1. Introduction
The environmental characteristics of high altitude areas are hypoxia, low pressure, low temperature, dry air, and strong ultraviolet radiation, in which hypoxia plays a major role in the changes of physiological functions [
1]. Under a plateau environment, inadequate oxygen makes people breathe less oxygen, reducing the level of oxygen metabolism and energy supply in the body [
2,
3]. Subsequently, the peripheral circulation, the contractile efficiency of myocardial cells, the pump of the bloodstream, the flow rate of blood in various tissues, and the excretion rate of waste in the body could be greatly reduced, which causes metabolic disorders and mountain sickness [
4,
5,
6]. Studies have shown that hypoxia could also affect the activities of drug metabolic enzymes, transporters, renal blood flow and intestinal flora, etc., which might affect the metabolism and efficacy of drugs [
7,
8,
9,
10].
Acipimox (
Figure 1), a derivative of nicotinic acid, has been used for almost 50 years as a lipid-lowering drug. The target of acipimox is hydroxycarboxylic acid receptor 2 (HCAR2), which can inhibit fat mobilization and lipid synthesis by reducing intracellular cAMP concentrations [
11,
12]. Acipimox can rapidly reduce the levels of free fatty acid (FFA), triglyceride, cholesterol, and low-density lipoprotein (LDL), and increase the level of high-density lipoprotein (HDL) in plasma [
13,
14,
15]. Compared with nicotinic acid, acipimox has fewer side effects and is a clinically used first-line lipid-lowering drug, especially for the hyperlipidaemic patients that do not respond to other therapeutic regimens. Under the normoxic environment, acipimox is rapidly and almost completely absorbed from the gastrointestinal tract and hardly bound to plasma proteins [
16]. Drug transporters also do not affect the absorption of acipimox. The drug is not significantly metabolized and elimination occurs by urinary excretion of the unchanged drug [
17].
The previous study of our group found that the transcription level of the HCAR2 gene, the target of acipimox, in a plateau was 4.37 times higher than in a plain environment, but few researchers explored the changes in its pharmacokinetics and pharmacodynamics in the hypoxic environment. Based on the hypothesis, the expression changes of the target may affect drug pharmacokinetics, and this study attempted to explore the pharmacokinetics of acipimox in the hypoxic environment. In this paper, we first developed a novel, sensitive and specific method for the quantification of acipimox in rat plasma and tissues, and then successfully applied it to study the pharmacokinetics of acipimox in normoxic and hypoxic rats. The results provide important and valuable information for discovering and developing novel anti-hypoxia drugs, as well as a better understanding of the safety and efficacy of acipimox.
3. Materials and Methods
3.1. Chemicals and Reagents
Reference standards of acipimox and acetylsalicylic acid (IS) with purity (>99.00%) were purchased from the National Institute for the Control of Pharmaceuticals and Biological Products (Beijing, China). HPLC-grade acetonitrile was obtained from Thermo Fisher Scientific (Waltham, MA, USA). The ammonia of HPLC grade was obtained from Sigma-Aldrich (St. Louis, MO, USA). The water used in the experiment was double distilled. All other chemicals were of the highest commercially available grade.
3.2. Animals
Male Sprague–Dawley rats aged seven to eight weeks, weighing 250 ± 20 g, were purchased from the Charles River (Beijing, China). All rats were housed internally in flawless animal rooms maintained at constant temperature (23 °C ± 2 °C) and humidity (55% ± 10%), and a 12 h light/dark cycle. The rats had unrestricted access to food and water. Animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Health Ministry of China.
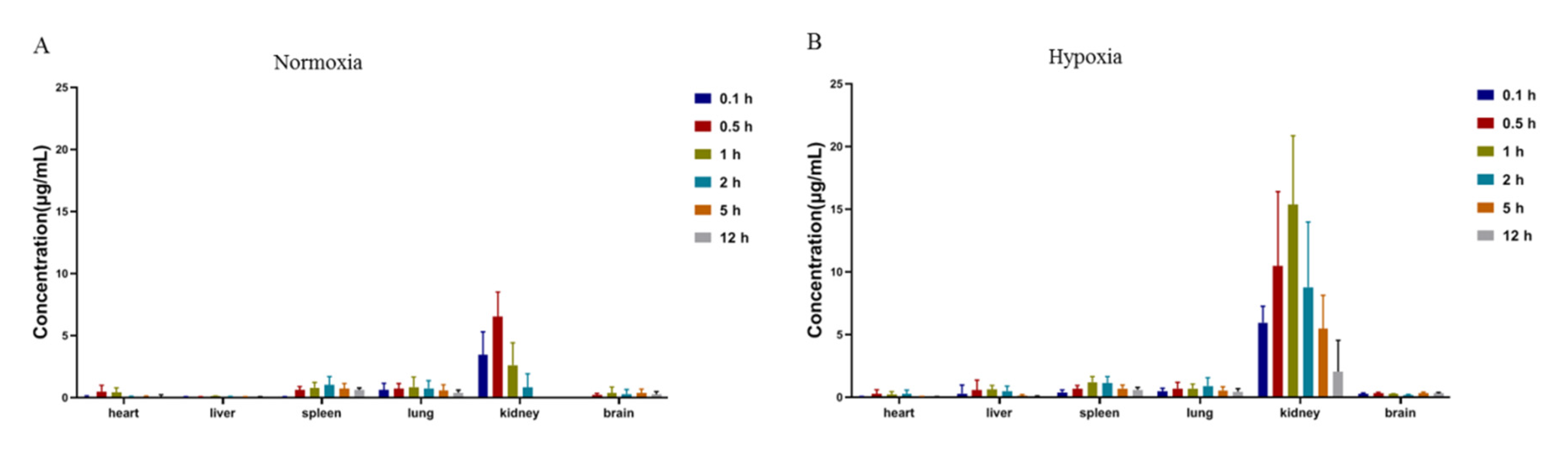
Protocols of animal experiments had been approved by Animal Center of Beijing Institute of Radiation Medicine. Twelve rats were randomly divided into two groups: (1) Normoxia group (n = 6): exposed to the normal air condition (21% O2) and received 80 mg/Kg acipimox; (2) Hypoxia group (n = 6): simulated the hypoxic air condition of 4300 m altitude (13% O2) and received 80 mg/Kg acipimox after seven d. The blood samples were collected in tubes containing sodium heparin at 0, 0.0167, 0.0833, 0.167, 0.333, 0.667, 1, 2, 4, 6, 8, 12 and 24 h after dosing; the tissue samples were collected at 0.1, 0.5, 1, 2, 5 and 12 h after dosing.
3.3. LC-MS/MS Conditions
Quantitative analysis was achieved using a Shiseido liquid chromatograph (NANOSPACE SI-2 3301) consisting of a quaternary pump, temperature controlled column compartment and flow-through needle autosampler. Chromatographic separation was conducted using a Shiseido Capcell PAK C18 MG III column (100 mm × 2.1 mm, 5 μm) at 30 °C through a column oven. The mobile phase solutions were composed of water containing 0.1% (v/v) ammonia (A) and acetonitrile (B). Initial gradient conditions were 85:15 (A:B). From the 2 to 2.5 min the B was increased to 80% and reverted the initial gradient conditions from 3.5 min. The total run time was 5 min. The mobile phase was returned to the initial conditions and reequilibration for a period of time. The flow rate was 0.2 mL/min. The sample injection volume was 2 μL.
The AB-4000 triple quadrupole mass spectrometer (Applied Biosystem, Waltham, MA, USA) was combined with an electrospray ionization (ESI) source. The parameters of the mass spectrometer are as follows: capillary voltage: 4.5 kV, drying gas temperature: 350 °C, drying gas flow: 12 L/min, nebulizer pressure: 45 psi, corona current:10 nA, sheath gas flow:10 L/min and sheath gas temperature: 350 °C. Multireaction monitoring (MRM) analysis using a negative ion ESI mode was used to monitor ion transitions of m/z 153.0 → 109.1 for acipimox, 178.9 → 137.3 for acetylsalicylic acid. The fragmentor (Frag) values set for acipimox and IS were 120 and 100 V, respectively. The collision energy values set for acipimox and IS were −13.5 and −9 eV, respectively. The software Analyst was used to acquire and process data (Analyst 1.6.3, Applied Biosystem, Waltham, MA, USA).
3.4. Stock Solutions, Calibration Standards and Quality Control Samples
The stock standard solutions of acipimox (1 mg/mL) and IS (1 mg/mL) were prepared by acetonitrile and then stored in the dark at −20 °C. The stock solution of acipimox was diluted with acetonitrile to prepare the series of working solutions for the analytes, and the IS stock solution was also diluted to the concentration of 50 μg/mL with acetonitrile. Working solutions for calibration standards were prepared by spiking the appropriate amount of the standard working solution into the blank rat plasma to give the nominal concentration range of 0.1–50 μg/mL for acipimox. The linearity of acipimox in different tissue homogenates (heart, liver: 0.05–5 μg/mL; spleen, lung: 0.1–25 μg/mL; kidney: 0.1–50 μg/mL; brain: 0.05–10 μg/mL) were also assessed using a seven-point calibration curve of the diluted standards. Three levels of QC samples in plasma and different tissue homogenates were prepared in the same way as the calibration standards.
3.5. Samples Preparation
100 μL of plasma sample (or tissue homogenate), spiked with 10 μL of IS solution (5 μg/mL), was spun for 30 s at room temperature. Next, 1 mL of acetonitrile was added into the mixture. After vortex mixing for 30 s, the mixed sample was centrifuged at 12,000 rpm for 15 min at 4 °C. 800 μL of the supernatant was transferred to another centrifuge tube and dried with nitrogen at 30 °C. The residue was redissolved with 100 μL acetonitrile and centrifuged at 12,000 rpm for 15 min at 4 °C after being vortexed for 3 min. The supernatant was then transferred into an autosampler vial, and 2 μL aliquot was injected into the LC-MS/MS system for analysis.
3.6. Method Validation
The method validation procedure was conducted according to the guiding principles of the US FDA and EMA.
3.6.1. Selectivity and Specificity
The selectivity of the method was evaluated with the retention time of analytes and IS by comparing chromatograms of blank plasma and plasma-spiked analytes for excluding endogenous material interference. To evaluate the specificity, the samples of blank plasma were analyzed by comparing with pharmacokinetic samples and the plasma-spiked analytes. The average response of endogenous interference is less than 20% of the analytes, and then the method was considered to be specific.
3.6.2. Linearity and Lower Limit of Quantification
A calibration curve was set up by calculating the peak area ratios (y) of the analyte to IS versus the concentrations of the analytes (x) using a 1/x2 weighted linear least squares regression. The concentration of each analyte was determined by using the equations of linear regression obtained from the calibration curves. LLOQ was determined as the lowest concentration of the calibration curve with the signal/noise ratio being not less than 10. The accuracy and precision of each LLOQ sample should be less than 20%.
3.6.3. Precision and Accuracy
Accuracy and precision were assessed in six replicates of four QC samples (LLOQ, low, middle and high) in a single day (intra-day) and four batches in three days in a row (inter-day), respectively. Acceptable limits for intra- and inter-day precision and accuracy were set at ±15%.
3.6.4. Recovery and Matrix Effect
Recoveries of acipimox were determined by comparing the peak areas of the extracted QC samples with the peak areas of post-extracted blank plasma spiked at the corresponding concentrations. The matrix effects were assessed by the ratio of the peak area of extracted blank plasma spiked post extraction with analytes to those for the clean standard solutions at three concentration levels. The recovery and matrix effect of IS were also investigated at 5 μg/mL.
3.6.5. Stability
A stability analysis of QC samples (low, medium and high) was performed by analyzing four conditions under different conditions: at room temperature for 12 h after preparation, extracted samples were kept at 4 °C for 24 h, samples after three freeze–thaw cycles, and samples stored at −80 °C for 30 days. An acceptable stability was defined as ≤15% loss of the initial drug concentration.
3.7. Pharmacokinetic and Tissue Distribution Comparison
All samples were subsequently treated as in the biological sample preparation procedure (3.5) and assayed by the LC–MS/MS method. The pharmacokinetic analysis was performed using the noncompartmental model analysis. The pharmacokinetic analysis was evaluated through Drug and Statistics version 2.2 (DAS 2.2, Shaanxi Energy Institute, Xianyang, China). The tissue distribution comparison used One-way ANOVA for statistical comparison between different groups with Prism software (GraphPad Software Inc. Version 8, San Diego, CA, USA).
The time to peak plasma concentration (Tmax) and the maximum plasma concentration (Cmax) could be obtained directly by observation. The total area under the plasma concentration-time curve (AUC) was calculated by the linear trapezoidal rule. The half-life (t1/2) is calculated as follows: t1/2 = ln2/ke. The clearance (CL) of the drug was obtained by the following formula: CL = Dose(oral)/AUC.
3.8. Western Blot Analysis
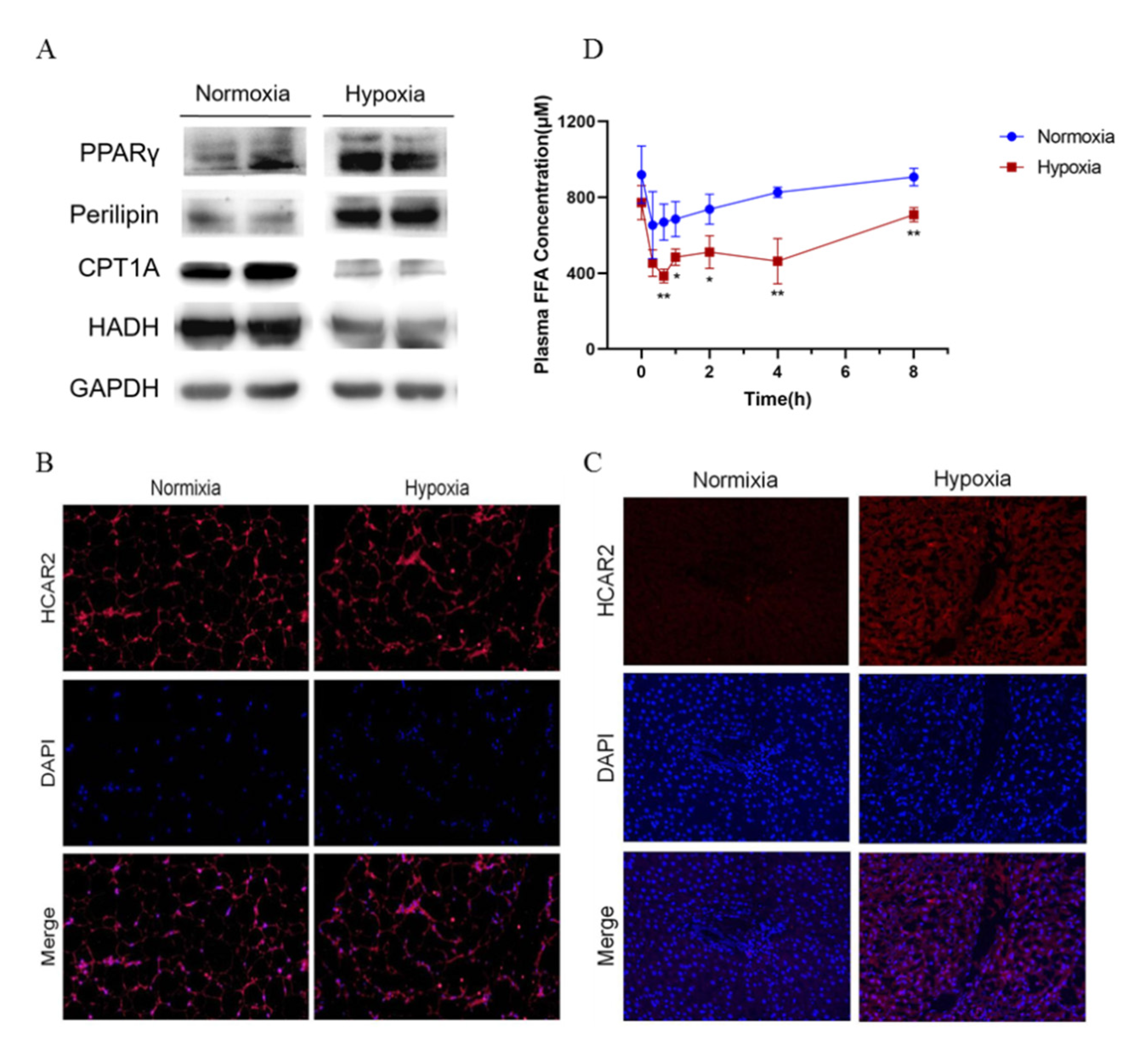
After treatment, rats in each group were used for the preparation of protein extracts. Total proteins were extracted from liver or adipose tissues samples using the radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) supplemented with phenylmethanesulfonyl fluoride (1 mmol/L; Sigma) and a protease and phosphatase inhibitor cocktail (100×; Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein samples were separated using 10% (v/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidenedifluoride (PVDF) membrane. After washing, the membranes were incubated overnight at 4 °C with one of the following primary antibodies: rabbit polyclonal antibodies against CPT1A (1:1000; Abcam, Cambridge, UK), HADH (1:1000; Proteintech, Rosemont, IL, USA), PPARγ, Perilipin, GAPDH (1:1000; Cell Signaling Technology, Danvers, MA, USA). After further washing, the membranes were incubated for 1 h with corresponding horseradish peroxidase conjugated secondary antibodies (anti-rabbit IgG or anti-mouse IgG, 1:10,000; Abcam, UK). The immunoreactive bands were visualized using an enhanced chemiluminescent substrate (MILLIPORE, Burlington, MA, USA) with a GE ImageQuant LAS 500 (GE Healthcare, USA). The intensity of the protein bands was quantitated using a Gel Doc XR system (Bio-Rad, Hercules, CA, USA).
3.9. Confocal Laser Scanning Fluorescence Microscopy
At the end of treatment, a part of the liver and adipose tissue was fixed with 4% paraformaldehyde, embedded in paraffin, and sequentially sectioned. The tissues were permeabilized with 1% (v/v) TritonX-100 in PBS for 10 min at room temperature, and then blocked with goat serum for 1 h. After washing, the tissues were incubated overnight with anti-HCAR2 antibody (1:100) in goat serum and then incubated for 1 h with fluorescein isothiocyanate conjugated secondary antibody (Cy3 Conjugated, 1:100, Abcam, UK). The coverslips were mounted on glass slides with antifade mounting media (Invitrogen, Carlsbad, CA, USA), and the images were collected using an Olympus confocal microscope model FV1000 at 800 × 600 pixel resolution with a 60× objective lens (Carl Zeiss, Oberkochen, Germany). No fluorescence crossover was observed between the channels, and images were collected separately using the appropriate laser excitation wavelength and then merged.
3.10. Serum FFA Analyses
After seven days of hypoxia, blood samples in each group were collected to separate the serum and blood after received 80 mg/Kg acipimox. Fasting serum levels of free fatty acids (FFAs)in each rat were assessed using the corresponding commercial kits (MEIMIAN, Guangzhou, China). After processing the samples according to the instructions, we measured the absorbance at 450 nm using a microplate spectrophotometer (PerkinElmer, Waltham, MA, USA), and calculated the FFA content.
3.11. Cell Culture
The HepG2 and HK2 cells were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained at 37 °C in the presence of 95% O2 and 5% CO2 in basic Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. The culture media was routinely changed after every two days, and the cells were passaged by trypsinization before reaching confluence.
The cells were also divided into two groups for different treatments: (1) Normoxia group (n = 6): exposed to the normal culture condition (95% O2 and 5% CO2)and treated with acipimox; (2) Hypoxia group (n = 6): simulated the hypoxic condition (1% O2 and 5% CO2) and treated with acipimox after 24 h.
3.12. Cell Viability Assay
Cell viability was detected using a Cell Counting kit-8 assay (CCK-8, Dojindo, Japan) according to the manufacturer’s instructions. 6000 cells were seeded in a 96-well plate per well. After growing for 24 h, cells were treated with gradient concentrations of acipimox (0–100 μg/mL) and treated with different oxygen concentrations. After 24 h, cells were washed with PBS buffer gently and incubated with DMEM medium containing 10% CCK-8 solution for 2 h in an incubator at 37 °C. The absorbance of each well was then determined with a microplate reader at 450 nm (Multiskan MK3, Thermo Fisher Scientific, Waltham, MA, USA). The cell viability of the treatment group was expressed as a percentage relative to that of the control group.
3.13. Statistical Analysis
All results were presented as mean ± standard deviation (SD) and generated from at least three independent experiments. Comparisons between the groups of rats were performed using an independent-samples Tukey’s test. p < 0.05 was considered statistically significant.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}