
Cyclometallated Palladium(II) Complexes: An Approach to the First Dinuclear Bis(iminophosphorane)phosphane-[C,N,S] Metallacycle
 , , ,
, , ,
Abstract
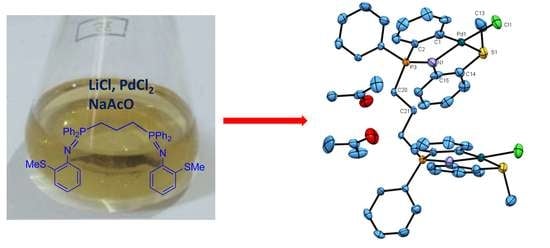
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
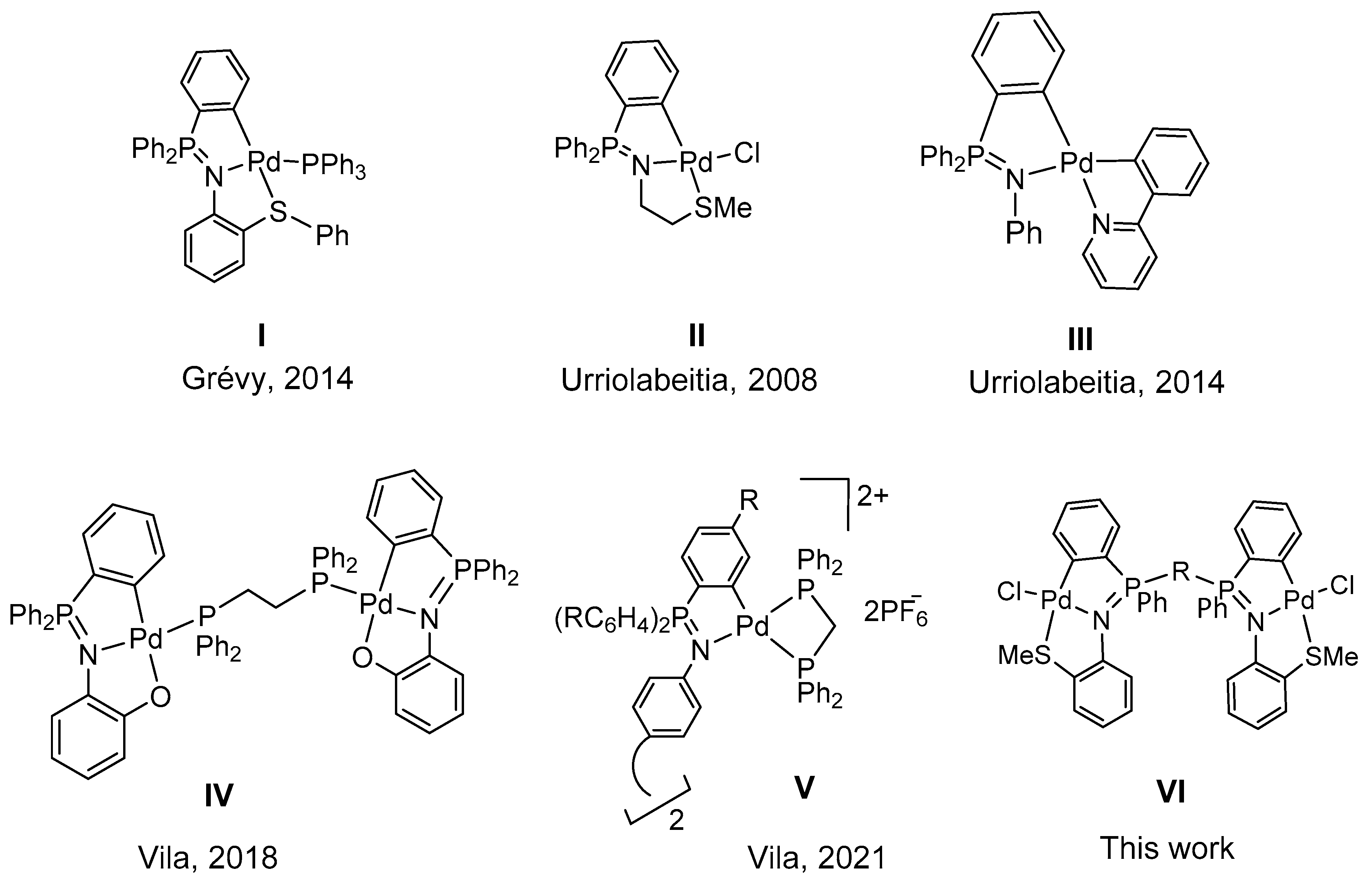
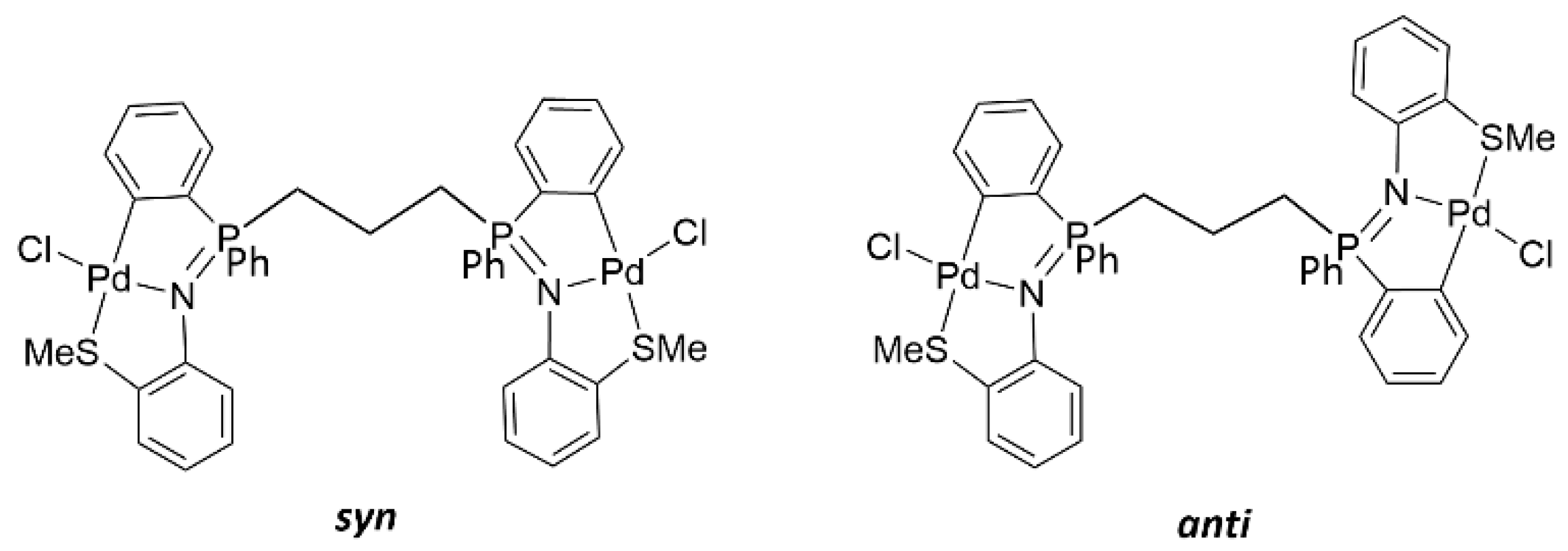
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. General Procedures
4.2. Preparation of the Ligands
4.3. Preparation of the Complexes
4.4. Crystal Structure Analysis and Details on Data Collection and Refinement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dharani, S.; Kalaiarasi, G.; Sindhuja, D.; Lynch, V.M.; Shankar, R.; Karvembu, R.; Prabhakaran, R. Tetranuclear palladacycles of 3-acetyl-7-methoxy-2H-chromen-2-one derived schiff bases: Efficient catalysts for suzuki–miyaura coupling in an aqueous medium. Inorg. Chem. 2019, 58, 8045–8055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, H.; Yuan, J.; Guo, S. Palladacycles incorporating a carboxylate-functionalized phosphine ligand: Syntheses, characterization and their catalytic applications toward suzuki couplings in water. Transit. Met. Chem. 2017, 42, 727–738. [Google Scholar] [CrossRef]
- Maji, A.; Singh, O.; Singh, S.; Mohanty, A.; Maji, P.K.; Ghosh, K. Palladium-based catalysts supported by unsymmetrical xyc–1 type pincer ligands: C5 arylation of imidazoles and synthesis of octinoxate utilizing the mizoroki–heck reaction. Eur. J. Inorg. Chem. 2020, 2020, 1596–1611. [Google Scholar] [CrossRef]
- López-Mosquera, C.; Grabulosa, A.; Rocamora, M.; Font-Bardia, M.; Muller, G. Cyclopalladated compounds with polyhalogenated benzylphosphanes for the mizoroki-heck reaction. Eur. J. Inorg. Chem. 2020, 2020, 2470–2484. [Google Scholar] [CrossRef]
- Carreira, M.; Calvo-Sanjuán, R.; Sanaú, M.; Marzo, I.; Contel, M. Organometallic palladium complexes with a water-soluble iminophosphorane ligand as potential anticancer agents. Organometallics 2012, 31, 5772–5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, T.A.; Abu El-Reash, G.M. Synthesis, and biological evaluation of complexes based on thiosemicarbazone ligand. J. Mol. Struct. 2020, 1201, 127180. [Google Scholar] [CrossRef]
- Pike, S.; Lord, R.; Kergreis, A. Influence of ligand and nuclearity on the cytotoxicity of cyclometallated C^ N^ C platinum (II) complexes. Chem. Eur. J. 2020, 26, 14938–14946. [Google Scholar] [CrossRef]
- Yasuda, J.; Inoue, K.; Mizuno, K.; Arai, S.; Uehara, K.; Kikuchi, A.; Yan, Y.-N.; Yamanishi, K.; Kataoka, Y.; Kato, M.; et al. Photooxidation reactions of cyclometalated palladium(ii) and platinum(II) complexes. Inorg. Chem. 2019, 58, 15720–15725. [Google Scholar] [CrossRef]
- Iliş, M.; Micutz, M.; Cîrcu, V. Luminescent palladium(ii) metallomesogens based on cyclometalated schiff bases and n-benzoyl thiourea derivatives as co-ligands. J. Organomet. Chem. 2017, 836–837, 81–89. [Google Scholar] [CrossRef]
- Fernández-Figueiras, A.; Lucio-Martínez, F.; Munín-Cruz, P.; Ortigueira, J.M.; Polo-Ces, P.; Reigosa, F.; Pereira, M.T.; Vila, J.M. From chemical serendipity to translational chemistry: New findings in the reactivity of palladacycles. Chem. Open 2018, 7, 754–763. [Google Scholar] [CrossRef]
- Lucio-Martínez, F.; Adrio, L.A.; Polo-Ces, P.; Ortigueira, J.M.; Fernández, J.J.; Adams, H.; Pereira, M.T.; Vila, J.M. Palladacycle catalysis: An innovation to the Suzuki–Miyaura cross-coupling reaction. Dalton Trans. 2016, 45, 17598–17601. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Cabaleiro-Lago, E.M.; Ortigueira, J.M.; Pereira, M.T.; Frieiro, P.; Lucio, F.; Vila, J.M. Synthesis and reactivity of thiosemicarbazone palladacycles. Crystal structure analysis and theoretical calculations. Inorg. Chim. Acta 2016, 449, 20–30. [Google Scholar] [CrossRef]
- Lucio-Martínez, F.; Bermúdez, B.; Ortigueira, J.M.; Adams, H.; Fernández, A.; Pereira, M.T.; Vila, J.M. A highly effective strategy for encapsulating potassium cations in small crown ether rings on a dinuclear palladium complex. Chem. Eur. J. 2017, 23, 6255–6258. [Google Scholar] [CrossRef] [PubMed]
- Frieiro-Gomis, P.; Lucio-Martínez, F.; Munín-Cruz, P.; Ortigueira, J.M.; Pereira, M.T.; Polo-Ces, P.; Vázquez-García, D.; Vila, J.M. The chelate-to-bridging shift of phosphane dipalladacycles: Convenient synthesis of double a-frame tetranuclear complexes. Chem. Comm. 2018, 54, 2662–2665. [Google Scholar] [CrossRef]
- Adrio, L.; Antelo, J.M.; Ortigueira, J.M.; Fernández, J.J.; Fernández, A.; Teresa Pereira, M.; Vila, J.M. The chemistry of n-benzylidene-1,4-phenylenediamine palladacycles: The crystal and molecular structure of the first tetranuclear palladacycle with bridging Ph2PCH2PPh2 ligands. J. Organomet. Chem. 2009, 694, 1273–1282. [Google Scholar] [CrossRef]
- Aguilar, D.; Gonzalez, G.; Villuendas, P.; Urriolabeitia, E.P. Bis-cyclometalated complexes of pd(ii) and pd(iv) from iminophosphoranes: Synthesis, structure and reactivity. J. Organomet. Chem. 2014, 767, 27–34. [Google Scholar] [CrossRef]
- León, T.; Parera, M.; Roglans, A.; Riera, A.; Verdaguer, X. P-stereogenic secondary iminophosphorane ligands and their rhodium (i) complexes: Taking advantage of nh/ph tautomerism. Angew Chem. Int. Ed. 2012, 124, 7057–7061. [Google Scholar] [CrossRef]
- Cheisson, T.; Mazaud, L.; Auffrant, A. Ruthenium complexes featuring cooperative phosphine–pyridine–iminophosphorane (pnn) ligands: Synthesis, reactivity and catalytic activity. Dalton Trans. 2018, 47, 14521–14530. [Google Scholar] [CrossRef]
- Martínez-De-León, C.G.; Flores Vallejo, R.d.C.; Rodríguez-Álvarez, A.; Villareal, M.L.; Grévy, J.-M. Synthesis, characterization and cytotoxic activity of cationic half-sandwich Ru(II) complexes stabilized by iminophosphorane N,N,S and N,N,Se tridentate ligands. New J. Chem. 2020, 44, 20676–20687. [Google Scholar] [CrossRef]
- Staudinger, H.; Meyer, J. New organic compounds of phosphorus. III. Phosphinemethylene derivatives and phosphinimines. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef]
- Aguilar, D.; Bielsa, R.; Contel, M.; Lledós, A.; Navarro, R.; Soler, T.; Urriolabeitia, E.P. Regioselective ortho palladation of stabilized iminophosphoranes in exo positions: Scope, limitations, and mechanistic insights. Organometallics 2008, 27, 2929–2936. [Google Scholar] [CrossRef]
- Dyer, H.; Picot, A.; Vendier, L.; Auffrant, A.; Le Floch, P.; Sabo-Etienne, S. Tridentate and tetradentate iminophosphorane-based ruthenium complexes in catalytic transfer hydrogenation of ketones. Organometallics 2011, 30, 1478–1486. [Google Scholar] [CrossRef]
- Picot, A.; Dyer, H.; Buchard, A.; Auffrant, A.; Vendier, L.; Le Floch, P.; Sabo-Etienne, S. Interplay between hydrido/dihydrogen and amine/amido ligands in ruthenium-catalyzed transfer hydrogenation of ketones. Inorg. Chem. 2010, 49, 1310–1312. [Google Scholar] [CrossRef]
- Alper, H. Cyclopalladation of phosphine imides. J. Organomet. Chem. 1977, 127, 385–389. [Google Scholar] [CrossRef]
- Aguilar, D.; Navarro, R.; Soler, T.; Urriolabeitia, E.P. Regioselective functionalization of iminophosphoranes through Pd-mediated C–H bond activation: C–C and C–X bond formation. Dalton Trans. 2010, 39, 10422–10431. [Google Scholar] [CrossRef] [PubMed]
- Tárraga, A.; Molina, P.; Curiel, D.; López, J.L.; Velasco, M.D. Aza-wittig reactions of iminophosphoranes derived from ferrocenylazido ketones: Preparation and electrochemical study of novel ferrocenyl-substituted azaheterocycles. Tetrahedron 1999, 55, 14701–14718. [Google Scholar] [CrossRef]
- Nie, Y.-B.; Wang, L.; Ding, M.-W. Synthesis of 1,2,4,5-tetrasubstituted imidazoles by a sequential aza-Wittig/Michael/isomerization reaction. J. Org. Chem. 2012, 77, 696–700. [Google Scholar] [CrossRef]
- Krawczyk, H.; Dzięgielewski, M.; Deredas, D.; Albrecht, A.; Albrecht, Ł. Chiral iminophosphoranes—An emerging class of superbase organocatalysts. Chem. Eur. J. 2015, 21, 10268–10277. [Google Scholar] [CrossRef]
- Frik, M.; Fernández-Gallardo, J.; Gonzalo, O.; Mangas-Sanjuan, V.; González-Alvarez, M.; Serrano del Valle, A.; Hu, C.; González-Alvarez, I.; Bermejo, M.; Marzo, I. Cyclometalated iminophosphorane gold (III) and platinum (II) complexes. A highly permeable cationic platinum (II) compound with promising anticancer properties. J. Med. Chem. 2015, 58, 5825–5841. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Rave, S.; Ramírez-Apan, M.T.; Tlahuext, H.; Morales-Morales, D.; Toscano, R.A.; Grévy, J.-M. Non-symmetric CNS-Pt(II) pincer complexes including thioether functionalized iminophosphoranes. Evaluation of their in vitro anticancer activity. J. Organomet. Chem. 2016, 814, 16–24. [Google Scholar] [CrossRef]
- Ramirez-Rave, S.; Estudiante-Negrete, F.; Toscano, R.A.; Hernandez-Ortega, S.; Morales-Morales, D.; Grevy, J.-M. Synthesis and characterization of new Pd (II) non-symmetrical pincer complexes derived from thioether functionalized iminophosphoranes. Evaluation of their catalytic activity in the Suzuki–Miyaura couplings. J. Organomet. Chem. 2014, 749, 287–295. [Google Scholar] [CrossRef]
- Fernández-Figueiras, A.; Lucio-Martínez, F.; Munín-Cruz, P.; Polo-Ces, P.; Reigosa, F.; Adams, H.; Pereira, M.T.; Vila, J.M. Palladium iminophosphorane complexes: The pre-cursors to the missing link in triphenylphosphane chalcogenide metallacycles. Dalton Trans. 2018, 47, 15801–15807. [Google Scholar] [CrossRef] [PubMed]
- Munín-Cruz, P.; Reigosa, F.; Rúa-Sueiro, M.; Ortigueira, J.M.; Pereira, M.T.; Vila, J.M. Chemistry of tetradentate [C,N : C,N] iminophosphorane palladacycles: Preparation, reactivity and theoretical calculations. Chem. Open 2020, 9, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, P.; Rodríguez-Rey, J.L.; Sousa, A. Deactivation of the coordinating ability of the iminophosphorane group by effect of ortho-carborane. Dalton Trans. 2017, 46, 257–593. [Google Scholar] [CrossRef]
- Aleksanyan, D.V.; Churusova, S.G.; Klemenkova, Z.S.; Aysin, R.R.; Rybalkina, E.Y.; Nelyubina, Y.V.; Artyushin, O.I.; Peregudov, A.S.; Kozlov, V.A. Extending the application scope of organophosphorus(V) compounds in palladium(II) pincer chemistry. Organometallics 2019, 38, 1062–1080. [Google Scholar] [CrossRef]
- Kilpin, K.J.; Henderson, W.; Nicholson, B.K. Synthesis and reactivity of gold(III) complexes containing cycloaurated iminophosphorane ligands. Inorg. Chim. Acta 2009, 362, 3669–3676. [Google Scholar] [CrossRef] [Green Version]
- Armarego, W.L. Purification of Laboratory Chemicals; Butterworth-Heinemann: Amsterdam, Netherlands, 2017; ISBN 978-185-617-567-8. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallographica Section A: Foundations of Crystallography. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2011, 44, 339–341. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rúa-Sueiro, M.; Munín-Cruz, P.; Fernández, A.; Ortigueira, J.M.; Pereira, M.T.; Vila, J.M. Cyclometallated Palladium(II) Complexes: An Approach to the First Dinuclear Bis(iminophosphorane)phosphane-[C,N,S] Metallacycle. Molecules 2022, 27, 7043. https://doi.org/10.3390/molecules27207043
Rúa-Sueiro M, Munín-Cruz P, Fernández A, Ortigueira JM, Pereira MT, Vila JM. Cyclometallated Palladium(II) Complexes: An Approach to the First Dinuclear Bis(iminophosphorane)phosphane-[C,N,S] Metallacycle. Molecules. 2022; 27(20):7043. https://doi.org/10.3390/molecules27207043
Chicago/Turabian StyleRúa-Sueiro, Marcos, Paula Munín-Cruz, Adolfo Fernández, Juan M. Ortigueira, María Teresa Pereira, and José M. Vila. 2022. "Cyclometallated Palladium(II) Complexes: An Approach to the First Dinuclear Bis(iminophosphorane)phosphane-[C,N,S] Metallacycle" Molecules 27, no. 20: 7043. https://doi.org/10.3390/molecules27207043