

Post-Functionalization of Organometallic Complexes via Click-Reaction

 ,
, 
Abstract
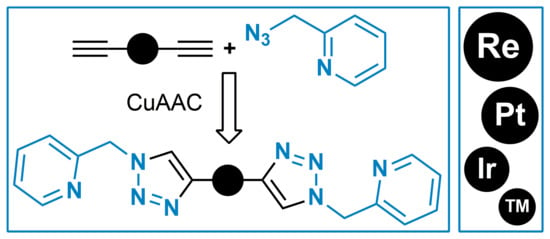
1. Introduction
2. Results
2.1. Synthesis of Functionalized NN Compounds 1P−3P
2.2. Synthesis and Post-Functionalization of Re(I) Complexes
2.3. Synthesis and Post-Functionalization of Ir(III) Complexes
2.4. Synthesis and Post-functionalization of Pt(II) Complexes
2.5. Photophysical Properties of Functionalized Complexes
3. Discussion
4. Materials and Methods
4.1. General Comments
4.2. Synthetic Methods
4.3. X-ray Crystal Structure Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ## | Metal | Substrate | Conditions; yield | Reference |
|---|---|---|---|---|
| 1 | Re(I), Mo(I) |  | Cu(SO4)2 × 5H2O, NaAsc, THF/H2O, r.t., 120 h; up to 88% | [6] |
| 2 | Re(I), Tc(I) |  | Cu(MeCOO)2, PBS, NaAsc, tBuOH/H2O, r.t., 1.5 h; 2–84% | [7] |
| 3 |  | Cu(MeCOO)2, NaAsc, tBuOH/H2O, 70 °C, 1.5 h; 81% | [8] | |
| 4 | Re(V), Tc(V) |  azido-modified angiotensin-II peptide | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/MeOH/H2O, r.t., 5 h; 51% | [9] |
| 5 | Re(I) |  | Cu(SO4)2 × 5H2O, NaAsc, DMF/H2O, r.t., 24 h; 84% | [10] |
| 6 |  | Cu(SO4)2 × 5H2O, NaAsc, THF/H2O, 60 °C, 18 h; 93% | [11] | |
| 7 | Ir(III) |  | Cu(SO4)2 × 5H2O, NaAsc, DMF/H2O, r.t., 12 h; 71–88% | [12] |
| 8 | Au(PR3)N3 | DIPEA, DMF, r.t., 48 h; 20% | [13] | |
| 9 |  | CuBr, MeCN, 70 °C, 12 h | [14] | |
| 10 |  | CuI, iPr2EtN, MeCN/MeOH/HCl, r.t., 12 h | [15] | |
| 11 | Pt(II) |  | Cu(SO4)2 ×5H2O, NaAsc, DMF/H2O, r.t., up to 16 h; 62–81% | [16,17] |
| 12 |  | Cu(SO4)2 × 5H2O, Hasc, DMF/H2O, r.t., 15 h; 62–54% | [18] | |
| 13 |  | Cu(MeCN)4PF6, Cu, DIPEA, CH2Cl2/MeOH, r.t., 72 h; 73–84% | [19] | |
| 14 |  | Cu(SO4)2 × 5H2O, Hasc, CH2Cl2/H2O, reflux, 48 h; 75% | [20] | |
| 15 |  | Cu(MeCN)4PF6, Cu, DIPEA, CH2Cl2/MeOH, r.t., up to 192 h; 17-84% | [21] | |
| 16 |  | Cu(MeCN)4PF6, Cu, Et(iPr)2N, CH2Cl2/MeOH, reflux, 312 h; 15% | [22] | |
| 17 | Pt(IV) | azido-functionalized enterobactin | Cu(MeCN)4PF6, TBTA, DMF/H2O, r.t., 3 h | [24] |
| 18 | Ru(II, III) |  | Cu(SO4)2 × 5H2O, NaAsc, tBuOH/H2O, r.t., 12 h; 63% | [25] |
| 19 | azido-containing poly(benzylether) dendrons | Cu(SO4)2 × 5H2O, NaAsc, THF/H2O, r.t., 12 h; ca. 50% | [26,27] | |
| 20 | Ru(II) |  | Cu(SO4)2 × 5H2O, NaAsc, (iPr)2EtN, tBuOH/MeOH/H2O, r.t., 18 h; 80% | [28] |
| 21 |  | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/H2O, r.t., 20 h; 58–81% | [29] | |
| 22 |  | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/H2O, r.t., 12 h; 67–69% | [30] | |
| 23 |  | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/H2O, r.t., 20 h; 61% | [31] | |
| 24 |  | Cu(SO4)2 × 5H2O, NaAsc, acetone/H2O, r.t., 1h; quantitative | [32] | |
| 25 |  | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/H2O, r.t., 48 h; 55% | [33] | |
| 26 | Fe(II) |  | Cu(SO4)2 × 5H2O, NaAsc, CH2Cl2/NCMe/H2O, r.t., 12 h; 79% | [35] |
| 27 |  | Cu(MeCOO)2, toluene, 70 °C, 2 h; 67% | [36] | |
| 28 | ferrocene |  | Cu(SO4)2 × 5H2O, NaAsc, THF/H2O, r.t, 12 h; up to 84% | [37] |
| 29 | azido-functionalized siloxanes | various | [38] | |
| 30 |  | various | [39] | |
| 31 |  | Cu(SO4)2×5H2O, NaAsc, MeOH/H2O, TTTA, r.t., 20 h; 17-86% | [40] | |
| 32 |  | Cu(SO4)2×5H2O, NaAsc, THF/H2O, 60°C, 6 h; 69% | [41] | |
 | Cu(SO4)2×5H2O, NaAsc, THF/H2O, r.t., 24 h; 75% | |||
| 33 | Mn(I) |  | Cu(SO4)2×5H2O, NaAsc, tBuOH/ H2O, r.t., 24 h; 61% | [42] |
| 34 | Ni(II) |  | CuI, Et3N, DMSO, 70°C, 5 h; 31-92% | [43] |
| 35 | Zn-porphyrin |  | CuI, DBU, toluene, 70°C, 4 h; 90% | [44] |
| 36 |  | CuI, DIPEA, CHCl3, reflux, 12 h; 87% | [45] | |
| 37 | Cr(0), W(0) | various | various | [48] |
| 38 | Au(I) | azido-amphiphilic oligopeptide | Cu(SO4)2×5H2O, NaAsc, TBTA, THF/H2O, 24 h, 45°C; up to 87% | [49] |
| 39 | Tb(III) |  | Cu(SO4)2×5H2O, NaAsc, THPTA, H2O/MeCN, r.t., 12 h, 37% | [50] |
| 40 | Ln(III) | various, including azido-functionalized Ln(III) complexes | various | [51,52,53,54,55,56] |
| 41 | {MC3B7H9}, M = Fe, Ru |  | Cu(SO4)2×5H2O, NaAsc, tBuOH/H2O, r.t., up to 19 h; up to 82% | [57] |
| 42 | M-bis(1,2-dicarbollide); M = Fe, Co | azido-cholesterol | CuI, DIPEA, EtOH, reflux, 12 h | [58] |
| 43 | POM |  | CuI, DIPEA, MeCN, r.t., 3 h; up to 97% | [59] |
| 44 | Au(PR3)N3 | DIPEA, DMF, 35°C, 50 h; 60% | [60] |
References
- Neto, J.S.S.; Zeni, G. A decade of advances in the reaction of nitrogen sources and alkynes for the synthesis of triazoles. Coord. Chem. Rev. 2020, 409, 213217. [Google Scholar] [CrossRef]
- Meldal, M.; Tomøe, C.W. Cu-catalyzed azide—Alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Scattergood, P.A.; Sinopoli, A.; Elliott, P.I.P. Photophysics and photochemistry of 1,2,3-triazole-based complexes. Coord. Chem. Rev. 2017, 350, 136–154. [Google Scholar] [CrossRef]
- Elliott, P.I.P. (Ed.) Organometallic complexes with 1,2,3-triazole-derived ligands. In Organometallic Chemistry: Volume 39; The Royal Society of Chemistry: Cambridge, UK, 2014; pp. 1–25. [Google Scholar]
- Álvarez, C.M.; García-Escudero, L.A.; García-Rodríguez, R.; Miguel, D. Schiff plus click: One-pot preparation of triazole-substituted iminopyridines and ring opening of the triazole ring. Dalt. Trans. 2013, 42, 2556–2561. [Google Scholar] [CrossRef]
- Moore, A.L.; Bučar, D.-K.; MacGillivray, L.R.; Benny, P.D. “Click” labeling strategy for M(CO)3 (M = Re, 99mTc) prostate cancer targeted Flutamide agents. Dalt. Trans. 2010, 39, 1926. [Google Scholar] [CrossRef]
- Bottorff, S.C.; Moore, A.L.; Wemple, A.R.; Bučar, D.-K.; MacGillivray, L.R.; Benny, P.D. pH-Controlled Coordination Mode Rearrangements of “Clickable” Huisgen-Based Multidentate Ligands with [M(CO)3]+ (M = Re, 99mTc). Inorg. Chem. 2013, 52, 2939–2950. [Google Scholar] [CrossRef]
- Castillo Gomez, J.D.; Hagenbach, A.; Abram, U. Propargyl-Substituted Thiocarbamoylbenzamidines of Technetium and Rhenium: Steps towards Bioconjugation with Use of Click Chemistry. Eur. J. Inorg. Chem. 2016, 2016, 5427–5434. [Google Scholar] [CrossRef]
- Gillam, T.A.; Caporale, C.; Brooks, R.D.; Bader, C.A.; Sorvina, A.; Werrett, M.V.; Wright, P.J.; Morrison, J.L.; Massi, M.; Brooks, D.A.; et al. Neutral Re(I) Complex Platform for Live Intracellular Imaging. Inorg. Chem. 2021, 60, 10173–10185. [Google Scholar] [CrossRef]
- Koenig, J.D.B.; Dubrawski, Z.S.; Rao, K.R.; Willkomm, J.; Gelfand, B.S.; Risko, C.; Piers, W.E.; Welch, G.C. Lowering Electrocatalytic CO2 Reduction Overpotential Using N-Annulated Perylene Diimide Rhenium Bipyridine Dyads with Variable Tether Length. J. Am. Chem. Soc. 2021, 143, 16849–16864. [Google Scholar] [CrossRef]
- Passays, J.; Rubay, C.; Marcelis, L.; Elias, B. Synthesis and Photophysical Properties of Triazolyl Ir III Nucleosides. Eur. J. Inorg. Chem. 2017, 2017, 623–629. [Google Scholar] [CrossRef]
- Luengo, A.; Marzo, I.; Reback, M.; Daubit, I.M.; Fernández-Moreira, V.; Metzler-Nolte, N.; Gimeno, M.C. Luminescent Bimetallic Ir III/Au I Peptide Bioconjugates as Potential Theranostic Agents. Chem.–A Eur. J. 2020, 26, 12158–12167. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Das, R.; Gupta, P.; Mukhopadhyay, B. Synthesis of a sugar-functionalized iridium complex and its application as a fluorescent lectin sensor. Tetrahedron Lett. 2012, 53, 3915–3918. [Google Scholar] [CrossRef]
- Wang, J.; Xue, J.; Yan, Z.; Zhang, S.; Qiao, J.; Zhang, X. Photoluminescence Lifetime Imaging of Synthesized Proteins in Living Cells Using an Iridium-Alkyne Probe. Angew. Chem. Int. Ed. 2017, 56, 14928–14932. [Google Scholar] [CrossRef]
- Gauthier, S.; Weisbach, N.; Bhuvanesh, N.; Gladysz, J.A. “Click” Chemistry in Metal Coordination Spheres: Copper(I)-Catalyzed 3+2 Cycloadditions of Benzyl Azide and Platinum Polyynyl Complexes trans -(C6F5)(p-tol3P)2Pt(C≡C)nH (n = 2−6). Organometallics 2009, 28, 5597–5599. [Google Scholar] [CrossRef]
- Amini, H.; Weisbach, N.; Gauthier, S.; Kuhn, H.; Bhuvanesh, N.; Hampel, F.; Reibenspies, J.H.; Gladysz, J.A. Trapping of Terminal Platinapolyynes by Copper(I) Catalyzed Click Cycloadditions; Probes of Labile Intermediates in Syntheses of Complexes with Extended sp Carbon Chains, and Crystallographic Studies. Chem.–A Eur. J. 2021, 27, 12619–12634. [Google Scholar] [CrossRef]
- Clough, M.C.; Zeits, P.D.; Bhuvanesh, N.; Gladysz, J.A. Toward Permetalated Alkyne/Azide 3+2 or “Click” Cycloadducts. Organometallics 2012, 31, 5231–5234. [Google Scholar] [CrossRef]
- Stengel, I.; Strassert, C.A.; Plummer, E.A.; Chien, C.-H.; De Cola, L.; Bäuerle, P. Postfunctionalization of Luminescent Bipyridine PtII Bisacetylides by Click Chemistry. Eur. J. Inorg. Chem. 2012, 2012, 1795–1809. [Google Scholar] [CrossRef]
- Li, Y.; Tsang, D.P.; Chan, C.K.; Wong, K.M.; Chan, M.; Yam, V.W. Synthesis of Unsymmetric Bipyridine–Pt II –Alkynyl Complexes through Post-Click Reaction with Emission Enhancement Characteristics and Their Applications as Phosphorescent Organic Light-Emitting Diodes. Chem.–A Eur. J. 2014, 20, 13710–13715. [Google Scholar] [CrossRef]
- Stengel, I.; Strassert, C.A.; De Cola, L.; Bäuerle, P. Tracking Intramolecular Interactions in Flexibly Linked Binuclear Platinum(II) Complexes. Organometallics 2014, 33, 1345–1355. [Google Scholar] [CrossRef]
- Stengel, I.; Götz, G.; Weil, M.; Bäuerle, P. A Dinuclear (bpy)PtII-Decorated Crownophane. Eur. J. Org. Chem. 2015, 2015, 3887–3893. [Google Scholar] [CrossRef]
- Sutton, E.C.; McDevitt, C.E.; Yglesias, M.V.; Cunningham, R.M.; DeRose, V.J. Tracking the cellular targets of platinum anticancer drugs: Current tools and emergent methods. Inorg. Chim. Acta 2019, 498, 118984. [Google Scholar] [CrossRef]
- Guo, C.; Nolan, E.M. Heavy-Metal Trojan Horse: Enterobactin-Directed Delivery of Platinum(IV) Prodrugs to Escherichia coli. J. Am. Chem. Soc. 2022, 144, 12756–12768. [Google Scholar] [CrossRef]
- Xu, G.-L.; Ren, T. Covalent Modification of Diruthenium Alkynyl Compounds: Novel Application of Click Reactions in Organometallic Chemistry. Organometallics 2005, 24, 2564–2566. [Google Scholar] [CrossRef]
- Chen, W.-Z.; Fanwick, P.E.; Ren, T. Dendronized Diruthenium Compounds via the Copper(I)-Catalyzed Click Reaction. Inorg. Chem. 2007, 46, 3429–3431. [Google Scholar] [CrossRef]
- Forrest, W.P.; Cao, Z.; Chen, W.-Z.; Hassell, K.M.; Kharlamova, A.; Jakstonyte, G.; Ren, T. Decorating Diruthenium Compounds with Fréchet Dendrons via the Click Reaction. Inorg. Chem. 2011, 50, 9345–9353. [Google Scholar] [CrossRef]
- Chitre, K.P.; Guillén, E.; Yoon, A.S.; Galoppini, E. Synthesis of Homoleptic Ruthenium “Star” Complexes by Click Reaction for TiO2 Sensitization. Eur. J. Inorg. Chem. 2012, 2012, 5461–5464. [Google Scholar] [CrossRef]
- Baron, A.; Herrero, C.; Quaranta, A.; Charlot, M.-F.; Leibl, W.; Vauzeilles, B.; Aukauloo, A. Click Chemistry on a Ruthenium Polypyridine Complex. An Efficient and Versatile Synthetic Route for the Synthesis of Photoactive Modular Assemblies. Inorg. Chem. 2012, 51, 5985–5987. [Google Scholar] [CrossRef]
- Wintergerst, P.; Witas, K.; Nauroozi, D.; Schmid, M.; Dikmen, E.; Tschierlei, S.; Rau, S. Minimizing Side Product Formation in Alkyne Functionalization of Ruthenium Complexes by Introduction of Protecting Groups. Z. Anorg. Allg. Chem. 2020, 646, 842–848. [Google Scholar] [CrossRef]
- Gotico, P.; Tran, T.; Baron, A.; Vauzeilles, B.; Lefumeux, C.; Ha-Thi, M.; Pino, T.; Halime, Z.; Quaranta, A.; Leibl, W.; et al. Tracking Charge Accumulation in a Functional Triazole-Linked Ruthenium-Rhenium Dyad Towards Photocatalytic Carbon Dioxide Reduction. ChemPhotoChem 2021, 5, 654–664. [Google Scholar] [CrossRef]
- Busemann, A.; Araman, C.; Flaspohler, I.; Pratesi, A.; Zhou, X.-Q.; van Rixel, V.H.S.; Siegler, M.A.; Messori, L.; van Kasteren, S.I.; Bonnet, S. Alkyne Functionalization of a Photoactivated Ruthenium Polypyridyl Complex for Click-Enabled Serum Albumin Interaction Studies. Inorg. Chem. 2020, 59, 7710–7720. [Google Scholar] [CrossRef] [PubMed]
- Herrero, C.; Batchelor, L.; Baron, A.; El Ghachtouli, S.; Sheth, S.; Guillot, R.; Vauzeilles, B.; Sircoglou, M.; Mallah, T.; Leibl, W.; et al. Click Chemistry as a Convenient Tool for the Incorporation of a Ruthenium Chromophore and a Nickel–Salen Monomer into a Visible-Light-Active Assembly. Eur. J. Inorg. Chem. 2013, 2013, 494–499. [Google Scholar] [CrossRef]
- Mede, T.; Jäger, M.; Schubert, U.S. “Chemistry-on-the-complex”: Functional Ru II polypyridyl-type sensitizers as divergent building blocks. Chem. Soc. Rev. 2018, 47, 7577–7627. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.S.; Prikhod’ko, A.I.; Novikov, V.V.; Vologzhanina, A.V.; Bubnov, Y.N.; Voloshin, Y.Z. First “Click” Synthesis of the Ribbed-Functionalized Metal Clathrochelates: Cycloaddition of Benzyl Azide to Propargylamine Iron(II) Macrobicycle and the Unexpected Transformations of the Resulting Cage Complex. Eur. J. Inorg. Chem. 2012, 2012, 4507–4514. [Google Scholar] [CrossRef]
- Zelinskii, G.E.; Belov, A.S.; Vologzhanina, A.V.; Limarev, I.P.; Pavlov, A.A.; Olshevskaya, V.A.; Makarenkov, A.V.; Dorovatovskii, P.V.; Lebed, E.G.; Voloshin, Y.Z. Iron(II) Clathrochelate with Terminal Triple C≡C Bond and Its Carboranoclathrochelate Derivative with a Flexible Linker between the Polyhedral Cages: Synthesis and X-ray Structure. ChemistrySelect 2019, 4, 11572–11577. [Google Scholar] [CrossRef]
- Rapakousiou, A.; Mouche, C.; Duttine, M.; Ruiz, J.; Astruc, D. Click Synthesis and Redox Chemistry of Mono- and Heterobimetallic Triazolyl and Triazolium-Ferrocene and Cobalticinium Complexes. Eur. J. Inorg. Chem. 2012, 2012, 5071–5077. [Google Scholar] [CrossRef]
- Yu, G.; Suzaki, Y.; Abe, T.; Osakada, K. Introduction of ferrocene-containing [2]rotaxanes onto siloxane, silsesquioxane and polysiloxanes via click chemistry. Dalt. Trans. 2013, 42, 1476–1482. [Google Scholar] [CrossRef]
- Sun, R.; Wang, H.; Hu, J.; Zhao, J.; Zhang, H. Pyridine-phosphinimine ligand-accelerated Cu(i)-catalyzed azide–alkyne cycloaddition for preparation of 1-(pyridin-2-yl)-1,2,3-triazole derivatives. Org. Biomol. Chem. 2014, 12, 5954. [Google Scholar] [CrossRef]
- Wieczorek, A.; Błauż, A.; Makal, A.; Rychlik, B.; Plażuk, D. Synthesis and evaluation of biological properties of ferrocenyl–podophyllotoxin conjugates. Dalt. Trans. 2017, 46, 10847–10858. [Google Scholar] [CrossRef]
- Biegański, P.; Kovalski, E.; Israel, N.; Dmitrieva, E.; Trzybiński, D.; Woźniak, K.; Vrček, V.; Godel, M.; Riganti, C.; Kopecka, J.; et al. Electronic Coupling in 1,2,3-Triazole Bridged Ferrocenes and Its Impact on Reactive Oxygen Species Generation and Deleterious Activity in Cancer Cells. Inorg. Chem. 2022, 61, 9650–9666. [Google Scholar] [CrossRef]
- Aucott, B.J.; Ward, J.S.; Andrew, S.G.; Milani, J.; Whitwood, A.C.; Lynam, J.M.; Parkin, A.; Fairlamb, I.J.S. Redox-Tagged Carbon Monoxide-Releasing Molecules (CORMs): Ferrocene-Containing [Mn(C^N)(CO)4] Complexes as a Promising New CORM Class. Inorg. Chem. 2017, 56, 5431–5440. [Google Scholar] [CrossRef] [PubMed]
- Larionov, V.A.; Adonts, H.V.; Gugkaeva, Z.T.; Smol’yakov, A.F.; Saghyan, A.S.; Miftakhov, M.S.; Kuznetsova, S.A.; Maleev, V.I.; Belokon, Y.N. The Elaboration of a General Approach to the Asymmetric Synthesis of 1,4-Substituted 1,2,3-Triazole Containing Amino Acids via Ni(II) Complexes. ChemistrySelect 2018, 3, 3107–3110. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.; Flood, A.H.; Liu, C.; Liu, H.; Li, Y. Nanometer-Sized Reactor-A Porphyrin-Based Model System for Anion Species. Chem.—A Eur. J. 2011, 17, 7499–7505. [Google Scholar] [CrossRef]
- Husain, A.; Ganesan, A.; Sebastian, M.; Makhseed, S. Large ultrafast nonlinear optical response and excellent optical limiting behaviour in pyrene-conjugated zinc(II) phthalocyanines at a near-infrared wavelength. Dye. Pigment. 2021, 184, 108787. [Google Scholar] [CrossRef]
- Ladomenou, K.; Nikolaou, V.; Charalambidis, G.; Coutsolelos, A.G. “Click”-reaction: An alternative tool for new architectures of porphyrin based derivatives. Coord. Chem. Rev. 2016, 306, 1–42. [Google Scholar] [CrossRef]
- Araújo, A.R.L.; Tomé, A.C.; Santos, C.I.M.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Simões, M.M.Q.; Moura, N.M.M.; Abu-Orabi, S.T.; Cavaleiro, J.A.S. Azides and Porphyrinoids: Synthetic Approaches and Applications. Part 1—Azides, Porphyrins and Corroles. Molecules 2020, 25, 1662. [Google Scholar] [CrossRef]
- Baeza, B.; Casarrubios, L.; Sierra, M.A. A Click Approach to Polymetallic Chromium(0) and Tungsten(0) Fischer-Carbene Complexes and Their Use in the Synthesis of Functionalized Polymetallic Metal ðCarbene Complexes. Chem. —A Eur. J. 2013, 19, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Lewe, V.; Preuss, M.; Woźnica, E.A.; Spitzer, D.; Otter, R.; Besenius, P. A clickable NHC–Au(i)-complex for the preparation of stimulus-responsive metallopeptide amphiphiles. Chem. Commun. 2018, 54, 9498–9501. [Google Scholar] [CrossRef]
- O’Malley, W.I.; Abdelkader, E.H.; Aulsebrook, M.L.; Rubbiani, R.; Loh, C.-T.; Grace, M.R.; Spiccia, L.; Gasser, G.; Otting, G.; Tuck, K.L.; et al. Luminescent Alkyne-Bearing Terbium(III) Complexes and Their Application to Bioorthogonal Protein Labeling. Inorg. Chem. 2016, 55, 1674–1682. [Google Scholar] [CrossRef]
- Szíjjártó, C.; Pershagen, E.; Borbas, K.E. Functionalisation of lanthanide complexes via microwave-enhanced Cu(i)-catalysed azide–alkyne cycloaddition. Dalt. Trans. 2012, 41, 7660. [Google Scholar] [CrossRef]
- Goswami, L.N.; Ma, L.; Chakravarty, S.; Cai, Q.; Jalisatgi, S.S.; Hawthorne, M.F. Discrete Nanomolecular Polyhedral Borane Scaffold Supporting Multiple Gadolinium(III) Complexes as a High Performance MRI Contrast Agent. Inorg. Chem. 2013, 52, 1694–1700. [Google Scholar] [CrossRef] [PubMed]
- Tropiano, M.; Kenwright, A.M.; Faulkner, S. Lanthanide Complexes of Azidophenacyl-DO3A as New Synthons for Click Chemistry and the Synthesis of Heterometallic Lanthanide Arrays. Chem. —A Eur. J. 2015, 21, 5697–5699. [Google Scholar] [CrossRef] [PubMed]
- Tropiano, M.; Kilah, N.L.; Morten, M.; Rahman, H.; Davis, J.J.; Beer, P.D.; Faulkner, S. Reversible Luminescence Switching of a Redox-Active Ferrocene–Europium Dyad. J. Am. Chem. Soc. 2011, 133, 11847–11849. [Google Scholar] [CrossRef] [PubMed]
- Tropiano, M.; Record, C.J.; Morris, E.; Rai, H.S.; Allain, C.; Faulkner, S. Synthesis and Spectroscopic Study of d–f Hybrid Lanthanide Complexes Derived from triazolylDO3A. Organometallics 2012, 31, 5673–5676. [Google Scholar] [CrossRef]
- Sørensen, T.J.; Tropiano, M.; Blackburn, O.A.; Tilney, J.A.; Kenwright, A.M.; Faulkner, S. Preparation and study of an f,f,f′,f′′ covalently linked tetranuclear hetero-trimetallic complex—A europium, terbium, dysprosium triad. Chem. Commun. 2013, 49, 783–785. [Google Scholar] [CrossRef]
- Perez-Gavilan, A.; Carroll, P.J.; Sneddon, L.G. Efficient and Systematic Click-Based Synthetic Routes to Amino Acid Functionalized Metallatricarbadecaboranes. Organometallics 2012, 31, 2741–2748. [Google Scholar] [CrossRef]
- Druzina, A.A.; Kosenko, I.D.; Zhidkova, O.B.; Ananyev, I.V.; Timofeev, S.V.; Bregadze, V.I. Novel Cobalt Bis(dicarbollide) Based on Terminal Alkynes and Their Click-Reactions. Eur. J. Inorg. Chem. 2020, 2020, 2658–2665. [Google Scholar] [CrossRef]
- Linnenberg, O.; Kondinski, A.; Stöcker, C.; Monakhov, K.Y. The Cu(i)-catalysed Huisgen 1,3-dipolar cycloaddition route to (bio-)organic functionalisation of polyoxovanadates. Dalt. Trans. 2017, 46, 15636–15640. [Google Scholar] [CrossRef]
- Petrovskii, S.K.; Khistiaeva, V.V.; Sizova, A.A.; Sizov, V.V.; Paderina, A.V.; Koshevoy, I.O.; Monakhov, K.Y.; Grachova, E.V. Hexavanadate-Organogold(I) Hybrid Compounds: Synthesis by the Azide-Alkyne Cycloaddition and Density Functional Theory Study of an Intriguing Electron Density Distribution. Inorg. Chem. 2020, 59, 16122–16126. [Google Scholar] [CrossRef]
- Zabarska, N.; Stumper, A.; Rau, S. CuAAC click reactions for the design of multifunctional luminescent ruthenium complexes. Dalt. Trans. 2016, 45, 2338–2351. [Google Scholar] [CrossRef]
- Wirth, R.; White, J.D.; Moghaddam, A.D.; Ginzburg, A.L.; Zakharov, L.N.; Haley, M.M.; DeRose, V.J. Azide vs Alkyne Functionalization in Pt(II) Complexes for Post-treatment Click Modification: Solid-State Structure, Fluorescent Labeling, and Cellular Fate. J. Am. Chem. Soc. 2015, 137, 15169–15175. [Google Scholar] [CrossRef] [PubMed]
- Salmain, M.; Fischer-Durand, N.; Rudolf, B. Bioorthogonal Conjugation of Transition Organometallic Complexes to Peptides and Proteins: Strategies and Applications. Eur. J. Inorg. Chem. 2020, 2020, 21–35. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Kimyonok, A.; Weck, M. Functionalization of polymers with phosphorescent iridium complexes via click chemistry. Chem. Commun. 2006, 37, 3933. [Google Scholar] [CrossRef] [PubMed]
- Baranovskii, E.M.; Khistiaeva, V.V.; Deriabin, K.V.; Petrovskii, S.K.; Koshevoy, I.O.; Kolesnikov, I.E.; Grachova, E.V.; Islamova, R.M. Re(I) Complexes as Backbone Substituents and Cross-Linking Agents for Hybrid Luminescent Polysiloxanes and Silicone Rubbers. Molecules 2021, 26, 6866. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Zhao, Y. Click chemistry as a versatile reaction for construction and modification of metal-organic frameworks. Coord. Chem. Rev. 2019, 380, 484–518. [Google Scholar] [CrossRef]
- González Cabrera, D.; Koivisto, B.D.; Leigh, D.A. A metal-complex-tolerant CuAAC ‘click’ protocol exemplified through the preparation of homo- and mixed-metal-coordinated [2]rotaxanes. Chem. Commun. 2007, 41, 4218. [Google Scholar] [CrossRef]
- Grosshenny, V.; Romero, F.M.; Ziessel, R. Construction of Preorganized Polytopic Ligands via Palladium-Promoted Cross-Coupling Reactions. J. Org. Chem. 1997, 62, 1491–1500. [Google Scholar] [CrossRef]
- Ponce, J.; Aragó, J.; Vayá, I.; Magenti, J.G.; Tatay, S.; Ortí, E.; Coronado, E. Photophysical Properties of Oligo (phenylene ethynylene) Iridium(III) Complexes Functionalized with Metal-Anchoring Groups. Eur. J. Inorg. Chem. 2016, 2016, 1851–1859. [Google Scholar] [CrossRef]
- Ni, J.; Wu, Y.-H.; Zhang, X.; Li, B.; Zhang, L.-Y.; Chen, Z.-N. Luminescence Vapochromism of a Platinum(II) Complex for Detection of Low Molecular Weight Halohydrocarbon. Inorg. Chem. 2009, 48, 10202–10210. [Google Scholar] [CrossRef]
- Guha, R.; Defayay, D.; Hepp, A.; Müller, J. Targeting Guanine Quadruplexes with Luminescent Platinum(II) Complexes Bearing a Pendant Nucleobase. Chempluschem 2021, 86, 662–673. [Google Scholar] [CrossRef]
- Ma, D.; Qiu, Y.; Duan, L. New Insights into Tunable Volatility of Ionic Materials through Counter-Ion Control. Adv. Funct. Mater. 2016, 26, 3438–3445. [Google Scholar] [CrossRef]
- Suhr, K.J.; Bastatas, L.D.; Shen, Y.; Mitchell, L.A.; Frazier, G.A.; Taylor, D.W.; Slinker, J.D.; Holliday, B.J. Phenyl substitution of cationic bis-cyclometalated iridium(iii) complexes for iTMC-LEECs. Dalt. Trans. 2016, 45, 17807–17823. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tong, X.; Yin, Y.; Yan, H.; Lu, C.; Huang, W.; Zhao, Q. Using highly emissive and environmentally sensitive o-carborane-functionalized metallophosphors to monitor mitochondrial polarity. Chem. Sci. 2017, 8, 5930–5940. [Google Scholar] [CrossRef] [PubMed]
- Kurz, P.; Probst, B.; Spingler, B.; Alberto, R. Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction. Eur. J. Inorg. Chem. 2006, 2006, 2966–2974. [Google Scholar] [CrossRef]
- Chan, S.-C.; Chan, M.C.W.; Wang, Y.; Che, C.-M.; Cheung, K.-K.; Zhu, N. Organic Light-Emitting Materials Based on Bis(arylacetylide)platinum(II) Complexes Bearing Substituted Bipyridine and Phenanthroline Ligands: Photo- and Electroluminescence from 3MLCT Excited States. Chem. —A Eur. J. 2001, 7, 4180–4190. [Google Scholar] [CrossRef]
- Wei, Q.-H.; Yin, G.-Q.; Zhang, L.-Y.; Chen, Z.-N. Luminescent Hepta- and Tetradecanuclear Complexes of 5,5′-Diethynyl-2,2′-bipyridine Capped with Triangular Trinuclear Cu3/Ag3 Cluster Units. Inorg. Chem. 2006, 45, 10371–10377. [Google Scholar] [CrossRef] [PubMed]
- Hissler, M.; Connick, W.B.; Geiger, D.K.; McGarrah, J.E.; Lipa, D.; Lachicotte, R.J.; Eisenberg, R. Platinum Diimine Bis(acetylide) Complexes: Synthesis, Characterization, and Luminescence Properties. Inorg. Chem. 2000, 39, 447–457. [Google Scholar] [CrossRef]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly phosphorescent bis-cyclometalated iridium complexes: Synthesis, photophysical characterization, and use in organic light emitting diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef]
- CrysAlisPro, Rigaku Oxford Diffraction; Version 1.171.39.35a; Agilent Technologies: Santa Clara, CA, USA, 2017.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrovskii, S.; Khistiaeva, V.; Paderina, A.; Abramova, E.; Grachova, E. Post-Functionalization of Organometallic Complexes via Click-Reaction. Molecules 2022, 27, 6494. https://doi.org/10.3390/molecules27196494
Petrovskii S, Khistiaeva V, Paderina A, Abramova E, Grachova E. Post-Functionalization of Organometallic Complexes via Click-Reaction. Molecules. 2022; 27(19):6494. https://doi.org/10.3390/molecules27196494
Chicago/Turabian StylePetrovskii, Stanislav, Viktoria Khistiaeva, Aleksandra Paderina, Evgenia Abramova, and Elena Grachova. 2022. "Post-Functionalization of Organometallic Complexes via Click-Reaction" Molecules 27, no. 19: 6494. https://doi.org/10.3390/molecules27196494
APA StylePetrovskii, S., Khistiaeva, V., Paderina, A., Abramova, E., & Grachova, E. (2022). Post-Functionalization of Organometallic Complexes via Click-Reaction. Molecules, 27(19), 6494. https://doi.org/10.3390/molecules27196494