
Anti-Electrostatic Pi-Hole Bonding: How Covalency Conquers Coulombics


Abstract
:
1. Introduction
2. Methods
3. Results
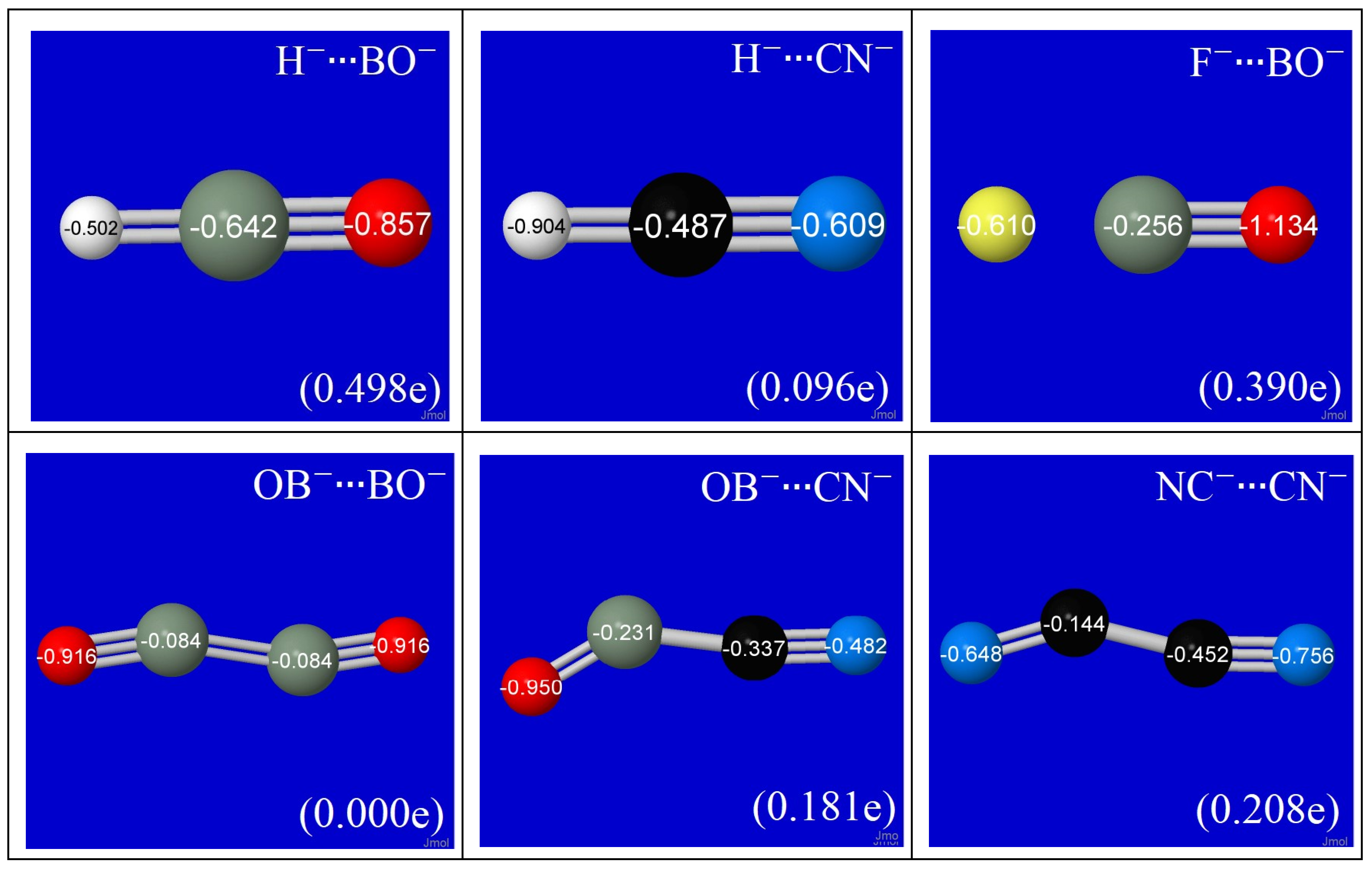
3.1. Structural, Energetic, and NBO Properties of Various Binary Complexes of H−, F−, BO−, CN− Anions
3.2. Unique Associations of Binding Properties with Specific Donor-Acceptor Interactions
4. Discussion
5. Concluding Summary
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bürgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Dunitz, J.D.; Lehn, J.; Wipff, G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Chichester, UK, 2010; pp. 214–215. [Google Scholar]
- DeRider, M.L.; Wilkens, S.J.; Waddell, M.J.; Bretscher, L.E.; Weinhold, F.; Raines, R.T.; Markley, J.L. Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J. Am. Chem. Soc. 2002, 124, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.J.; Choudhary, A.; Raines, R.T.; Woolfson, D.N. n→π* interactions in proteins. Nat. Chem. Biol. 2010, 6, 615–620. [Google Scholar] [CrossRef]
- Choudhary, A.; Newberry, R.W.; Raines, R.T. n→π* interactions engender chirality in carbonyl groups. Org. Lett. 2014, 16, 3421–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newberry, R.W.; Raines, R.T. The n→π* interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [Green Version]
- Newberry, R.W.; Raines, R.T. Secondary forces in protein folding. ACS Chem. Biol. 2019, 14, 16771686. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO analysis and how is it useful? Int. Rev. Phys. Chem. 2016, 35, 399–440. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital theory: Discovering chemistry with NBO7. In Complementary Bonding Analysis; Grabowsky, S., Ed.; De Gruyer: Amsterdam, The Netherlands, 2021; pp. 129–156. [Google Scholar]
- Weinhold, F. Resonance character of hydrogen-bonding interactions in water and other H-bonded species. In Peptide Solvation and HBonds: Advances in Protein Chemistry; Baldwin, R.L., Baker, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 72, pp. 121–155. [Google Scholar]
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Resonance covalency in the supramolecular domain. Chem. Educ. Res. Pract. 2014, 15, 276–285. [Google Scholar] [CrossRef]
- Light, S.H.; Minasov, G.; Duban, M.-E.; Angerson, W.F. Adherence to Bürgi–Dunitz stereochemical principles requires significant structural rearrangements in Schiff-base formation: Insights from transaldolase complexes. Acta Crystallogr. Sec. D Biol. Crystallogr. 2014, 70, 544–552. [Google Scholar] [CrossRef]
- Breton, G.W.; Crasto, C.J. Substituted 2(dimethylamino)biphenyl-2′-carboxaldehydes as substrates for studying n→π* interactions and as a promising framework for tracing the Bürgi–Dunitz trajectory. J. Org. Chem. 2015, 80, 7375–7384. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E. Hydrogen bond seen, halogen bond defined and carbon bond proposed: Intermolecular bonding, a field that is maturing! Curr. Sci. 2013, 105, 892–894. [Google Scholar]
- Michalczyk, M.; Zierkiewicz, W.; Wysokinski, R.; Scheiner, S. Theoretical studies of IR and NMR spectral change induced by sigma-hole hydrogen, halogen, chalcogen, pnicogen, and tetrel bonds in a model protein environment. Molecules 2019, 24, 3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. Sigma-holes, pi-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Solimannejad, M.; Ramezani, V.; Trujillo, C.; Alkorta, I.; SanchezSanz, G.; Elguero, J. Competition and interplay between sigma-hole and pi-hole interactions: A computational study of 1:1 and 1:2 complexes of nitryl halides (O2NX) with ammonia. J. Phys. Chem. A 2012, 116, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.R.; Wang, H.; Jin, W.J. The competition of C–X⋯O=P halogen bond and π-hole⋯O=P bond between halopentafluorobenzenes C6F5X (X = F, Cl, Br, I) and triethylphosphine oxide. J. Mol. Model. 2013, 19, 5007–5014. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Directionality of pi-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef] [Green Version]
- McDowell, S.A.C.; Joseph, J.A. A comparative study of model halogen-bonded, pi-hole-bonded and cationic complexes involving NCX and H2O (X = F, Cl, Br). Mol. Phys. 2015, 113, 16–21. [Google Scholar] [CrossRef]
- Gao, L.; Zeng, Y.; Zhang, X.; Meng, L. Comparative studies on group III σ-hole and π-hole interactions. J. Comput. Chem. 2016, 37, 1321–1327. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-hole bond vs. π-hole bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A.; Mooibroek, T.J. Pi-hole interactions involving nitro compounds: Directionality of nitrate esters. Cryst. Growth Des. 2016, 16, 5520–5524. [Google Scholar] [CrossRef]
- Echeverria, J. Alkyl groups as electron density donors in pi-hole bonding. CrystEngComm 2017, 19, 6289–6296. [Google Scholar] [CrossRef]
- Zhang, J.R.; Li, W.Z.; Cheng, J.B.; Liu, Z.B.; Li, Q.Z. Cooperative effects between pi-hole triel and pi-hole chalcogen bonds. RSC Adv. 2018, 8, 26580–26588. [Google Scholar] [CrossRef] [Green Version]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F. Chemical bonding as a superposition phenomenon. J. Chem. Educ. 1999, 76, 1141–1146. [Google Scholar] [CrossRef]
- Shahi, A.; Arunan, E. Hydrogen bonding, halogen bonding and lithium bonding: An atoms in molecular and natural bond orbital perspective towards conservation of total bond order, inter- and intra-molecular bonding. Phys. Chem. Chem. Phys. 2014, 16, 22935–22952. [Google Scholar] [CrossRef]
- Jiao, Y.; Weinhold, F. What is the nature of supramolecular bonding? Comprehensive NBO/NRT picture of halogen and pnicogen bonding in RPH2⋯IF/FI complexes (R = CH3, OH, CF3, CN, NO2). Molecules 2019, 24, 2090. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F.; Klein, R.A. Anti-electrostatic hydrogen bonds. Angew. Chem. Int. Ed. 2014, 53, 11214–11217. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F. Theoretical prediction of robust 2nd-row oxyanion clusters in the metastable domain of anti-electrostatic hydrogen bonding. Inorg. Chem. 2018, 57, 2035–2044. [Google Scholar] [CrossRef]
- Knorr, A.; Ludwig, R. Cation-cation clusters in ionic liquids: Cooperative hydrogen bonding overcomes like-charge repulsion. Sci. Rep. 2015, 5, 17505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knorr, A.; Stange, P.; Fumino, K.; Weinhold, F.; Ludwig, R. Spectroscopic evidence for clusters of like-charged ions in ionic liquids stabilized by cooperative hydrogen bonding. ChemPhysChem 2015, 17, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Fatila, E.M.; Twum, E.B.; Sengupta, A.; Pink, M.; Karty, J.A.; Raghavachari, K.; Flood, A.H. Anions stabilize each other inside macrocyclic hosts. Angew. Chem. Int. Ed. 2016, 55, 14057–14062. [Google Scholar] [CrossRef] [PubMed]
- Strate, A.; Niemann, T.; Michalik, D.; Ludwig, R. When like charged ions attract in ionic liquids: Controlling the formation of cationic clusters by the interaction strength of the counterions. Angew. Chem. Int. Ed. 2017, 56, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. Like-likes-like: Cooperative hydrogen bonding overcomes Coulomb repulsion in cationic clusters with net charges up to Q = +6e. Chem. Phys. Chem. 2018, 19, 1691–1695. [Google Scholar] [CrossRef] [Green Version]
- Fatila, E.M.; Pink, M.; Twum, E.B.; Karty, J.A.; Flood, A.H. Phosphate–phosphate oligomerization drives higher order co-assemblies with stacks of cyanostar macrocycles. Chem Sci. 2018, 9, 2863–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prohens, R.; Portell, A.; Font-Bardia, M.; Bauza, A.; Frontera, A. H-bonded anion-anion complex trapped in a squaramidobased receptor. Chem. Commun. 2018, 54, 1841–1844. [Google Scholar] [CrossRef]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. When hydrogen bonding overcomes Coulomb repulsion: From kinetic to thermodynamic stability of cationic dimers. Phys. Chem. Chem. Phys. 2019, 21, 8215–8220. [Google Scholar] [CrossRef]
- Cullen, D.A.; Gardner, M.G.; White, N.G. A three dimensional hydrogen bonded organic framework assembled through antielectrostatic hydrogen bonds. Chem. Commun. 2019, 55, 12020–12023. [Google Scholar] [CrossRef] [PubMed]
- Barbas, R.; Prohens, R.; Bauza, A.; Franconetti, A.; Frontera, A. H-Bonded anion–anion complexes in fentanyl citrate polymorphs and solvates. Chem. Commun. 2019, 55, 115–118. [Google Scholar] [CrossRef]
- White, N.G. Anti-electrostatically hydrogen bonded anion dimers: Counter-intuitive, common and consistent. Cryst. Eng. Commun. 2019, 21, 4855–5858. [Google Scholar] [CrossRef]
- Gogoi, A.; Dutta, D.; Verma, A.K.; Nath, H.; Frontera, A.; Guha, A.K.; Bhattacharyya, M.K. Energetically favorable anti-electrostatic hydrogen bonded cationic clusters in Ni(II) 3,5-dimethylpyrazone complexes: Anticancer evaluation and theoretical studies. Polyhedron 2019, 168, 113–126. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Engelage, E.; Weiss, R.; Huber, S.M. “Anti-electrostatic” halogen bonding. Angew. Chem. Int. Ed. 2020, 59, 11150–11157. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Tropp, J.; Qiao, B.; Pink, M.; Axoulay, J.D.; Flood, A.H. Tunable adhesion from stoichiometry-controlled and sequence-defined supramolecular polymers emerges hierarchically from cyanostar-stabilized anion-anion linkages. J. Am. Chem. Soc. 2020, 142, 2579–2591. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Flood, A.H.; White, N.G. Recognition and applications of anion-anion dimers based on anti-electrostatic hydrogen bonds. Chem. Soc. Rev. 2020, 49, 7893–7906. [Google Scholar] [CrossRef] [PubMed]
- Wysokinski, R.; Zierkiewicz, W.; Michalczk, M.; Scheiner, S. Anion-anion attraction in complexes of MCl3− (M = Zn, Cd, Hf) with CN−. ChemPhysChem 2020, 21, 1119–1125. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Wyozikinski, R.; Michalczyk, M.; Scheiner, S. On the stability of interactions between pairs of anions—Complexes of MCl3− (M = Be, Mg, Ca, Sr, Ba) with pyridine and CN−. ChemPhysChem 2020, 21, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S.; Wysokinski, R.; Michalczyk, M.; Zierkiewicz, W. Pnicogen bonds pairing anionic Lewis acid with neutran and anionic bases. J. Phys. Chem. A 2020, 124, 4998–5006. [Google Scholar] [CrossRef]
- Azofra, L.M.; Elguero, J.; Alkorta, I. A conceptual DFT study of phosphonate dimers: Dianions supported by H-bonds. J. Phys. Chem. A 2020, 124, 2207–2214. [Google Scholar] [CrossRef]
- Scheiner, S. Understanding noncovalent bonds and their controlling forces. J. Chem. Phys. 2020, 153, 140901. [Google Scholar] [CrossRef]
- Zapata, F.; Gonzalez, L.; Bastida, A.; Bautista, D.; Caballero, A. Formation of self-assembled supramolecular polymers by anti-electrostatic anon-anion and halogen bonding interactions. Chem. Commun. 2020, 56, 7084–7087. [Google Scholar] [CrossRef] [PubMed]
- Quinonero, D.; Alkorta, I.; Elguero, J. Metastable dianions and dications. ChemPhysChem 2020, 21, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Azofra, L.M.; Elguero, J.; Alkorta, I. Stabilisation of dianion dimers trapped inside cyanostar macrocycles. Phys. Chem. Chem. Phys. 2020, 22, 11348–11353. [Google Scholar] [CrossRef]
- Khorrami, F.; Kowsari, M.H. Tracing local nanostructure of the aqueous solutions of the biocompatible [Cho][Gly] ionic liquid: Importance of hydrogen bond attraction between like-charged ions. J. Phys. Chem. B 2020, 124, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.O.; Duarte, D.J.R.; Alkorta, I. Anion-anion complexes established between aspartate dimers. ChemPhysChem 2020, 21, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qian, K.; Xiao, K.; Luo, J.; Li, H.; Ma, T.; Kortz, U.; Tsige, M.; Liu, T. Co-ion effects in the self-assembly of macroions: From co-ions to co-macroions and to the unique feature of self-recognition. Langmuir 2020, 36, 10519–10527. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.S.C.; Feringa, B.L.; Price, W.S.; Wezenberg, S.J.; Beves, J.E. Controlled Diffusion of Photoswitchable Receptors by Binding Anti-electrostatic Hydrogen-Bonded Phosphate Oligomers. J. Am. Chem. Soc. 2020, 142, 20014–20020. [Google Scholar] [CrossRef] [PubMed]
- Philipp, J.K.; Ludwig, R. Clusters of hydroxyl functionalized cations stabilized by cooperative hydrogen bonds: The role of polarizability and alkyl chain length. Molecules 2020, 25, 4972. [Google Scholar] [CrossRef]
- Wysokinski, R.; Zierkiewidz, W.; Michalczyk, M.; Scheiner, S. Crystallographic and theoretical evidences of anion···anion interaction. ChemPhysChem 2021, 22, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Michalczyk, M.; Zierkiewicz, W.; Scheiner, S. Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment. Molecules 2021, 26, 2116. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.D.P.; Xu, G.; Zhu, X.; Chamoreau, L.-M.; Zhang, Y.; Bistri-Aslanoff, O.; Roland, S.; Alabugin, I.V.; Sollogoub, M. Mapping C-H⋯M interactions in confined spaces: (α-ICyDMe)Au, Ag, Cu complexes reveal “contra-electrostatic H bonds” masquerading as anagostic interactions. Chem. Eur. J. 2021, 27, 8127–8142. [Google Scholar] [CrossRef] [PubMed]
- Wysokinski, R.; Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Anion···anion (MX3−)2 dimers (M = Zn, Cd, Hg; X = Cl, Br, I) in different environments. Phys. Chem. Chem. Phys. 2021, 23, 13853–13861. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Weiss, R.; Rosokha, S.V.; Huber, S.M. “Anti-electrostatic” halogen bonding between ions of like charge. Chem. Eur. J. 2021; in press. [Google Scholar]
- Ludwig, R.; Weinhold, F.; Farrar, T.C. Quantum cluster equilibrium theory of liquids: Molecular clusters and thermodynamics of liquid ethanol. Mol. Phys. 1999, 97, 465–477. [Google Scholar] [CrossRef]
- Yatsuhashi, T.; Nakashima, N. Multiple ionization and Coulomb explosion of molecules, molecular complexes, clusters and solid surfaces. J. Photochem. Photobio. C Photochem. Rev. 2018, 34, 52–84. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Foresman, J.B.; Frisch, Æ. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian, 3rd ed.; Gaussian Inc.: Pittsburgh, PA, USA, 2015. [Google Scholar]
- Glendening, E.D.; Wright, S.J.; Weinhold, F. Efficient optimization of natural resonance theory weightings and bond orders by Gram-based convex programming. J. Comput. Chem. 2019, 40, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Resonance theory reboot. J. Am. Chem. Soc. 2019, 141, 4156–4166. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Weinhold, F.; Phillips, D.; Glendening, E.D.; Foo, Z.Y.; Hanson, R.M. NBOPro7@Jmol; Theoretical Chemistry Institute, University Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Mulliken, R.S. Quelques aspects de la théorie des orbitales moléculaires. J. Chim. Phys. 1949, 46, 497–542. [Google Scholar] [CrossRef]
- Nori-Shargh, D.; Weinhold, F. Natural bond orbital theory of pseudo Jahn-Teller effects. J. Phys. Chem. A 2018, 122, 4490–4498. [Google Scholar] [CrossRef]
- NBO Program Manual. Sec. B.5. Available online: https://nbo7.chem.wisc.edu/nboman.pdf (accessed on 29 December 2021).
- Jagau, T.-C.; Bravaya, K.B.; Krylov, A. Extending quantum chemistry of bound states to electronic resonances. Annu. Rev. Phys. Chem. 2017, 68, 525–553. [Google Scholar] [CrossRef] [Green Version]
- Feynman, R.P. Forces in molecules. Phys. Rev. 1939, 56, 340–343. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Hellmann-Feynman theorem: A perspective. J. Mol. Model. 2018, 24, 266. [Google Scholar] [CrossRef] [PubMed]
- Salem, L.; Wilson, E.B., Jr. Reliability of the Hellmann-Feynman theorem for approximate charge densities. J. Chem. Phys. 1962, 36, 3421–3427. [Google Scholar] [CrossRef]
- Kato, T. On the eigenfunctions of many-particle systems in quantum mechanics. Commun. Pure Appl. Math. 1957, 10, 151–177. [Google Scholar] [CrossRef]
- Weinhold, F.; Klain, R.A. What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions. Mol. Phys. 2012, 110, 565–579. [Google Scholar] [CrossRef]
- Stone, A.J. Natural bond orbitals and the nature of the hydrogen bond. J. Phys. Chem. A 2017, 121, 1531–1534. [Google Scholar] [CrossRef]
- Weinhold, F.; Glendening, E.D. Comment on “Natural bond orbitals and the nature of the hydrogen bond”. J. Phys. Chem. A 2018, 122, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Wong-Ekkabut, J.; Karttunen, M. The good, the bad and the user in soft matter simulations. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 18, 4490–4498. [Google Scholar] [CrossRef]
- Mallada, B.; Gallardo, A.; Lamanec, M.; Torre, B.; Spirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | R12 | R23 | R34 | A123 | A234 |
|---|---|---|---|---|---|
| H−∙∙∙BO− | 1.130 | 1.217 | - | 180.0 | - |
| H−∙∙∙CN− | 1.061 | 1.171 | - | 180.0 | - |
| F−∙∙∙BO− | 1.303 | 1.219 | - | 180.0 | - |
| OB−∙∙∙BO− | 1.240 | 1.631 | 1.240 | 162.9 | 162.9 |
| OB−∙∙∙CN− | 1.257 | 1.515 | 1.172 | 141.9 | 171.3 |
| NC−∙∙∙CN− | 1.206 | 1.427 | 1.193 | 149.5 | 166.7 |
| Species | Property | DFT | MP2 |
|---|---|---|---|
| H−···BO− | ΔEbind | −16.45 | −24.96 |
| [−ΔEdissoc] | [+87.72] | [+81.24] | |
| νi(eq) | 756,825,1771,2974 | 827,835,1771,3020 | |
| ν1‡ | 837i | 869i | |
| H−···CN− | ΔEbind | −26.80 | −37.24 |
| [−ΔEdissoc] | [+95.46] | [+89.94] | |
| νi(eq) | 923(2),2181,3531 | 900(2),2263,3463 | |
| ν1‡ | 1373i | 1517i | |
| F−···BO− | ΔEbind | −11.01 | −15.37 |
| [−ΔEdissoc] | [+88.26] | [+85.94] | |
| νi(eq) | 362(2),891,1970 | 428(2),896,1967 | |
| ν1‡ | 258i | 301i | |
| F−···CN− | ΔEbind | NA | NA |
| OB−···BO− | ΔEbind | −6.50 | −3.55 |
| [−ΔEdissoc] | [+91.64] | [+97.72] | |
| νi(eq) | 211,227,345, 601,1656,1831 | 207,245,438, 572,1591,1829 | |
| ν1‡ | 299i | 321i | |
| OB−···CN− | ΔEbind | −8.78 | −10.40 |
| [−ΔEdissoc] | [+100.97] | [+107.42] | |
| νi(eq) | 195,339,487, 687,1653,2107 | 237,355,556, 680,1563,2775 | |
| ν1‡ | 436i | 549i | |
| NC−···CN− | ΔEbind | −7.44 | −12.71 |
| [−ΔEdissoc] | [+145.13] | [+151.23] | |
| νi(eq) | 148,230,287, 881,1737,2161 | 248,489,528, 896,1909,2597 | |
| ν1‡ | 979i | 1166i |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinhold, F. Anti-Electrostatic Pi-Hole Bonding: How Covalency Conquers Coulombics. Molecules 2022, 27, 377. https://doi.org/10.3390/molecules27020377
Weinhold F. Anti-Electrostatic Pi-Hole Bonding: How Covalency Conquers Coulombics. Molecules. 2022; 27(2):377. https://doi.org/10.3390/molecules27020377
Chicago/Turabian StyleWeinhold, Frank. 2022. "Anti-Electrostatic Pi-Hole Bonding: How Covalency Conquers Coulombics" Molecules 27, no. 2: 377. https://doi.org/10.3390/molecules27020377
APA StyleWeinhold, F. (2022). Anti-Electrostatic Pi-Hole Bonding: How Covalency Conquers Coulombics. Molecules, 27(2), 377. https://doi.org/10.3390/molecules27020377