficacy. Org. Biomol. Chem. 2006, 4, 3815–3821. [Google Scholar] [CrossRef]
- Ol’shevskaya, V.A.; Zaytsev, A.V.; Savchenko, A.N.; Shtil, A.A.; Cheong, C.S.; Kalinin, V.N. Boronated porphyrins and chlorins as potential anticancer drugs. Bull. Korean Chem. Soc. 2007, 28, 1910–1916. [Google Scholar]
- Bhupathiraju, N.V.S.D.K.; Vicente, M.G.H. Synthesis and cellular studies of polyamine conjugates of a mercaptomethyl-carboranylporphyrin. Bioorg. Med. Chem. 2013, 21, 485–495. [Google Scholar] [CrossRef]
- Nakamura, H.; Shoji, A.; Takeuchi, A.; Ban, H.S.; Lee, J.D.; Yamori, T.; Kang, S.O. Discovery of ortho-carborane-conjugated triazines as selective topoisomerase I/II inhibitors. Aust. J. Chem. 2011, 64, 1430–1437. [Google Scholar] [CrossRef]
- Azev, Y.A.; Dülcks, T.; Gabel, D. Cyanuric and thiocyanuric esters as carriers of boron-containing fragments and their fragmentation in mass spectrometry. Tetrahedron Lett. 2003, 44, 8689–8691. [Google Scholar] [CrossRef]
- Lee, C.H.; Lim, H.G.; Nakamura, H.; Kang, S.O. o-Carboranyl derivatives of 1,3,5-s-triazines: Structures, properties and in vitro activities. Appl. Organomet. Chem. 2003, 17, 539–548. [Google Scholar] [CrossRef]
- Ronchi, S.; Prosperi, D.; Compostella, F.; Panza, L. Synthesis of novel carborane-hybrids based on a triazine scaffold for boron neutron capture therapy. Synlett 2004, 6, 1007–1010. [Google Scholar]
- Kellert, M.; Worm, D.J.; Hoppenz, P.; Sárosi, M.B.; Lönnecke, P.; Riedl, B.; Koebberling, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Modular triazine-based carborane-containing carboxylic acids–synthesis and characterisation of potential boron neutron capture therapy agents made of readily accessible building blocks. Dalton Trans. 2019, 48, 10834–10844. [Google Scholar] [CrossRef]
- Kruper, W.J.; Chamberlin, T.A.; Kochanny, M. Regiospecific aryl nitration of meso-substituted tetraarylporphyrins: A simple route to bifunctional porphyrins. J. Org. Chem. 1989, 54, 2753–2756. [Google Scholar] [CrossRef]
- Luechai, A.; Gasiorowski, J.; Petsom, A.; Neugebauer, H.; Sariciftci, N.S.; Thamyongkit, P. Photosensitizing porphyrin–triazine compound for bulk heterojunction solar cells. J. Mater. Chem. 2012, 22, 23030–23037. [Google Scholar] [CrossRef]
- Krajčiová, D.; Pecher, D.; Garaj, V.; Mikuš, P. Optimization and comparison of synthetic procedures for a group of triazinyl-substituted benzene-sulfonamide conjugates with amino acids. Molecules 2017, 22, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blotny, G. Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron 2006, 62, 9507–9522. [Google Scholar] [CrossRef]
- Carofiglio, T.; Lubian, E.; Menegazzo, I.; Saielli, G.; Varotto, A. Melamine-bridged bis(porphyrin-ZnII) receptors: Molecular recognition properties. J. Org. Chem. 2009, 74, 9034–9043. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, T.; Korkmaz, M.; Kutluay, T.; Bekaroğlu, Ö. Synthesis, characterization and EPR spectroscopy of novel s-triazine bearing three oxygen-linked phthalocyanines. J. Porphyr. Phthalocyanines 2004, 8, 1383–1389. [Google Scholar] [CrossRef]
- Xiao, S.C.; Liu, C.Z.; Liu, W.K.; Xie, W.Z.; Lin, W.Y.; Jiang, G.F.; Guo, C.C. Selective synthesis and biological activity of triazine-porphyrins as potential anti-cancer agents. J. Porphyr. Phthalocyanines 2010, 14, 123–127. [Google Scholar] [CrossRef]
- Ringot, C.; Saad, N.; Granet, R.; Bressollier, P.; Sol, V.; Krausz, P. Meso-functionalized aminoporphyrins as efficient agents for photo-antibacterial surfaces. J. Porphyr. Phthalocyanines 2010, 14, 925–931. [Google Scholar] [CrossRef]
- Kim, E.M.; Choi, J.H. Preparation of novel iron phthalocyanine containing reactive groups and its deodorizing property on cellulose. Text. Color. Finish. 2013, 25, 247–253. [Google Scholar] [CrossRef]
- Carofiglio, T.; Varotto, A.; Tonellato, U. One-pot synthesis of cyanuric acid-bridged porphyrin-porphyrin dyads. J. Org. Chem. 2004, 69, 8121–8124. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmánek, S. Chemistry of 9-mercapto-1,7-dicarba-closo-dodecaborane. Collect. Czech. Chem. Commun. 1978, 43, 1332–1338. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Zhigareva, G.G. Synthesis and some reactions of mercapto derivatives of bareness. Izv. Akad. Nauk SSSR Ser. Khim. 1967, 16, 1358–1360. [Google Scholar]
- Zakharkin, L.I.; Kalinin, V.N.; Gedymin, V.V. Synthesis and some reactions of 3-amino-o-carboranes. J. Organomet. Chem. 1969, 16, 371–379. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Kalinin, V.N.; Snyakin, A.P.; Kvasov, B.A. Effect of solvents on the electronic properties of 1-o-, 3-o- and 1-m-carboranyl groups. J. Organometal. Chem. 1969, 18, 19–26. [Google Scholar] [CrossRef]
- Ol’shevskaya, V.A.; Alpatova, V.M.; Makarenkov, A.V.; Kononova, E.G.; Smol’yakov, A.F.; Peregudov, A.S.; Rys, E.G. Synthesis of maleimide-functionalized carboranes and their utility in Michael addition reactions. New J. Chem. 2021, 45, 12159–12167. [Google Scholar]
- Ol’shevskaya, V.A.; Zaitsev, A.V.; Petrova, A.S.; Arkhipova, A.Y.; Moisenovich, M.M.; Kostyukov, A.A.; Egorov, A.E.; Koroleva, O.A.; Golovina, G.V.; Volodina, Y.L.; et al. The synthetic fluorinated tetracarboranylchlorin as a versatile antitumor photoradiosensitizer. Dyes Pigment. 2021, 186, 108993. [Google Scholar] [CrossRef]
- Wawrzyńska, M.; Kałas, W.; Biały, D.; Zioło, E.; Arkowski, J.; Mazurek, W.; Leon Strządała, L. In vitro photodynamic therapy with chlorin e6 leads to apoptosis of human vascular smooth muscle cells. Arch. Immunol. Ther. Exp. 2010, 58, 67–75. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpatova, V.M.; Rys, E.G.; Kononova, E.G.; Khakina, E.A.; Markova, A.A.; Shibaeva, A.V.; Kuzmin, V.A.; Ol’shevskaya, V.A. Multicomponent Molecular Systems Based on Porphyrins, 1,3,5-Triazine and Carboranes: Synthesis and Characterization. Molecules 2022, 27, 6200. https://doi.org/10.3390/molecules27196200
Alpatova VM, Rys EG, Kononova EG, Khakina EA, Markova AA, Shibaeva AV, Kuzmin VA, Ol’shevskaya VA. Multicomponent Molecular Systems Based on Porphyrins, 1,3,5-Triazine and Carboranes: Synthesis and Characterization. Molecules. 2022; 27(19):6200. https://doi.org/10.3390/molecules27196200
Chicago/Turabian StyleAlpatova, Victoria M., Evgeny G. Rys, Elena G. Kononova, Ekaterina A. Khakina, Alina A. Markova, Anna V. Shibaeva, Vladimir A. Kuzmin, and Valentina A. Ol’shevskaya. 2022. "Multicomponent Molecular Systems Based on Porphyrins, 1,3,5-Triazine and Carboranes: Synthesis and Characterization" Molecules 27, no. 19: 6200. https://doi.org/10.3390/molecules27196200
APA StyleAlpatova, V. M., Rys, E. G., Kononova, E. G., Khakina, E. A., Markova, A. A., Shibaeva, A. V., Kuzmin, V. A., & Ol’shevskaya, V. A. (2022). Multicomponent Molecular Systems Based on Porphyrins, 1,3,5-Triazine and Carboranes: Synthesis and Characterization. Molecules, 27(19), 6200. https://doi.org/10.3390/molecules27196200