3. Materials and Methods
3.1. Chemistry General Section
Commercially available chemicals were provided as reagent grade and used as received. Some reactions requiring anhydrous conditions were carried out using oven-dried glassware and under an atmosphere of dry Argon. All anhydrous solvents were provided from commercial sources as very dry reagents. The reactions were monitored by thin layer chromatography (TLC) analysis using silica gel precoated plates (Kieselgel 60F254, E. Merck). Compounds were visualized by UV irradiation and/or spraying with sulfuric acid (H2SO4 5% in ethanol) stain followed by charring at average 150 °C. Flash column chromatography was performed on Silica Gel 60 M (0.040–0.063 mm, E. Merck). The infrared spectra were measured with Perkin–Elmer Spectrometer. The 1H and 13C NMR spectra were recorded on BrukerAvance DPX 250 or BrukerAvance 400 Spectrometers. Chemical shifts are given in ppm and are referenced to the deuterated solvent signal or to TMS as internal standard and multiplicities are reported as s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Carbon multiplicities were assigned by distortion less enhancement by polarization transfer (DEPT) experiments. 1H and 13C signals were attributed on the basis of H–H and H–C correlations. High Resolution Mass spectra were performed on a Bruker Q-TOF MaXis spectrometer by the “Fédération de Recherche” ICOA/CBM (FR2708) platform. LC-MS data were acquired on a Thermo-Fisher UHPLC-MSQ system equipped with an electron spray ionization source (ESI). The temperature of the source was maintained at 350 °C. Initially, the cone voltage was set at 35 V and after 5 min was increased to 75V. In full scan mode, data were acquired between 100 and 1000 m/z in the positive mode with a 1.00 S scan time. In addition, a UV detection was performed with a Diode array detector at three wavelengths 273, 254 and 290 nm, respectively. A water/methanol (70%/30%) solution mixture with 0.1% formic acid was used as mobile phase. The composition of the mobile phase was increased to 100% methanol with 0.1% formic acid with a 7% ramp. The flow rate was set at 0.300 mL.min−1. Samples diluted in the mobile phase were injected (3 μL) on a C18 column (X-terra, Waters), 2.1 mm internal diameter, and 100 mm length placed into an oven at 40 °C. Electronic extraction of ions was performed and the subsequent areas under the corresponding chromatographic peaks determined.
3.2. General Synthetic Procedure 1 for Strecker-Ugi Cyclization
A mixture of amino pyridine derivatives (200 mg) and ethyl glyoxalate (50% solution in toluene) (1 eq.) was stirred at 25 °C for 2 min. THF (4.5 mL) and 1,4-diazabicyclo [2.2.2] octane (1 eq.) were subsequently added. The reaction mixture was cooled to 0–5 °C and cyanotrimethylsilane (1 eq.) was added. The mixture was heated under microwave irradiation at 120 °C for 15 min. The solvent was evaporated under reduced pressure. The crude product was dissolved with EtOAc, washed with K2CO3, dried over MgSO4, filtrated and concentrated under reduced pressure.
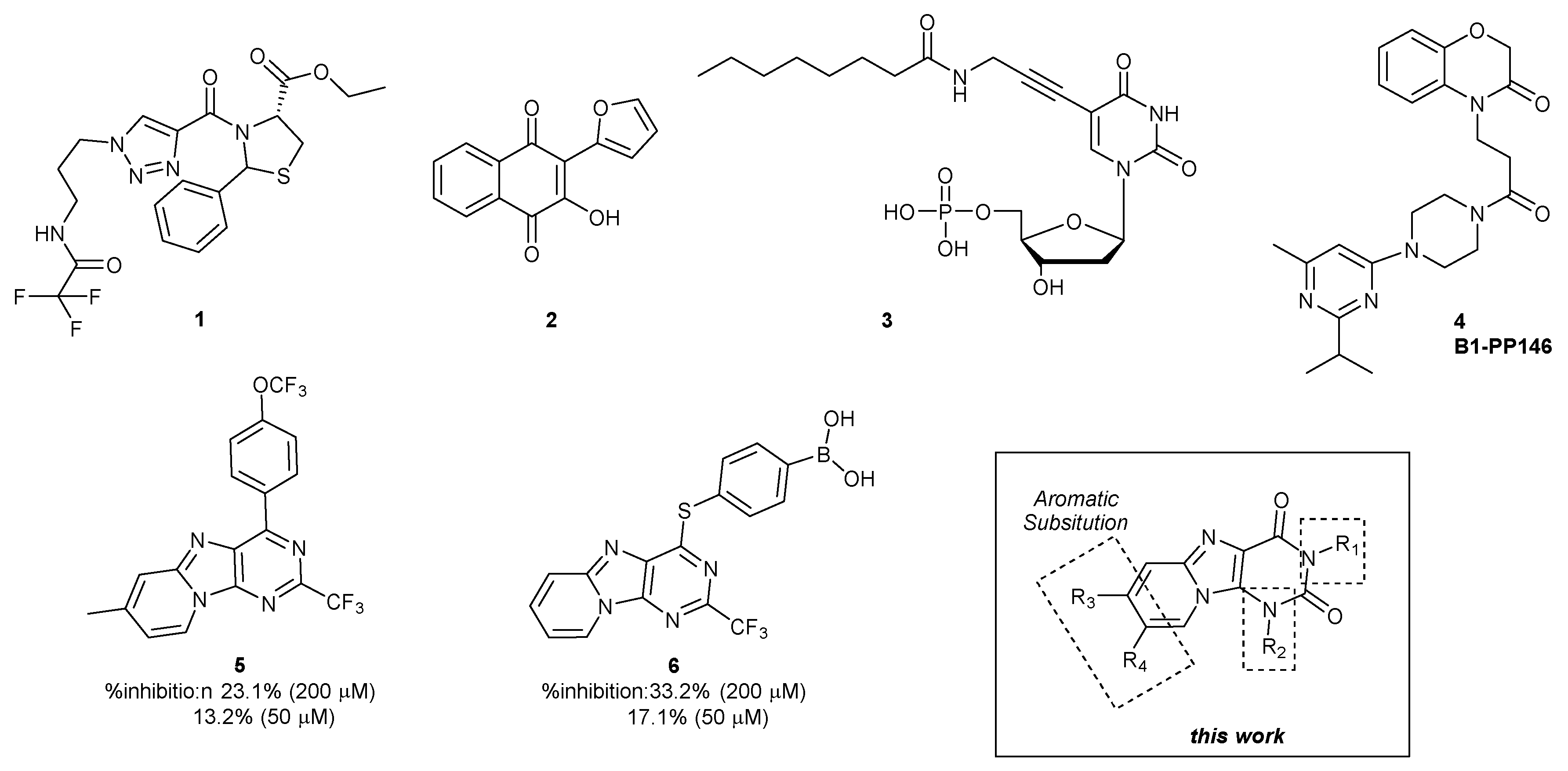
Ethyl 3-aminoimidazo [1,2-a]pyridine-2-carboxylate (8). The title compound was prepared from commercially available 7 to afford after purification the desired product as a yellow solid (50%). CAS # 1487454-00-1. 1H NMR (250 MHz, DMSO-d6) δ 8.17 (dt, 1H, J = 7.1, 1,1 Hz, H4), 7.31 (dt, 1H, J = 9.3, 1,1 Hz, H7), 7.05 (ddd, 1H, J = 9.3, 6.5, 1.1 Hz, H6), 6.79 (ddd, 1H, J = 7.1, 6.5, 1.1 Hz, H5), 6.35 (bs, 2H, NH2), 4.27 (q, 2H, J = 7.1 Hz, O–CH2), 1.31 (t, 3H, J = 7.1 Hz CH2–CH3) ppm.
Ethyl 3-amino-7-methylimidazo [1,2-a]pyridine-2-carboxylate (16a). The title compound was prepared from commercially available 14a to afford after purification the desired product as an orange solid (45%). CAS # 1216262-11-1. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (s, 1H, H4), 7.24 (d, 1H, J = 9.4 Hz, H7), 6.93 (d, 1H, J = 9.4 Hz, H6), 6.35 (bs, 2H, NH2), 4.26 (q, 2H, J = 7.0, Hz, O–CH2), 2.22 (s, 3H, C–CH3), 1.30 (t, 3H, J = 7.0, Hz CH2–CH3) ppm.
Ethyl 3-amino-6-methylimidazo [1,2-a]pyridine-2-carboxylate (16b). The title compound was prepared from commercially available 14b to afford after purification the desired product as an orange solid (34%). CAS # 1498691-03-4. 1H NMR (400 MHz, Acetone-d6) δ 7.62 (d, 1H, J = 7.1 Hz, H4), 6.62 (s, 1H, H7), 6.19 (d, 1H, J = 7.1 Hz, H5), 5.82 (bs, 2H, NH2), 3.80 (q, 2H, J = 7.0, Hz, O–CH2),1.82 (s, 3H, C–CH3), 0.85 (t, 3H, J = 7.0, Hz CH2–CH3) ppm.
Ethyl 3-amino-7-bromoimidazo [1,2-a]pyridine-2-carboxylate (17a). The title compound was prepared from commercially available 15a to afford after purification the desired product as a yellow solid (47%). CAS # 1536009-01-4. 1H NMR (400 MHz, DMSO-d6) δ 8.16 (d, 1H, J = 7.4 Hz, H4), 7.86–7.71 (m, 2H, H7), 6.96 (dd, 1H, J = 7.4, 2.1 Hz, H6), 6.47 (bs, 2H, NH2), 4.27 (q, 2H, J = 7.1 Hz, O–CH2), 1.30 (t, 3H, J = 7.1 Hz, CH2–CH3) ppm.
Ethyl 3-amino-6-bromoimidazo [1,2-a]pyridine-2-carboxylate (17b). The title compound was prepared from commercially available 15b to afford after purification the desired product as a yellow solid (44%). CAS # 82193-31-5. 1H NMR (250 MHz, DMSO-d6) δ 8.55 (s, 1H, J = 1.8, 0.9 Hz, H7), 7.32 (dd, 1H, J = 9.7, 0.9 Hz, H4), 7.13 (dd, 1H, J = 9.7, 1.8 Hz, H5), 6.47 (bs, 2H, NH2), 4.27 (q, 2H, J = 7.1 Hz, O–CH2), 1.30 (t, 3H, J = 7.1 Hz, CH2–CH3) ppm.
3.3. General Synthetic Procedure 2 for Isocyanate Cyclisation
To a solution of amino ester compound (350 mg) in anhydrous ethanol (4.5 mL), were added subsequently isocyanate (2 eq.) and sodium ethoxide (2 eq.) under inert atmosphere. The reaction mixture was heated at 120 °C under microwave irradiation for 20 min. The solvent was evaporated under vacuum. Pure compounds were obtained after purification by flash column chromatography with DCM/MeOH (92:8) as eluent.
3-(4-Fluorophenyl)pyrido [1,2-
e]purine-2,4(
1H,3H)-dione (
9a). The title compound was prepared from commercially available
8 to afford after purification the desired product as a light yellow solid (75%). CAS # 1842362-44-0.
1H NMR (400 MHz, Methanol-
d4) 8.36 (d, 1H,
J = 7.0 Hz, H
6), 7.55 (d, 1H,
J = 9.4 Hz, H
9), 7.41–7.37 (m, 1H, H
8), 7.35–7.28 (m, 2H, 2 × H
arom), 7.26–7.18 (m, 2H, 2 × H
arom), 7.00 (t, 1H,
J = 7.0, Hz, H
7) ppm (see
Supplementary Materials).
3-(4-Chlorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9b). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light-yellow solid (39%). CAS # 1842362-41-7. 1H NMR (250 MHz, DMSO-d6) δ 8.47 (d, 1H, J = 6.9 Hz, H6), 7.56–7.48 (m, 3H, H9, 2 × Harom), 7.34–7.23 (m, 3H, H8, 2 × Harom), 7.00 (t, 1H, J = 6.9 Hz, H7) ppm.
3-(4-Bromophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9c). The title compound was prepared from commercially available 8 to afford after purification the desired product as a white/grey solid (23%). 1H NMR (250 MHz, DMSO-d6) δ 8.54 (d, 1H, J = 7.0 Hz, H6), 7.70–7.61 (m, 2H, 2 × Harom), 7.52 (d, 1H, J = 9.4 Hz, H9), 7.25 (td, 3H, J = 6.2, 2.4 Hz, H8, 2 × Harom), 6.96 (t, 1H, J = 7.0 Hz, H7) ppm. 13C NMR (63 MHz, DMSO-d6) δ 158.9 (C=O), 151.7 (C=O), 141.5 (Cquat), 136.5 (Cquat), 135.7 (Cquat), 131.8 (4 × Carom), 126.1 (Carom), 124.2 (Carom), 120.4 (Cquat), 118.1 (Carom), 117.6 (Cquat), 112.3 (Carom) ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C15H9BrN4O2: 356.9981 found: 356.9980. λabs: 263 nm λem: 527nm λexc: 286 nm. Rf 0.43 (DCM/MeOH, 9:1).
3-(p-Tolyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9d). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light yellow solid (24%). CAS # 1842362-40-6. 1H NMR (250 MHz, DMSO-d6) δ 8.52 (d, 1H, J = 7.0 Hz, H6), 7.55 (d, 1H, J = 9.4 Hz, H9), 7.33–7.22 (m, 3H, H8, 2 × Harom), 7.14 (d, 2H, J = 8.2 Hz, 2 × Harom), 6.99 (t, 1H, J = 7.0 Hz, H7), 2.36 (s, 3H, C–CH3) ppm.
3-(4-(Trifluoromethyl)phenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9e). The title compound was prepared from commercially available 8 to afford after purification the desired product as a yellow solid (58%). 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, 1H, J = 6.9 Hz, H6), 7.80 (d, 2H, J = 8.2 Hz, 2 × Harom), 7.49–7.39 (m, 3H, H9, 2 × Harom), 7.23–7.14 (m, 1H, H8), 6.85 (t, 1H, J = 6.9 Hz, H7) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.1 (C=O), 154.5 (C=O), 143.0 (Cquat), 141.7 (Cquat), 131.0 (Carom), 127.9 (d, JC–F = 31.6 Hz, Cquat), 126.2 (d, JC–F = 22.7 Hz, Carom), 126.1 (Cquat), 125.9 (d, JC–F = 3.7 Hz, 2 × Carom), 124.5 (Carom), 123.4 (Cquat), 118.6 (2 × Carom), 117.6 (Cquat), 111.8 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ –60.86 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C16H9F3N4O2: 347.0750 found: 347.0751. λabs: 264 nm λem: 486 nm λexc: 266 nm. Rf 0.49 (DCM/MeOH, 9:1).
3-(p-Methoxyphenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9f). The title compound was prepared from commercially available 8 to afford after purification the desired product as a beige solid (34%). CAS # 1842362-42-8. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (d, 1H, J = 6.8 Hz, H6), 7.55 (d, 1H, J = 9.3 Hz, H9), 7.33–7.25 (t, 1H, J = 9.3 Hz, H8), 7.17 (d, 2H, J = 8.7 Hz, 2 × Harom), 7.06–6.96 (m, 3H, H7, 2 × Harom), 3.81 (s, 3H, O–CH3) ppm.
3-(3-Fluorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9g). The title compound was prepared from commercially available 8 to afford after purification the desired product as a yellow solid (19%). 1H NMR (400 MHz, DMSO-d6) δ 8.50 (d, 1H, J = 6.2 Hz, H6), 7.55–7.49 (m, 2H, H9, Harom), 7.33–7.24 (m, 3H, H8, 2 × Harom), 7.20 (d, 1H, J = 9.7 Hz, Harom), 7.14 (d, 1H, J = 7.3 Hz, Harom), 6.97 (t, 1H, J = 6.2 Hz, H7) ppm. 13C NMR (101 MHz, DMSO-d6) δ 163.3 (C=O), 160.8 (Cquat), 158.9 (d, J = 11.1 Hz, Cquat), 151.7 (C=O), 141.6 (Cquat), 130.0 (Cquat), 129.9 (d, JC–F = 9.3 Hz, Carom), 126.2 (Carom), 125.7 (d, JC–F = 3.03 Hz, Carom), 124.1 (Carom), 118.3 (Carom), 117.6 (Cquat), 116.7 (d, JC–F = 22.7 Hz, Carom), 114.4 (d, JC–F = 20.2 Hz, Carom), 112.4 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −113.60 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C15H9FN4O2: 297.0782, found: 297.079. λabs: 263 nm λem: 498 nm λexc: 266 nm. Rf 0.29 (DCM/MeOH 9:1).
3-(3,4-Difluorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9h). The title compound was prepared from commercially available 8 to afford after purification the desired product as a white/yellow solid (38%). 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, 1H, J = 6.2 Hz, H6), 7.55–7.47 (m, 2H, 2 × Harom), 7.44 (dd, 1H, J = 7.4, 2.3 Hz, Harom), 7.25 (dd, 1H J = 9.4, 6.6 Hz, H8), 7.17–7.11 (m, 1H, Harom), 6.93 (t, 1H, J = 6.2 Hz, H7) ppm. 13C NMR (101 MHz, DMSO-d6) δ 159.2 (C=O), 152.5 (C=O), 150.1 (dd, J = 38.2, 12.9 Hz, Cquat), 147.7 (dd, JC–F = 38.4, 12.8 Hz, Cquat), 141.5 (Cquat), 137.1 (Cquat), 134.3 (dd, JC–F = 8.6, 3.5 Hz, Cquat), 126.6 (dd, JC–F = 6.6, 3.4 Hz, Carom), 126.0 (Carom), 124.0 (Carom), 118.9 (d, JC–F = 18.2 Hz, Carom), 118.2 (Carom), 117.5 (Cquat), 117.0 (d, JC–F = 17.9 Hz, Carom), 112.1 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −140.24, −138.54 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C15H9F2N4O2: 315.0688, found: 315.0687. λabs: 261 nm λem: 49 nm λexc: 266 nm. Rf 0.27 (DCM/MeOH 9:1).
3-(2,4-Difluorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9i). The title compound was prepared from commercially available 8 to afford after purification the desired product as a yellow solid (64%).1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, 1H, J = 6.9 Hz, H6), 7.58–7.36 (m, 3H, H9, 2 × Harom), 7.28 (t, 1H, J = 6.8 Hz, H8), 7.20 (t, 1H, J = 8.4 Hz, Harom), 6.95 (t, 1H, J = 6.9 Hz, H7) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.8 (dd, J = 46.5, 11.7 Hz, C=O), 158.5 (Cquat), 159.6-156.3 (dd, J = 251.49, 13.2 Hz, Cquat), 151.7 (C=O), 141.7 (Cquat), 136.7 (Cquat), 132.7 (dd, JC–F = 10.3, 2.4 Hz, Carom), 126.3 (Carom), 124.1 (Carom), 121.3 (dd, JC–F = 13.6, 4.0 Hz, Cquat), 118.2 (Carom), 117.2 (Cquat), 112.3 (Carom), 111.6 (dd, JC–F = 22.4, 3.6 Hz, Carom), 104.4 (dd, JC–F = 26.9, 24.6 Hz, Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −110.21, −117.12 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C15H9F2N4O2: 315.0687, found: 315.0688. λabs: 264 nm λem: 490 nm λexc: 266 nm. Rf 0.32 (DCM/MeOH 9:1).
3-(3,4-Dimethylphenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (9j). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light-yellow solid (56%). 1H NMR (400 MHz, DMSO-d6) δ 8.46 (d, 1H, J = 6.9 Hz, H6), 7.54 (d, 1H, J = 9.3 Hz, H9), 7.28 (t, 1H, J = 8.0 Hz, Harom), 7.21 (d, 1H, J = 9.3 Hz, H8), 7.00–6.92 (m, 3H, H7, 2 × Harom), 2.27 (s, 3H, C–CH3). 2.24 (s, 3H, C–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 159.0 (C=O), 151.7 (C=O), 141.5 (Cquat), 136.5 (Cquat), 135.6 (Cquat), 134.4 (Cquat), 129.9 (Carom), 129.6 (Carom), 129.3 (Carom), 126.1 (Carom), 124.0 (Carom), 118.3 (Carom), 117.8 (Cquat), 112.5 (Carom), 19.2 (C–CH3), 19.0 (C–CH3) ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C17H15N4O2: 307.1189, found: 307.1190. λabs: 263 nm λem: 496 nm λexc: 266 nm. Rf 0.68 (DCM/MeOH, 9:1).
3-(4-Fluorophenyl)-2-thioxo-2,3-dihydropyrido [1,2-e]purin-4(1H)-one (11). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light-yellow solid (74%). 1H NMR (400 MHz, DMSO-d6) δ 8.43 (d, 1H, J = 6.8 Hz, H6), 7.47 (d, 1H, J = 9.2 Hz, H9), 7.30–7.23 (m, 1H, H8), 7.20 (t, 2H, J = 8.5 Hz, 2 × Harom), 7.07–7.06 (m, 2H, 2 × Harom), 6.88 (t, 1H, J = 6.8 Hz, H7) ppm. 13C NMR (101 MHz, DMSO-d6) δ 175.6 (C=S), 161.9 (C=O), 159.5 (d, J = 10.9 Hz, Cquat), 142.3 (Cquat), 142.1 (Cquat), 139.0 (Cquat), 131.4 (d, JC–F = 8.6 Hz, 2 × Carom), 126.8 (Carom), 124.2 (Carom), 121.0(Cquat), 118.0 (Carom), 115.1 (d, JC–F = 22.5 Hz, 2 × Carom), 111.4 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −116.93 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C15H10FN4OS: 313.0553, found: 313.0553. λabs: 340 nm λem: 492 nm λexc: 341 nm. Rf 0.11 (DCM/MeOH 9:1).
3-(Benzyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (10a). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light yellow solid (71%). CAS # 1842362-38-2. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, 1H, J = 7.0 Hz, H6), 7.52 (dd, 1H, J = 8.3, 5.5 Hz, Harom), 7.41 (d, 1H, J = 9.3 Hz, H9), 7.35 (dd, 2H, J = 8.3, 5.5 Hz, 2 × Harom), 7.24 (t, 1H, J = 8.3 Hz, Harom), 7.16 (dd, 1H, J = 9.3, 6.75 Hz, H8), 7.08 (t, 2H, J = 8.8 Hz, 2× Harom), 6.81 (t, 1H, J = 6.7 Hz, H7), 5.09 (s, 2H, N–CH2) ppm.
3-(p-Fluorobenzyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (10b). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light-yellow solid (52%). 1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, 1H, J = 7.0 Hz, H6), 7.52 (dd, 1H, J = 8.3, 7.5 Hz, Harom), 7.41 (d, 1H, J = 9.3 Hz, H9), 7.35 (dd, 1H, J = 8.3, 5.7 Hz, Harom), 7.24 (t, 1H, J = 7.1 Hz, Harom), 7.21–7.12 (m, 1H, H8), 7.08 (t, 1H, J = 8.8 Hz, Harom), 6.81 (t, 1H, J = 7.0 Hz, H7), 5.09 (s, 2H, N–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.1 (C=O), 154.9 (C=O), 141.6 (Cquat), 135.8 (Cquat), 131.6 (d, JC–F = 8.4 Hz, Carom), 131.1 (Cquat), 130.0 (d, JC–F = 8.1 Hz, 2 × Carom), 129.0 (Cquat), 126.0 (Carom), 124.4 (Carom), 118.6 (Carom), 117.4 (Cquat), 115.8 (d, JC–F = 21.1 Hz, Carom), 115.1 (d, JC–F = 21.5 Hz, Carom), 111.7 (Carom), 42.7 (d, JC–F = 91.0 Hz, N–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ −116.00 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C16H12FN4O2: 311.0938, found: 311.0939. λabs: 259 nm λem: 501 nm λexc: 268 nm. Rf 0.37 (DCM/MeOH, 9:1).
3-(p-(Trifluoromethyl)benzyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (10c). The title compound was prepared from commercially available 8 to afford after purification the desired product as a white/yellow solid (46%). 1H NMR (400 MHz, DMSO-d6) δ 8.54 (d, 1H, J = 6.9 Hz, H6), 7.66 (d, 2H, J = 8.1 Hz, 2 × Harom), 7.53 (dd, 3H, J = 14.5, 8.8 Hz, H9, 2 × Harom), 7.31–7.26 (m, 1H, H8), 6.98 (t, 1H, J = 6.9 Hz, H7), 5.20 (s, 2H, N–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 158.6 (C=O), 150.9 (C=O), 142.7 (Cquat), 141.7 (Cquat), 127.9 (2 × Carom), 127.6 (Cquat), 126.4 (Cquat), 126.4 (Carom), 125.1 (t, JC–F = 3.7 Hz, 2 × Carom), 124.2 (Carom), 118.3 (Carom), 117.4 (m, Cquat), 113.5 (Cquat), 112.6 (Carom), 43.1 (N–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ –60.72. HRMS-ESI (m/z) [M+H]+ calcd. for C17H12F3N4O2: 361.0906, found: 361.0906. λabs: 260 nm λem: 495 nm λexc: 265 nm. Rf 0.35 (DCM/MeOH, 9:1).
3-(p-Methylbenzyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (10d). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light yellow solid (44%). 1H NMR (400 MHz, DMSO-d6) δ 8.49 (d, 1H, J = 7.0 Hz, H6), 7.54 (d, 1H, J = 9.5 Hz, H9), 7.28 (dd, 1H, J = 9.5, 6.5 Hz, H8), 7.21 (d, 2H, J = 7.9 Hz, 2 × Harom), 7.15–7.04 (m, 2H, 2 × Harom), 6.97 (dd, 1H, J = 7.0, 6.5 Hz, H7), 5.07 (s, 2H, N–CH2), 2.25 (s, 3H, C-CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 161.7 (C=O), 158.7 (Cquat), 154.5 (C=O), 141.6 (Cquat), 135.9 (Cquat), 135.0 (Cquat), 128.6 (2 × Carom), 127.4 (2 × Carom), 126.2 (Carom), 124.2 (Carom), 118.2 (Carom), 117.6 (Cquat), 112.5 (Carom), 43.0 (N–CH2), 20.6 (C–CH3) ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C17H15N4O2: 307.1189, found: 307.1191. λabs: 260 nm λem: 496 nm λexc: 265 nm. Rf 0.46 (DCM/MeOH, 9:1).
3-Benzyl-2-thioxo-2,3-dihydropyrido [1,2-e]purin-4(1H)-one (12). The title compound was prepared from commercially available 8 to afford after purification the desired product as a light yellow solid (71%). CAS # 1842362-47-3. 1H NMR (250 MHz, DMSO-d6) δ 8.69 (d, 1H, J = 6.8 Hz, H6), 7.58 (d, 1H, J = 9.3 Hz, H9), 7.41–7.15 (m, 6H, H8, 5 × Harom), 7.01 (t, 1H, J = 6.8 Hz, H7), 5.79 (s, 2H, N–CH2) ppm.
3-(p-Fluorophenyl)-7-methylpyrido [1,2-e]purine-2,4(1H,3H)-dione (18a). The title compound was prepared from commercially available 16a to afford after purification the desired product as a yellow solid (66%). 1H NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H, H6), 7.41 (d, 1H, J = 9.4, Hz, H9), 7.26 (d, 4H, J = 6.5, Hz, 4 × Harom), 7.11 (d, 1H, J = 9.4 Hz, H8), 2.28 (s, 3H, C–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.8 (C=O), 159.2 (Cquat), 152.8 (Cquat), 152.9 (C=O), 141.8 (Cquat), 136.5 (Cquat), 133.7 (Cquat), 131.2 (d, JC–F = 18.6 Hz, 2 × Carom), 123.2 (Carom), 117.3 (Cquat), 115.8 (Carom), 115.6 (Carom), 114.7 (d, JC–F = 29.4 Hz, 2 × Carom), 21.0 (C–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.61 ppm. HRMS-ESI (m/z) [M+H]+ calcd for C16H12FN4O2: 311.0937, found: 311.0938. λabs: 260 nm λem: 497 nm λexc: 265 nm. Rf 0.28 (DCM/MeOH 9:1).
3-(p-Fluorophenyl)-8-methylpyrido [1,2-e]purine-2,4(1H,3H)-dione (18b). The title compound was prepared from commercially available 16b to afford after purification the desired product as a yellow solid (69%). 1H NMR (250 MHz, DMSO-d6) δ 8.31 (d, 1H, J = 7.0 Hz, H6), 7.31–7.24 (m, 5H, H9, 4 × Harom), 6.81 (d, 1H, J = 7.0 Hz, H7), 2.35 (s, 3H, C-CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 162.4 (C=O), 159.3 (d, J = 121.2 Hz, Cquat), 151.4 (C=O), 140.9 (Cquat), 133.7 (Cquat), 133.0 (d, J = 3.0 Hz, Cquat), 131.3 (d, JC–F = 8.7 Hz, 2 × Carom), 129.5 (Carom), 121.8 (Cquat), 120.8 (Carom), 117.8 (Cquat), 117.6 (Carom), 115.5 (d, JC-F = 22.6 Hz, 2 × Carom), 17.8 (C–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.29 ppm. HRMS-ESI (m/z) [M+H]+ calcd for C16H12FN4O2: 311.0938, found: 311.0938. λabs: 263 nm λem: 465 nm λexc: 269 nm. Rf 0.28 (DCM/MeOH 9:1).
7-Bromo-3-(p-fluorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (19a). The title compound was prepared from commercially available 17a to afford after purification the desired product as a yellow/orange solid (46%). 1H NMR (400 MHz, DMSO-d6) δ 8.36 (d, 1H, J = 7.4 Hz, H9), 7.85–7.77 (m, 1H, H6), 7.34–7.20 (m, 4H, 4 × Harom), 7.05 (dd, 1H, J = 7.4, 1.9 Hz, H8) ppm. 13C NMR (101 MHz, DMSO-d6) δ 163.0 (C=O), 159.4 (d, J = 17.2 Hz, Cquat), 153.7 (C=O), 141.0 (Cquat), 139.1 (Cquat), 134.0 (d, JC–F = 3.1 Hz, Cquat), 131.2 (d, JC–F = 8.7 Hz, 2 × Carom), 125.0 (Carom), 120.0 (Carom), 118.8 (Cquat), 117.8 (Cquat), 115.3 (d, JC–F = 22.6 Hz, 2 × Carom), 115.2 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.56 ppm. HRMS-ESI (m/z) [M+H]+ calcd for C15H9BrFN4O2 374.9887, found: 374.9887. λabs: 267 nm λem: 492 nm λexc: 269 nm. Rf 0.32 (DCM/MeOH 9:1).
8-Bromo-3-(p-fluorophenyl)pyrido [1,2-e]purine-2,4(1H,3H)-dione (19b). The title compound was prepared from commercially available 17b to afford after purification the desired product as a yellow/green light solid (51%). 1H NMR (400 MHz, DMSO-d6) δ 8.74 (s, 1H, H9), 7.49 (d, 1H, J = 9.7 Hz, H6), 7.32 (d, 1H, J = 9.7 Hz, H7), 7.30–7.21 (m, 4H, 4 × Harom) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.8 (C=O), 159.7 (d, J = 70.7 Hz, Cquat), 153.5 (C=O), 140.1 (Cquat), 138.0 (Cquat), 134.1 (d, JC–F = 2.0 Hz, Cquat), 131.7 (d, JC–F = 8.8 Hz, 2 × Carom), 129.1 (Carom), 124.6 (Carom), 119.9 (Carom), 118.6 (Cquat), 115.9 (d, JC–F = 22.6 Hz, 2 × Carom), 106.3 (Cquat) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.29 ppm. HRMS-ESI (m/z) [M+Na]+ calcd for C15H9BrFNaN4O2 396.9706, found: 396.9693. λabs: 262 nm λem: 496 nm λexc: 267 nm. Rf 0.30 (DCM/MeOH 9:1).
3.4. General Synthetic Procedure 3 for the N1-Alkylation
To a solution of fluorobenzyl compound (150 mg) in anhydrous DMF (4 mL), were added subsequently potassium carbonate (1.5 eq.) and bromide derivative (1.5 eq.) under inert atmosphere. The reaction mixture was heated at 120 °C under microwave irradiation for 20 min. The mixture was dissolved with EtOAc, washed twice with saturated NH4Cl, dried over MgSO4, filtrated and concentrated under vacuum. Pure compound was obtained after purification by flash column chromatography using an elution gradient of DCM/MeOH (from 98:2 to 95:5).
1-Benzyl-3-(4-fluorophenyl)purino [9,8-a]pyridine-2,4-dione (13a). The title compound was prepared from commercially available 9a to afford after purification the desired product as a light orange solid (36%). 1H NMR (400 MHz, DMSO-d6) δ 8.23 (d, 1H, J = 7.0 Hz, H6), 7.60 (d, 1H, J = 9.3 Hz, H9), 7.50–7.27 (m, 9H, 5 × Harom-N1, 4 × Harom-N3), 7.26–7.20 (m, 1H, H8), 6.82 (t, 1H, J = 7.0 Hz, H7), 5.73 (s, 2H, N–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.3 (C=O), 157.9 (Cquat), 151.1 (C=O), 142.5 (Cquat), 136.1 (Cquat), 132.6 (d, JC–F = 3.03 Hz, Cquat), 132.08 (Cquat), 131.1 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 129.0 (2 × Carom-N1), 127.6 (Carom), 126.0 (Carom), 125.7 (2 × Carom-N1), 125.0 (Carom), 120.1 (Cquat), 118.9 (Carom), 115.7 (d, JC–F = 22.8 Hz, 2 × Carom-N3), 113.4 (Carom), 46.9 (N–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.07 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H16FN4O4: 387.1251, found: 387.1249. λabs: 261 nm λem: 418 nm λexc: 267 nm. Rf 0.65 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-1-[(p-methylphenyl)methyl]purino[9,8-a]pyridine-2,4-dione (13b). The title compound was prepared from commercially available 9a to afford after purification the desired product as a light yellow solid (20%). 1H NMR (250 MHz, DMSO-d6) δ 8.20 (d, 1H, J = 7.3 Hz, H6), 7.56 (d, 1H, J = 9.3 Hz, H9), 7.41 (dd, 2H, J = 9.0, 5.2 Hz, 2 × Harom-N3), 7.38–7.25 (m, 4H, 2 × Harom-N1, 2 × Harom-N3), 7.24–7.09 (m, 3H, H8, 2 × Harom-N1), 6.79 (t, 1H, J = 7.3 Hz, H7), 5.64 (s, 2H, N–CH2), 2.23 (s, 3H, C–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 159.5 (C=O), 157.9 (Cquat), 151.1 (C=O), 142.5 (Cquat), 136.7 (Cquat), 133.0 (Cquat), 132.7 (d, JC–F = 3.1 Hz, Cquat), 132.0 (Cquat), 131.1 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 129.5 (2 × Carom-N1), 126.0 (Carom), 125.5 (2 × Carom-N1), 125.1 (Carom), 120.1 (Cquat), 118.9 (Carom), 115.7 (d, JC–F = 23.3 Hz, 2 × Carom-N3), 113.4 (Carom), 46.7 (N–CH2), 20.6 (C–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.08 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C23H18FN4O2: 401.1708, found: 401.1405. λabs: 260 nm λem: 418 nm λexc: 261 nm. Rf 0.5 (DCM/MeOH 95:5).
4-[[3-(p-Fluorophenyl)-2,4-dioxo-purino[9,8-a]pyridin-1-yl]methyl]benzonitrile (13c). The title compound was prepared from commercially available 9a to afford after purification the desired product as a light yellow solid (30%). 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, 1H, J = 7.3 Hz, H6), 7.86 (d, 2H, J = 8.1 Hz, 2 × Harom-N1), 7.69 (d, 2H, J = 8.1 Hz, 2 × Harom-N1), 7.62 (d, 1H, J = 9.3 Hz, H9), 7.45–7.43 (m, 2H, 2 × Harom-N3), 7.34 (t, 2H, J = 8.7 Hz, 2 × Harom-N3), 7.29–7.21 (m, 1H, H8), 6.83 (t, 1H, J = 7.3 Hz, H7), 5.80 (s, 2H, N–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.3 (C=O), 157.9 (Cquat), 151.1 (C=O), 142.5 (Cquat), 142.0 (Cquat), 132.8 (2 × Carom-N1), 132.6 (d, JC–F = 3.0 Hz, Cquat), 131.8 (Cquat), 131.1 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 126.9 (2 × Carom-N1), 126.1 (Carom), 124.7 (Carom), 120.3 (Cquat), 119.0 (Carom), 118.5 (Cquat), 115.7 (d, JC–F = 22.8 Hz, 2 × Carom-N3), 113.7 (Carom), 110.4 (Cquat), 47.0 (N–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.00 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C23H15FN5O2: 412.1203, found: 412.1204. λabs: 259 nm λem: 418 nm λexc: 259 nm. Rf 0.5 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-1-[(p-nitrophenyl)methyl]purino [9,8-a]pyridine-2,4-dione (13d). The title compound was prepared from commercially available 9a to afford after purification the desired product as a yellow solid (28%). 1H NMR (250 MHz, DMSO-d6) δ 8.19 (d, 2H, J = 8.8 Hz, 2 × Harom-N1), 7.95 (d, 1H, J = 7.3 Hz, H6), 7.77 (d, 2H, J = 8.8 Hz, 2 × Harom-N1), 7.63 (d, 1H, J = 9.3 Hz, H9), 7.50–7.20 (m, 5H, H8, 4 × Harom-N3), 6.84 (t, 1H, J = 7.3 Hz, H7), 5.85 (s, 2H, N-CH2) ppm. 13C NMR (63 MHz, DMSO-d6) δ 159.5 (C=O), 157.9 (Cquat), 151.1 (C=O), 147.0 (Cquat), 144.0 (Cquat), 142.5 (Cquat), 132.5 (d, JC–F = 3.1 Hz, Cquat), 131.8 (Cquat), 131.1 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 127.2 (2 × Carom-N1), 126.1 (Carom), 124.7 (Carom), 123.9 (2 × Carom-N1), 120.3 (Cquat), 119.06 (Carom), 115.7 (d, JC–F = 23.3 Hz, 2 × Carom-N3), 113.7 (Carom), 46.9 (N–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ −113.96 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H15FN5O4: 432.1002, found: 432.1100. λabs: 269.5 nm λem: 551 nm λexc: 275 nm. Rf 0.5 (DCM/MeOH 95:5).
Methyl 4-[[3-(p-fluorophenyl)-2,4-dioxo-purino [9,8-a]pyridin-1-l]methyl]benzoate (13e). The title compound was prepared from commercially available 9a to afford after purification the desired product as a white solid (65%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (d, 1H, J = 7.3 Hz, H6), 7.95 (d, 2H, J = 7.9 Hz, 2 × Harom-N1), 7.61 (d, 3H, J = 7.9 Hz, H9, 2 × Harom-N1), 7.45 (dd, 2H, J = 8.6, 5.3 Hz, 2 × Harom-N3), 7.35 (t, 2H, J = 8.6 Hz, 2 × Harom-N3), 7.28–7.20 (m, 1H, H8), 6.82 (t, 1H, J = 7.3 Hz, H7), 5.80 (s, 2H, N–CH2), 3.83 (s, 3H, O–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 165.8 (C=O), 157.9 (C=O), 151.1 (C=O), 142.5 (Cquat), 141.8 (Cquat), 132.6 (d, JC–F = 3.2 Hz, Cquat), 131.9 (Cquat), 131.1 (d, JC–F = 8.9 Hz, 2 × Carom-N3), 129.7 (2 × Carom-N1), 128.9 (Cquat), 126.2 (2 × Carom-N1), 126.1 (Carom), 125.0 (Cquat), 124.8 (Carom), 120.2 (Cquat), 119.0 (Carom), 115.7 (d, JC–F = 22.7 Hz, 2 × Carom-N3), 113.6 (Carom), 52.1 (O–CH2), 47.01 (N–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.03 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C24H18FN4O4: 445.1306 found: 445.1305. λabs: 259 nm λem: 492 nm λexc: 266 nm. Rf 0.45 (DCM/MeOH 95:5).
1-[[p-(Dimethoxyphosphorylmethyl)phenyl]methyl]-3-(4-fluorophenyl)purino [9,8-a]pyridine-2,4-dione (13f). The title compound was prepared from commercially available 9a to afford after purification the desired product as a yellow foam (16%). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (d, 1H, J = 7.3 Hz, H6), 7.60 (d, 1H, J = 9.3 Hz, H9), 7.46 (dd, 2H, J = 7.8, 5.3 Hz, 2 × Harom-N3), 7.41–7.31 (m, 4H, 2 × Harom-N1, 2 × Harom-N3), 7.31–7.20 (m, 3H, H8, 2 × Harom-N1), 6.81 (t, 1H, J = 7.3 Hz, H7), 5.71 (s, 2H, N–CH2), 3.55 (s, 3H, O–CH3), 3.53 (s, 3H, O–CH3), 3.33 (s, 2H, C–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.8 (C=O), 158.4 (Cquat), 151.6 (C=O), 143.0 (Cquat), 134.9 (d, J = 3.7 Hz, Cquat), 133.1 (d, JC–F = 3.0 Hz, Cquat), 132.5 (Cquat), 131.9 (d, JC–F = 9.1 Hz, Cquat), 131.6 (d, JC–F = 9.0 Hz, 2 × Carom-N3), 130.8 (d, JC–P = 6.5 Hz, 2 × Carom-N1), 126.5 (Carom), 126.2 (d, JC–P = 3.0 Hz, 2 × Carom-N1), 125.5 (Carom), 120.6 (Cquat), 119.4 (Carom), 116.2 (d, JC–F = 22.9 Hz, 2 × Carom-N3), 113.9 (Carom), 52.8 (O–CH3), 52.7 (O–CH3), 47.2 (N–CH2), 29.52 (d, J1C–P = 108.5 Hz, P–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ -114.07 ppm. 31P NMR (162 MHz, DMSO-d6) δ 28.89 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C24H21FN4O5P: 509.1384, found: 509.1380.λabs: 264 nm λem: 415 nm λexc: 265 nm. Rf 0.2 (DCM/MeOH 95:5).
3.5. General Synthetic Procedure 4 for the Sonogashira Coupling cross Coupling
Under inert atmosphere, to a solution of deprotected Br-compound (1 eq.) in dry DMF (0.082 M) were successively added copper iodide (0.2 eq.), triethylamine (3 eq.), alkynyl substrate (3 eq.), and Pd(PPh3)4 (10 mol%). The reaction mixture was heated under microwave irradiation at 110 °C for 15 min. The reaction was quenched with EtOAc and co-evaporated with heptane. Pure compounds were obtained after purification by flash column chromatography with DCM/MeOH as eluent.
3-(p-Fluorophenyl)-7-(4-phenylbut-1-ynyl)-1H-purino [9,8-a]pyridine-2,4-dione (20a). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange/yellow solid (70%). 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, 1H, J = 7.2 Hz, H9), 7.45 (s, 1H, H6), 7.35–7.30 (d, 4H, 4 × Harom), 7.29–7.22 (m, 5H, 4 × Harom-N3, Harom), 6.77 (dd, 1H, J = 7.2, 1.8 Hz, H8), 2.88 (t, 2H, J = 7.2 Hz, C–CH2), 2.76 (t, 2H, J = 7.2 Hz, CH2–CH2) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.3 (C=O), 159.9 (Cquat), 159.2 (Cquat), 153.2 (C=O), 140.7 (Cquat), 140.3 (Cquat), 134.0–133.6 (m, Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 128.5 (2 × Carom), 128.2 (2 × Carom), 126.2 (Carom), 123.9 (Carom), 120.4 (Cquat), 119.9 (Carom), 118.6 (Cquat), 115.3 (d, JC–F = 22.7 Hz, 2 × Carom-N3), 114.0 (Carom), 93.9 (CH2–C), 80.0 (C≡C), 34.0 (CH2–C), 21.0 (C–CH2) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.39 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C25H18FN4O2: 425.1408, found: 425.1409. λabs: 266 nm λem: 513 nm λexc: 278 nm. Rf 0.34 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-7-[2-(4-propylphenyl)ethynyl]-1H-purino [9,8-a]pyridine-2,4-dione (20b). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow solid (74%). 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H, H1), 8.39 (d, 1H, J = 7.3 Hz, H9), 7.71 (s, 1H, H6), 7.52 (d, 2H, J = 7.8 Hz, 2 × Harom), 7.28 (dd, 6H, J = 7.5, 3.5 Hz, 4 × Harom-N3, 2 × Harom), 6.99 (d, 1H, J = 7.3 Hz, H8), 2.60 (t, 2H, J = 7.5 Hz, C–CH2), 1.61 (h, 2H, J = 7.5 Hz, CH2–CH2), 0.90 (t, 3H, J = 7.5 Hz, CH2–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.3 (C=O), 159.9 (Cquat), 159.2 (Cquat), 154.3 (C=O), 143.6 (Cquat), 140.6 (Cquat), 133.6 (Cquat), 131.4 (2 × Carom), 131.2 (d, JC–F = 8.7 Hz, 2 × Carom-N3), 128.8 (2 × Carom), 123.9 (Carom), 120.9 (Carom), 119.0 (Cquat), 118.9 (Cquat), 115.3 (d, JC–F = 22.3 Hz, 2 × Carom-N3), 113.8 (Carom), 92.6 (Cquat), 90.1 (CH2–C), 87.6 (C≡C), 37.0 (C–CH2), 23.7 (CH2–CH2), 13.5 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.43 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C26H20FN4O2: 439.1564, found: 439.1563. λabs: 300 nm λem: 528 nm λexc: 307 nm. Rf 0.35 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-hex-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (20c). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow/orange solid (91%). 1H NMR (400 MHz, DMSO-d6) δ 12.95 (bs, 1H, H1), 8.40 (d, 1H, J = 7.2 Hz, H9), 7.61 (s, 1H, H6), 7.39–7.25 (m, 4H, 4 × Harom), 6.95 (dd, 1H, J = 7.2, 1.6 Hz, H8), 2.51–2.47 (m, 2H, CH2), 1.59–1.52 (m, 2H, CH2), 1.50–1.39 (m, 2H, CH2), 0.93 (t, 3H, J = 7.3 Hz, CH2–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 161.6 (C=O), 159.5 (Cquat), 150.0 (C=O), 141.1 (Cquat), 132.4 (Cquat), 131.3 (d, JC–F = 9.0 Hz, 2 × Carom), 123.8 (Carom), 120.7 (Cquat), 120.5 (Carom), 119.9 (Cquat), 115.6 (d, JC–F = 23.2 Hz, 2 × Carom), 115.3 (Carom), 113.7 (Cquat), 95.0 (CH2–C), 79.1 (C≡C), 30.0 (CH2–CH2), 21.4 (CH2–CH2), 18.4 (CH2–CH2), 13.4 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.41 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H18FN4O2: 377.1408, found: 377.1406. λabs: 263 nm λem: 513nm λexc: 278 nm. Rf 0.33 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-hept-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (20d). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange solid (95%). 1H NMR (250 MHz, DMSO-d6) δ 12.95 (s, 1H, H1), 8.43 (d, 1H, J = 7.3 Hz, H9), 7.62 (s, 1H, H5), 7.42–7.24 (m, 4H, 4 × Harom), 6.97 (dd, 1H, J = 7.3, 1.6 Hz, H8), 2.51–2.47 (m, 2H, CH2), 1.57 (q, 2H, J = 7.0 Hz, CH2–CH2), 1.46–1.28 (m, 4H, 2 × CH2), 0.90 (t, 3H, J = 7.0 Hz, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.4 (C=O), 158.2 (Cquat), 150.2 (C=O), 141.1 (Cquat), 132.3 (d, JC–F = 3.7 Hz, Cquat), 131.3 (d, JC–F = 8.8 Hz, 2 × Carom), 128.6 (Cquat), 123.8 (Carom), 120.8 (Carom), 120.5 (Cquat), 119.0 (Cquat), 115.9 (2 × Carom), 115.5 (Carom), 95.2 (CH2–C), 79.0 (C≡C), 30.4 (C–CH2), 27.5 (CH2–CH2), 21.5 (CH2–CH2), 18.8 (CH2–CH2), 13.8 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.36 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H20FN4O2: 391.1564, found: 391.1563. λabs: 267 nm λem: 517 nm λexc: 277 nm. Rf 0.33 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-7-oct-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (20e). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow solid (92%). 1H NMR (400 MHz, DMSO-d6) δ 8.41 (d, 1H, J = 7.7 Hz, H9), 7.61 (s, 1H, H6), 7.39–7.27 (m, 4H, 4 × Harom), 6.96 (dd, 1H, J = 7.7, 1.5 Hz, H8), 2.51–2.40 (m, 2H, CH2), 1.57 (p, 2H, J = 7.0 Hz, CH2), 1.43 (dt, 2H, J = 13.3, 7.0 Hz, CH2–CH2), 1.30 (dt, 4H, J = 7.0, 3.8 Hz, 2 × CH2), 0.89 (t, 3H, J = 7.0 Hz, CH2–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.7 (C=O), 158.4 (Cquat), 150.3 (C=O), 141.2 (Cquat), 132.4 (Cquat), 132.3 (d, JC–F = 3.1 Hz, Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom), 123.8 (Carom), 120.8 (Cquat), 120.5 (Carom), 118.9 (Cquat), 115.6 (d, JC–F = 22.7 Hz, 2 × Carom), 115.4 (Carom), 95.1 (CH2–C), 79.1 (C≡C), 30.7 (C–CH2), 27.9 (CH2–CH2), 27.8 (CH2–CH2), 21.9 (CH2–CH2), 18.7 (CH2–CH2), 13.9 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.38 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C23H22FN4O2: 405.1721, found: 405.1718. λabs: 268 nm λem: 512 nm λexc: 276 nm. Rf 0.33 (DCM/MeOH 95:5).
N-[3-[3-(p-fluorophenyl)-2,4-dioxo-1H-purino [9,8-a]pyridin-7-yl]prop-2-ynyl]octanamide (20f). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange solid (95%). 1H NMR (250 MHz, DMSO-d6) δ 8.43–8.31 (m, 1H, H9), 7.52 (s, 1H, H6), 7.26 (t, 4H, J = 7.1 Hz, 4 × Harom), 6.82 (t, 1H, J = 8.0 Hz, H8), 4.23 (d, 2H, J = 11.3 Hz, C–CH2), 2.10 (q, 2H, J = 12.5, 9.9 Hz, CO–CH2), 1.65 (q, 2H, J = 7.1 Hz, CH2–CH2), 1.24 (q, 8H, J = 9.6, 7.1 Hz, 4 × CH2), 0.85 (tq, 3H, J = 11.1, 6.5, 5.6 Hz, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 167.0 (C=O), 162.7 (C=O), 159.3 (Cquat), 152.03 (C=O), 136.1 (Cquat), 134.0 (Cquat), 133.8 (d, JC–F = 3.2 Hz, Cquat), 131.3 (d, JC–F = 11.6 Hz, 2 × Carom), 124.1 (Carom), 120.9 (Carom), 119.0 (Cquat), 118.8 (Cquat), 115.2 (2 × Carom), 113.7 (Carom), 90.9 (CH2–C), 79.6 (C≡C), 45.3 (N–CH2), 34.6 (CO–CH2), 31.0 (CH2–CH2), 28.5 (CH2–CH2), 28.2 (CH2–CH2), 24.9 (CH2–CH2), 22.1 (CH2-CH2), 13.7 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.58 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C26H27FN5O2: 476.2092, found: 476.2089. λabs: 271 nm λem: 510 nm λexc: 279 nm. Rf 0.32 (DCM/MeOH 95:5).
1-[3-[3-(p-Fluorophenyl)-2,4-dioxo-1H-purino [9,8-a]pyridin-7-yl]prop-2-ynyl]-3-hexyl-urea (20g). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange solid (92%). 1H NMR (250 MHz, DMSO-d6) δ 8.29 (d, 1H, J = 7.5 Hz, H9), 7.48 (s, 1H, H6), 7.41–7.11 (m, 4H, 4 × Harom-N3), 6.73 (t, 1H, J = 7.5 Hz, H8), 4.09 (d, 1H, J = 5.7 Hz, NH–CH2), 1.52–0.99 (m, 10H, 5 × CH2), 0.85 (t, 3H, J = 6.6 Hz, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.8 (C=O), 158.0 (C=O), 151.4 (C=O), 140.7 (Cquat), 136.9 (Cquat), 135.3 (Cquat), 131.6 (d, JC–F = 9.9 Hz, 2 × Carom), 124.3 (Carom), 121.2 (Carom), 118.9 (Cquat), 115.7 (Cquat), 115.2 (2 × Carom), 113.3 (Carom), 96.3 (Cquat), 92.1 (CH2–C), 80.6 (C≡C), 39.7 (N–CH2), 31.4 (CH2–CH2), 30.4 (CH2–CH2), 30.0 (CH2–CH2), 26.4 (CH2–CH2), 22.4 (CH2–CH2), 14.2 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −116.27 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C25H26FN6O3: 477.2044, found: 477.2042. λabs: 279 nm λem: 515 nm λexc: 278 nm. Rf 0.30 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-(4-phenylbut-1-ynyl)-1H-purino [9,8-a]pyridine-2,4-dione (21a). The title compound was prepared from commercially available 19b to afford after purification the desired product as an orange solid (80%). 1H NMR (250 MHz, DMSO-d6) δ 8.44 (s, 1H, H9), 7.45 (d, 1H, J = 9.6 Hz, H6), 7.33 (d, 4H, J = 4.3 Hz, 4 × Harom), 7.25 (h, 5H, J = 4.0 Hz, 4 × Harom-N3, Harom), 7.07 (d, 1H, J = 9.6 Hz, H7), 2.89 (t, 2H, J = 6.9 Hz, C–CH2), 2.75 (t, 2H, J = 6.9 Hz, CH2–CH2) ppm. 13C NMR (63 MHz, DMSO-d6) δ 162.4 (C=O), 159.3 (Cquat), 153.7 (C=O), 151.2 (Cquat), 140.3 (Cquat), 139.8 (Cquat), 133.9 (Cquat), 131.2 (2 × Carom), 128.5 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 128.2 (2 × Carom), 127.9 (Carom), 126.3 (Carom), 126.2 (Carom), 118.3 (Carom), 118.0 (Cquat), 115.3 (d, JC–F = 22.3 Hz, 2 × Carom-N3), 107.8 (Cquat), 91.7 (CH2–C), 77.4 (C≡C), 34.0 (C–CH2), 20.8 (CH2–C) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.57 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C25H18FN4O2: 425.1408, found: 425.1406. λabs: 272 nm λem: 443 nm λexc: 286 nm. Rf 0.33 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-[2-(4-propylphenyl)ethynyl]-1H-purino [9,8-a]pyridine-2,4-dione (21b). The title compound was prepared from commercially available 19b to afford after purification the desired product as a beige solid (67%). 1H NMR (250 MHz, DMSO-d6) δ 8.81 (s, 1H, H9), 7.86–7.72 (m, 1H, H6), 7.68–7.61 (m, 3H, H7, 2 × Harom), 7.55–7.48 (m, 2H, 2 × Harom), 7.44–7.23 (m, 4H, 4 × Harom-N3), 2.61 (t, 2H, J = 7.6 Hz, C–CH2), 1.59 (ddt, 2H, J = 12.8, 10.0, 6.7 Hz, CH2–CH2), 0.98–0.80 (m, 3H, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.4 (C=O), 159.6 (Cquat), 150.0 (C=O), 143.6 (Cquat), 140.4 (Cquat), 133.7 (Cquat), 132.2 (Cquat), 131.4 (2 × Carom), 131.3 (d, JC–F = 10.3 Hz, 2 × Carom-N3), 128.8 (2 × Carom), 128.8 (Carom), 126.9 (Carom), 118.7 (Carom), 118.5 (Cquat), 118.5 (Cquat), 115.5 (d, JC–F = 24.9 Hz, 2 × Carom-N3), 108.5 (Cquat), 91.2 (CH2–C), 84.7 (C≡C), 37.0 (CH2–CH2), 23.6 (CH2–CH2), 13.4 (CH2–CH3) ppm. 19F NMR (235 MHz, DMSO-d6) δ −114.29 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C26H20FN4O2: 439.1564, found: 439.1562. λabs: 302 nm λem: 454 nm λexc: 288 nm. Rf 0.35 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-hex-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (21c). The title compound was prepared from commercially available 19b to afford after purification the desired product as a beige solid (42%). 1H NMR (400 MHz, DMSO-d6) δ 12.80 (s, 1H, H1), 8.64 (s, 1H, H9), 7.56 (d, 1H, J = 9.5 Hz, H6), 7.38–7.27 (m, 4H, 4 × Harom), 7.23 (dd, 1H, J = 9.5, 1.6 Hz, H8), 2.48 (d, 2H, J = 7.0 Hz, C–CH2), 1.61–1.51 (m, 2H, CH2–CH2), 1.51–1.41 (m, 2H, CH2–CH2), 0.94 (t, 3H, J = 7.0 Hz, CH2–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.3 (C=O), 158.4 (Cquat), 150.2 (C=O), 140.3 (Cquat), 132.3 (Cquat), 132.2 (Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom), 128.7 (Carom), 126.5 (Carom), 118.3 (Cquat), 118.3 (Carom), 115.6 (d, JC–F = 22.8 Hz, 2 × Carom), 109.2 (Cquat), 92.8 (CH2–C), 76.4 (C≡C), 30.0 (CH2–CH2), 21.3 (CH2–CH2), 18.2 (CH2–CH2), 13.4 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.37 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H18FN4O2: 377.1408, found: 377.1404. λabs: 273nm λem: 498 nm λexc: 286 nm. Rf 0.33 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-hept-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (21d). The title compound was prepared from commercially available 19b to afford after purification the desired product as an orange solid (50%). 1H NMR (250 MHz, DMSO-d6) δ 8.56 (s, 1H, H9), 7.93–7.79 (m, 1H, H6), 7.57–7.41 (m, 1H, H7), 7.24 (dd, 4H, J = 28.7, 8.3 Hz, 4 × Harom), 2.60–2.45 (m, 2H, C–CH2), 1.58 (t, 2H, J = 7.2 Hz, CH2–CH2), 1.52–1.30 (m, 4H, 2 × CH2), 0.88 (dt, 3H, J = 13.5, 6.5 Hz, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.4 (C=O), 158.8 (Cquat), 151.7 (C=O), 146.8 (Cquat), 140.0 (Cquat), 133.1 (d, JC–F = 3.2 Hz, Cquat), 131.3 (d, JC–F = 11.7 Hz, 2 × Carom), 128.4 (Carom), 126.2 (Carom), 118.2 (Carom), 118.1 (Cquat), 115.5 (d, JC–F = 24.3 Hz, 2 × Carom), 108.3 (Cquat), 92.31 (CH2–C), 76.7 (C≡C), 30.4 (CH2–CH2), 27.6 (CH2–CH2), 21.5 (CH2–CH2), 18.6 (CH2–CH2), 13.8 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.97 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H20FN4O2: 391.1564, found: 391.1563. λabs: 271nm λem: 515 nm λexc: 284 nm. Rf 0.33 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-oct-1-ynyl-1H-purino [9,8-a]pyridine-2,4-dione (21e). The title compound was prepared from commercially available 19b to afford after purification the desired product as an orange solid (74%). 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 1H, H1), 8.63 (s, 1H, H9), 7.57 (d, 1H, J = 9.6 Hz, H6), 7.41–7.28 (m, 4H, 4 × Harom), 7.23 (dd, 1H, J = 9.6, 1.6 Hz, C–CH2), 2.48–2.42 (m, 2H, CH2–CH2), 1.62–1.52 (m, 2H, CH2–CH2), 1.48–1.38 (m, 2H, CH2–CH2), 1.32 (dq, 4H, J = 7.1, 3.3 Hz, 2 × CH2), 0.95–0.79 (m, 3H, CH2–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.7 (C=O), 160.3 (Cquat), 158.4 (Cquat), 150.2 (C=O), 140.3 (Cquat), 132.3 (d, J = 4.3 Hz, Cquat), 131.2 (d, JC–F = 9.9 Hz, 2 × Carom), 128.7 (Carom), 126.4 (Carom), 118.5 (Cquat), 118.3 (Carom), 115.6 (d, JC–F = 22.6 Hz, 2 × Carom), 109.2 (Cquat), 92.9 (CH2–C), 76.4 (C≡C), 30.7 (CH2–CH2), 27.9 (CH2–CH2), 27.9 (CH2–CH2), 21.9 (CH2–CH2), 18.5 (CH2–CH2), 13.9 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.34 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C23H22FN4O2: 405.1721, found: 405.1720. Rf 0.33 (DCM/MeOH 95:5). λabs: 275 nm λem: 503 nm λexc: 286 nm.
N-[3-[3-(p-fluorophenyl)-2,4-dioxo-1H-purino [9,8-a]pyridin-8-yl]prop-2-ynyl]octanamide (21f). The title compound was prepared from commercially available 19b to afford after purification the desired product as an orange solid (40%). 1H NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H, H9), 7.39 (d, 1H,J = 9.4 Hz, H6), 7.28–7.17 (m, 4H, 4 × Harom), 7.06 (d, 1H, J = 9.4 Hz, H7), 4.07 (s, 1H, NH–CH2), 3.11–2.87 (m, 2H, CO–CH2), 1.43–1.28 (m, 2H, CH2–CH2), 1.21 (bs, 8H, 4 × CH2), 0.84 (t, 3H, J = 6.8 Hz, CH2–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 175.0 (C=O), 162.0 (C=O), 159.5 (Cquat), 150.4 (C=O), 139.6 (Cquat), 134.9 (Cquat), 134.4 (Cquat), 131.2 (d, JC–F = 16.8 Hz, 2 × Carom), 127.4 (Carom), 126.9 (Carom), 118.2 (Carom), 117.5 (Cquat), 115.2 (d, JC–F = 14.4 Hz, 2 × Carom), 106.4 (Cquat), 89.7 (CH2–C), 77.8 (C≡C), 45.4 (N–CH2), 35.0 (C–CH2), 31.0 (CH2–CH2), 29.9 (CH2–CH2), 29.5 (CH2–CH2), 26.0 (CH2–CH2), 22.0 (CH2–CH2), 13.9 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.90 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C26H27FN5O2: 476.2092, found: 476.2091. λabs: 273 nm λem: 515 nm λexc: 284 nm. Rf 0.32 (DCM/MeOH 95:5).
1-[3-[3-(p-Fluorophenyl)-2,4-dioxo-1H-purino [9,8-a]pyridin-8-yl]prop-2-ynyl]-3-hexyl-urea (21g). The title compound was prepared from commercially available 19b to afford after purification the desired product as an orange solid (92%). 1H NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H, H9), 7.39 (d, 1H, J = 9.5 Hz, H6), 7.31–7.16 (m, 4H, 4 × Harom), 7.06 (d, 1H, J = 9.5 Hz, H7), 4.07 (d, 2H, J = 5.7 Hz, CH2), 1.45–1.03 (m, 10H, 5 × CH2), 0.84 (q, 3H, J = 5.7 Hz, CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.0 (C=O), 159.9(Cquat), 157.58 (C=O), 150.4 (C=O), 139.6 (Cquat), 134.8 (Cquat), 131.3 (Cquat), 131.1 (2 × Carom), 127.3 (Carom),126.9 (Carom), 118.2 (Carom), 117.74 (Cquat), 115.1 (2 × Carom), 106.4 (Cquat), 89.7 (CH2–C), 77.8 (C≡C), 45.4 (N–CH2), 31.0 (CH2–CH2), 29.9 (CH2–CH2), 28.5 (CH2–CH2), 26.0 (CH2–CH2), 22.0 (CH2–CH2), 14.2 (CH2–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −116.17 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C25H26FN6O3: 477.2045, found: 477.2045. λabs: 285 nm λem: 512 nm λexc: 286 nm. Rf 0.30 (DCM/MeOH 95:5).
3.6. General Synthetic Procedure 1 for the Suzuki-Miyaura Coupling cross Coupling
A mixture of Br-compound (1 eq.), boronic acid (1.5 eq.), cesium carbonate (2 eq.), Pd(PPh3)4 (0.1 eq.) in anhydrous DMF (0.1 M) under inert atmosphere was heated under microwave irradiation at 120 °C for 40 min. Pure compounds were obtained after purification by flash column chromatography with DCM/MeOH as eluent.
3-(p-Fluorophenyl)-7-phenyl-1H-purino [9,8-a]pyridine-2,4-dione (22a). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow solid (45%). 1H NMR (250 MHz, DMSO-d6) δ 8.52 (d, 1H, J = 7.3 Hz, H9), 7.85 (d, 3H, J = 6.9 Hz, H6, 2 × Harom), 7.60–7.34 (m, 4H, H8, 3 × Harom), 7.30 (d, 4H, J = 7.1 Hz, 4 × Harom-N3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.1 (C=O), 159.2 (Cquat), 159.0 (Cquat), 152.1 (C=O), 142.0 (Cquat), 137.2 (d, J = 14.2 Hz, Cquat), 135.7 (Cquat), 133.3 (d, JC–F = 9.3 Hz, Cquat), 131.2 (d, JC–F = 9.1 Hz, 2 × Carom-N3), 128.9 (2 × Carom), 128.4 (Carom), 126.3 (2 × Carom), 123.9 (Carom), 118.4 (Cquat), 115.4 (d, JC–F = 24.2 Hz, 2 × Carom-N3), 114.1 (Carom), 111.7 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.15 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H14FN4O2: 373.1095, found: 373.1099. λabs: 264 nm λem: 517 nm λexc: 285 nm. Rf 0.31 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-7-(p-tolyl)-1H-purino [9,8-a]pyridine-2,4-dione (22b). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange solid (44%). 1H NMR (250 MHz, DMSO-d6) δ 8.33 (d, 1H, J = 7.3 Hz, H9), 7.72 (d, 2H, J = 8.1 Hz, 2 × Harom), 7.67 (s, 1H, H6), 7.31 (d, 2H, J = 8.0 Hz, 2 × Harom), 7.26–7.17 (m, 5H, H8, 4 × Harom-N3), 2.37 (s, 3H, C–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 159.9 (C=O), 158.7 (Cquat), 152.3 (C=O), 141.5 (Cquat), 138.0 (Cquat), 137.7 (Cquat), 136.3 (Cquat), 135.1 (d, JC–F = 2.5 Hz, Cquat), 134.9 (Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 129.6 (2 × Carom), 126.4 (Cquat), 126.1 (2 × Carom), 123.8 (Carom), 115.0 (d, JC–F = 22.6 Hz, 2 × Carom-N3), 113.5 (Carom), 110.3 (Carom), 20.7 (C–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −116.31 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H16FN4O2: 387.1251, found: 387.1255. λabs: 276 nm λem: 519 nm λexc: 279 nm. Rf 0.32 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-7-(p-methoxyphenyl)-1H-purino [9,8-a]pyridine-2,4-dione (22c). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow solid (15%). 1H NMR (250 MHz, DMSO-d6) δ 8.50 (d, 1H, J = 7.2 Hz, H9), 7.81 (d, 2H, J = 8.4 Hz, 2 × Harom), 7.77 (s, 1H, H6), 7.40–7.06 (m, 5H, H8, 4 × Harom-N3), 7.07 (d, 2H, J = 8.4 Hz, 2 × Harom), 3.82 (s, 3H, O–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 159.6 (C=O), 159.1 (Cquat), 152.7 (C=O), 142.4 (Cquat), 140.1 (Cquat), 136.0 (Cquat), 133.9 (Cquat), 133.6 (d, JC–F = 4.4 Hz, Cquat), 131.2 (d, JC–F = 8.19 Hz, 2 × Carom-N3), 129.6 (Cquat), 127.7 (2 × Carom), 123.8 (Carom), 118.2 (Cquat), 115.3 (d, JC–F = 22.6 Hz, 2 × Carom-N3), 114.4 (2 × Carom), 112.8 (Carom), 111.5 (Carom), 55.2 (O–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −115.28 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H16FN4O3: 403.1200, found: 403.1202. λabs: 275 nm λem: 522 nm λexc: 278 nm. Rf 0.32 (DCM/MeOH 95:5).
3-(p-fluorophenyl)-7-[p-(trifluoromethoxy)phenyl]-1H-purino [9,8-a]pyridine-2,4-dione (22d). The title compound was prepared from commercially available 19a to afford after purification the desired product as an orange solid (15%). 1H NMR (250 MHz, DMSO-d6) δ 8.53 (d, 1H, J = 7.4 Hz, H9), 7.98 (d, 2H, J = 8.5 Hz, 2 × Harom), 7.88 (s, 1H, H6), 7.49 (d, 2H, J = 8.5 Hz, 2 × Harom), 7.39 (d, 1H, J = 7.4 Hz, H8), 7.30 (d, 4H, J = 7.0 Hz, 4 × Harom-N3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 163.1 (C=O), 159.3 (Cquat), 159.0 (Cquat), 152.2 (C=O), 148.4 (Cquat), 141.7 (Cquat), 136.7 (Cquat), 136.1 (Cquat), 135.6 (Cquat), 133.4 (Cquat), 131.2 (d, JC–F = 8.7 Hz, 2 × Carom-N3), 128.5 (2 × Carom), 124.1 (Carom), 121.4 (2 × Carom), 118.6 (Cquat), 115.3 (d, JC–F = 22.6 Hz, 2 × Carom-N3), 114.8 (Carom), 111.6 (Carom) ppm. 19F NMR (376 MHz, DMSO-d6) δ –56.71 (–OCF3), −115.18 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H13F4N4O3: 457.0918, found: 457.0917. λabs: 274 nm λem: 536 nm λexc: 279 nm. Rf 0.3 (DCM/MeOH 95:5).
3,7-Bis(p-fluorophenyl)-1H-purino [9,8-a]pyridine-2,4-dione (22e). The title compound was prepared from commercially available 19a to afford after purification the desired product as a yellow solid (45%). 1H NMR (250 MHz, DMSO-d6) δ 8.50 (d, 1H, J = 7.4 Hz, H9), 7.97–7.86 (m, 2H, 2 × Harom-N3), 7.82 (s, 1H, H6), 7.44–7.22 (m, 7H, H8, 2 × Harom-N3, 4 × Harom-N3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 160.3 (C=O), 159.1 (Cquat), 159.0 (Cquat), 152.4–152.1 (m, C=O, Cquat), 141.8 (Cquat), 136.0 (Cquat), 133.8 (d, JC–F = 3.1 Hz, Cquat), 133.5 (Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 128.6 (d, JC–F = 8.1 Hz, 2 × Carom), 124.5 (Cquat), 124.0 (Carom), 118.4 (Cquat), 115.8 (d, JC–F = 21.4 Hz, 2 × Carom-N3), 115.3 (d, JC–F = 22.6 Hz, 2 × Carom), 114.1 (Carom), 111.6 (Carom) ppm. 19F NMR (235 MHz, DMSO-d6) δ −113.66, −115.26 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H13F2N4O2: 391.1002, found: 391.1001. λabs: 274 nm λem: 539 nm λexc: 277 nm. Rf 0.26 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-phenyl-1H-purino [9,8-a]pyridine-2,4-dione (23a). The title compound was prepared from commercially available 19b to afford after purification the desired product as a yellow solid (50%). 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H, H9), 7.75–7.72 (m, 2H, 2 × Harom), 7.67 (qd, 2H, J = 9.7, 1.4 Hz, H6, Harom), 7.54 (t, 2H, J = 7.7 Hz, 2 × Harom), 7.47–7.41 (m, 1H, H7), 7.36–7.27 (m, 4H, 4 × Harom-N3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.5 (C=O), 160.1 (Cquat), 158.9 (Cquat), 151.5 (C=O), 140.8 (Cquat), 136.0 (Cquat), 134.0 (Cquat), 133.1 (d, JC–F = 2.8 Hz, Cquat), 131.3 (d, JC–F = 9.1 Hz, 2 × Carom-N3), 129.2 (2 × Carom), 128.1 (Carom), 126.2 (2 × Carom), 125.2 (Cquat), 120.9 (Carom), 118.3 (Carom), 115.4 (d, JC–F = 22.7 Hz, 2 × Carom-N3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.90 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H14FN4O2: 373.1095, found: 373.1094. λabs: 274 nm λem: 441 nm λexc: 288 nm. Rf 0.31 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-(p-tolyl)-1H-purino [9,8-a]pyridine-2,4-dione (23b). The title compound was prepared from commercially available 19b to afford after purification the desired product as a yellow/beige solid (31%). 1H NMR (250 MHz, DMSO-d6) δ 8.82 (s, 1H, H9), 7.62 (d, 5H, J = 7.9 Hz, H6, 4 × Harom), 7.33 (t, 7H, J = 6.7 Hz, H7, 2 × Harom, 4 × Harom-N3), 2.37 (s, 3H, C–CH3) ppm. 13C NMR (63 MHz, DMSO-d6) δ 158.9 (C=O), 157.0 (Cquat), 151.6 (C=O), 150.8 (Cquat), 140.8 (Cquat), 137.5 (Cquat), 135.0 (Cquat), 133.1 (d, JC–F = 3.7 Hz, Cquat), 133.0 (Cquat), 131.3 (d, JC–F = 8.8 Hz, 2 × Carom-N3), 129.7 (2 × Carom), 126.2 (Carom), 126.0 (2 × Carom), 125.0 (Cquat), 120.4 (Carom), 118.2 (Carom), 115.4 (d, JC–F = 22.6 Hz, 2 × Carom-N3), 20.6 (C–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.93 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H16FN4O2: 387.1251, found: 387.1253. λabs: 244 nm λem: 444 nm λexc: 274 nm. Rf 0.32 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-(p-methoxyphenyl)-1H-purino [9,8-a]pyridine-2,4-dione (23c). The title compound was prepared from commercially available 19b to afford after purification the desired product as a yellow/beige solid (31%). 1H NMR (400 MHz, DMSO-d6) δ 8.78 (s, 1H, H9), 7.70–7.57 (m, 4H, H6, H7, 2 × Harom), 7.33–7.30 (m, 4H, 4 × Harom-N3), 7.10 (d, 2H, J = 8.7 Hz, 2 × Harom), 3.82 (s, 3H, O–CH3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 162.5 (C=O), 162.5 (Cquat), 159.3 (Cquat), 151.6 (C=O), 140.7 (Cquat), 140.0 (Cquat), 133.1 (Cquat), 131.3 (d, JC–F = 9.0 Hz, 2 × Carom-N3), 128.2 (Cquat), 127.4 (2 × Carom), 126.3 (Carom), 124.9 (Cquat), 119.9 (Carom), 118.2 (Carom), 115.4 (d, JC–F = 22.2 Hz, 2 × Carom-N3), 114.6 (2 × Carom), 99.4 (Cquat), 55.2 (O–CH3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.96 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H16FN4O3: 403.1200, found: 403.1201. λabs: 274 nm λem: 438 nm λexc: 286 nm. Rf 0.32 (DCM/MeOH 95:5).
3-(p-Fluorophenyl)-8-[p-(trifluoromethoxy)phenyl]-1H-purino [9,8-a]pyridine-2,4-dione (23d). The title compound was prepared from commercially available 19b to afford after purification the desired product as a yellow/beige solid (43%). 1H NMR (250 MHz, DMSO-d6) δ 8.68 (s, 1H, H9), 7.94–7.84 (m, 2H, 2 × Harom), 7.48 (d, 1H, J = 9.7 Hz, H6), 7.39–7.22 (m, 7H, H7, 2 × Harom, 4 × Harom-N3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 164.6 (C=O), 159.3 (Cquat), 159.0 (Cquat), 152.5 (C=O), 146.2 (Cquat), 139.7 (Cquat), 138.9 (Cquat), 136.1 (2 × Carom), 133.3 (Cquat), 131.2 (d, JC–F = 3.1 Hz, Cquat), 131.9 (Cquat), 131.2 (d, JC–F = 8.8 Hz, 2 × Carom), 128.7 (Carom), 124.0 (Carom), 119.5 (d, JC–F = 5.5 Hz, 2 × Carom), 118.3 (Cquat), 115.3 (d, JC–F = 22.8 Hz, 2 × Carom), 106.0 (Cquat) ppm. 19F NMR (376 MHz, DMSO-d6) δ –56.72 (–O–CF3), −114.27 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C22H13F4N4O3: 457.0919, found: 457.0919. λabs: 273 nm λem: 510 nm λexc: 294 nm. Rf 0.3 (DCM/MeOH 95:5).
3,8-Bis(p-Fluorophenyl)-1H-purino [9,8-a]pyridine-2,4-dione (23e). The title compound was prepared from commercially available 19b to afford after purification the desired product as a yellow/beige solid (11%). 1H NMR (400 MHz, DMSO-d6) δ 8.90 (s, 1H, H9), 7.82–7.73 (m, 2H, 2 × Harom), 7.69–7.58 (m, 2H, H6, H7), 7.38 (t, 2H, J = 8.8 Hz, 2 × Harom), 7.32–7.22 (m, 4H, 4 × Harom-N3) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.8 (C=O), 160.0 (Cquat), 159.1 (Cquat), 151.9 (C=O), 140.7 (Cquat), 136.4 (Cquat), 135.9 (Cquat), 132.5 (d, JC–F = 3.5 Hz, Cquat), 131.5–13.4 (m, Cquat), 131.3 (d, JC–F = 8.4 Hz, 2 × Carom), 128.4 (d, JC–F = 8.3 Hz, 2 × Carom-N3), 126.1 (Carom), 124.0 (Cquat), 121.0 (Carom), 118.3 (Carom), 116.0 (d, JC–F = 21.8 Hz, 2 × Carom), 115.4 (d, JC–F = 22.7 Hz, 2 × Carom-N3) ppm. 19F NMR (376 MHz, DMSO-d6) δ −114.36, −115.10 ppm. HRMS-ESI (m/z) [M+H]+ calcd. for C21H13F2N4O2: 391.1001, found: 391.1006. λabs: 274 nm λem: 441 nm λexc: 273 nm. Rf 0.26 (DCM/MeOH 95:5).
3.7. Biological Assays
3.7.1. Mtb ThyX Protein Expression and Purification [30]
The M. tuberculosis ThyX enzyme was expressed in E. coli BL21(DE3)/pLysS strains containing the recombinant pET24d plasmid carrying the M. tuberculosis H37Rv thyX gene (Rv2754c) as previously described. Before the purification step, 200 μM of flavin-adenine dinucleotide (FAD) cofactor was added to the supernatant after the lysis step to increase the amount of FAD bound to Mtb ThyX protein. The solubilized protein extract was loaded on a Hi-Trap Talon 5 mL column (GE Healthcare) previously equilibrated with equilibration buffer containing 30 mM Hepes and 300 mM NaCl at pH 8.0. The His-tagged ThyX protein was eluted with elution buffer (30 mM Hepes pH 8.0, 300 mM NaCl, 500 mM imidazol). The fractions containing Mtb ThyX enzyme were pooled, buffer-exchanged on Econo-Pac PD-10 columns (Bio-rad) with the equilibration buffer, concentrated to a final concentration of 480 μM and stored at −20 °C for further use. The measured absorbance of FAD bound to Mtb ThyX at 450 nm showed a ratio FAD to ThyX of 1 to 3 for the purified Mtb ThyX chain.
3.7.2. M. tuberculosis ThyX NADPH Oxidase Assay
The NADPH oxidation assay for M. tuberculosis ThyX activity in 96-well plates was used to screen the synthesized compounds at a final concentration of 200 μM. All molecules were solubilized in dimethylsulfoxide (DMSO) and used at a 1% final concentration of DMSO during the test. One hundred microlitres of standard reaction mixture contained HEPES 50 mM pH 8, NaCl 30 mM, FAD 50 μM, β-mercaptoethanol 1.43 mM, dUMP 100 μM, NADPH 750 μM, and 10 μM of purified MtbThyX. Microtiter plates were prepared and transferred to the microplate reader Chameleon II (Hidex). Molecules at 200 μM were incubated with MtbThyX in the standard reaction mixture for 10 min at 25 °C before starting measurements. The reactions were started by automatically injecting NADPH into individual wells and ThyX activity was determined by following a decrease in absorbance at 340 nm for up to 20 min at 25 °C. The experiment was done in duplicates and samples with added DMSO and enzyme-free reactions were used as positive and negative controls, respectively. % of inhibition was calculated using the following equation: (1 − Vi/Vo)*100; Vo and Vi are, respectively, the initial rates of the reaction without or with addition of molecule in the assay.
3.7.3. M. tuberculosis ThyX Tritium Release Assays
Mtb ThyX tritium release assays consist to measure “deprotonation” of [5-3H]-dUMP in vitro. Reaction mixture included 10 mM MgCl2, 500 µM FAD, 10% (v/v) glycerol, 2 mM NADPH, 1 mM CH2H4folate, 10 mM β-mercaptoethanol, bovine serum albumin (200 µg/mL), 100 µM dUMP, [5-3H]dUMP and 2 µM Mtb ThyX in 50 mM HEPES pH 8. Molecules at 200 μM in DMSO were incubated with MtbThyX in the standard reaction mixture for 10 min at 25 °C before starting measurements. DMSO concentration was maintained constant at 1%. Reactions were initiated by addition of NADPH (1 mM) at 37 °C and were stopped after 20 min. The specific activity of tritiated [5-3H]dUMP (diammonium salt) stock was 15–30 Ci mmol−1 (Moravek Biochemicals, CA, USA). 700 μL of activated charcoal (10% (w/v), Norit A in 2% trichloroacetic acid) was added to the reaction mixture to stop the reaction and removal of radioactive nucleotides from the solution. The suspension was centrifuged at 12,000 rpm for 2 min, and 450 μL of the supernatant were collected before addition of 5 mL of scintillation solution. The radioactivity remaining in the supernatant was measured for scintillation counting.
3.7.4. Cytotoxicity Assays
Assays were performed in human peripheral blood mononuclear (PBM) cells via MTS assay using the CellTiter 96® Non-Radioactive Cell Proliferation (Promega) kit. Cytotoxicity was expressed as the concentration of test compounds that inhibited cell growth by 50% (CC50).
3.8. Virtual Docking
The MtbThyX protein structure from PDB code 3GWC26 was used to perform in silico molecular docking with the QuickVina 2 software [
31]. The A to D chains, water oxygen atoms and UFP cofactor were removed from the structure, keeping only chains F to G and their FAD molecule. Atomic partial charges were assigned and polar hydrogen atoms were added with the Pymol [
32] Vina plugin [
33]. A cubic search volume of 35 × 35 × 35 Å centered on each active site of the four chains was used. The Arg87, Gln103, Ser105, Arg107, Tyr108 and Arg199 amino acids were chosen to be flexible during the docking attempts. Ten docking poses were generated and the pose with the best score was used to further analysis.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




























