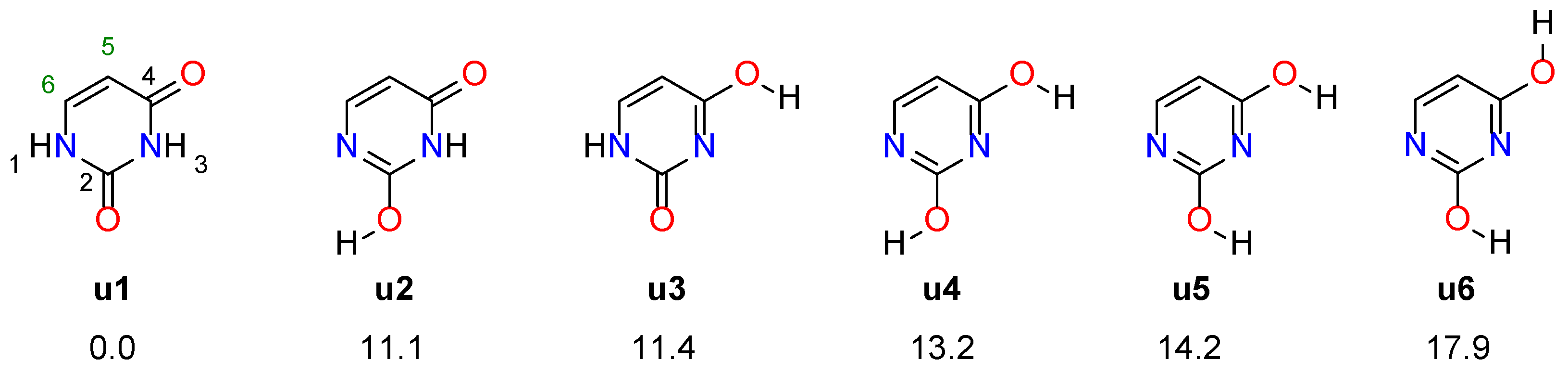
3.1. Electronic Properties of Substituents
The raw data generated in this study and used in statistical analyses are available in the
Supplementary Materials.
Table 2 presents the cSAR values of the substituents in all studied systems. In the case of amino derivatives, the NH
2 substituent in position 6 has more than twice, in the cSAR scale, stronger electron-donating properties than in position 5. In nitro derivatives, the NO
2 group in position 5 is more electron-withdrawing than in position 6. Therefore, the substitution position, i.e., the position in relation to the nitrogen atoms in the ring, has a decisive influence on the properties of the substituent. In contrast, the effect of the tautomeric form of uracil is clearly less significant. It is also worth noting that in polar solvents, the characteristic properties of both NO
2 and NH
2 groups are enhanced, as shown by the difference between cSAR(X) values in the water and gas phase (Δ).
In 5-NH2 derivatives, electron-donating strength of the amino group decreases in the sequence: u2 > u5~u4~u1 > u3 > u6. The clearly lower cSAR(X) for u6 is a consequence of the rotation of the NH2 group by 90 degrees and the formation of the hydrogen bond, H2N∙∙∙HO, which is discussed in more detail later in the paper. In this case, the large influence of the solvent on the cSAR(NH2) value is due to the rotation of the NH2 group to more planar conformation with respect to the ring in polar solvents. This strengthens the resonance effect.
In 6-NH
2 derivatives, electron-donating strength of the amino group decreases in the sequence:
u3 >
u1~
u5 >
u6~
u4 >
u2. Two systems containing the NH endocyclic group at the
ortho position,
u3 and
u1, have the greatest electron-donating properties. An interesting difference is present between the
u5 form and its rotamers:
u4 and
u6. Among them, the highest cSAR(NH
2) value and the lowest Δ occur in
u5, where the two OH groups are facing in the same direction. When they are in opposite directions, as in
u4 and
u6, the value of cSAR(X) is lower, while Δ is higher. This may be due to the differences in the dipole moments in these two cases, as the conformation of the OH groups has a significant impact on the value and direction of molecular dipole moment (
Table S1). By far the strongest solvent effect on cSAR(X) among the 6-NH
2 derivatives occurs in
u1 and
u3 (highest Δ). These systems also have the highest values of the dipole moment (
Table S1). All cSAR(NH
2) values in 5-NH
2 derivatives are lower than in aniline (0.094), while in 6-NH
2 they are higher.
Generally, in all 5-NO2 tautomers, the NO2 group is withdrawing electrons more strongly than in nitrobenzene, where the cSAR(X) is higher, −0.140. Its rotation by 90 degree increases cSAR(NO2) by about 0.4 units. The only exception is u6, where a decrease in cSAR is observed; however, this is caused by the hydrogen bonding between the NO2 and ortho OH groups. In 5-NO2 systems, electron-withdrawing strength of the nitro group decreases in the sequence: u3 > u4~u5 > u2 > u1 > u6. The systems with the strongest electron-withdrawing NO2 groups (u3, u4 and u5) have an electron-donating OH group in the ortho position, but its hydrogen atom is directed to the endocyclic N atom, so that NO∙∙∙OH interaction can be expected. When NO∙∙∙HO interaction is present (5-NO2 u6), the electron-withdrawing ability of the NO2 group is the weakest. Again, the greatest variability of cSAR(X) due to solvation occurs in the derivatives with the highest values of the dipole moments (u1 and u2).
In the 6-NO
2 derivatives, the cSAR(NO
2) values are high, indicating weak electron-withdrawing properties. This is caused by the disturbance of the resonance interactions by ring nitrogen atoms in
ortho and
para positions. Weak resonance is also evidenced by a smaller increase in cSAR due to the rotation of NO
2 by 90° as compared to the 5-NO
2 derivatives. This increase is by about 0.2 units, with the exception of
u1 and
u3 where cSAR(NO
2) is positive and its change due to rotation is smaller. Electron-withdrawing strength decreases in the sequence:
u2 >
u4 >
u6~
u5 >
u1 >
u3. The loss of electron-withdrawing properties (cSAR close to 0.0) of the 6-NO
2 group occurs in the
u1 and
u3 derivatives, where the NH group is in the
ortho position. Thus, apart from the relative position of the endo N atoms and the substituent, the NO∙∙∙HN through-space interaction has an effect as well. The summary of the cSAR analysis in the form of a bar chart is shown in
Figure 3.
In most cases, the dependences of cSAR(X) on 1/ε are well approximated by a linear function. The parameters of resulting cSAR (X) =
a∙(1/ε) +
b functions are summarized in
Table 3. The slope value,
a, informs about the sensitivity of the electronic properties of the substituent in a given derivative to the solvent effect. In general, except for
u6 5-NH
2, large absolute values of the coefficient occur in systems with a large dipole moment, and small ones in systems with a small dipole moment (
Table 3 and
Table S1). In 6-substituted systems (6-NO
2 and 6-NH
2), the values of
a in
u1 and
u3 (
ortho NH) clearly differ from other tautomers (
ortho N). This can be attributed to the influence of
ortho interactions with endocylic N/NH groups. It can be concluded that the repulsive
ortho interaction, NH∙∙∙HN for 6-NH
2 and NO∙∙∙N for 6-NO
2, causes high sensitivity of the substituent properties to the solvent effect, whereas the attractive interaction causes low sensitivity. A similar effect was observed in adenine and purine derivatives [
29,
30].
Properties of the =O/−OH groups of all studied forms of uracil, quantified by cSAR, are shown in
Figure 4. Negative values correspond to the electron-withdrawing =O group, whereas positive values to the electron-donating −OH. Both the interactions with the substituent and the type of tautomer can affect the electron-donating (−OH) or -withdrawing (=O) properties of these groups. The electron-withdrawing properties of the =O groups are greater in the amino derivatives than in the nitro derivatives, which is shown by the more negative cSAR(=O) values in the 5-NH
2 and 6-NH
2 derivatives. In turn, the electron-donating properties of the −OH groups are greater in the nitro than in the amino derivatives. This is due to charge transfer between groups with opposite electronic properties. Global ranges of variation of cSAR are 0.143 for the =O group and 0.107 for the −OH group. The ranges for the =O group in C4 and C2 positions are 0.096 and 0.078, respectively, while the average values are −0.134 for C4 and −0.115 for C2. In the case of the −OH group, the ranges are 0.099 for C4 and 0.103 for C2 positions; the average values are 0.159 for C4 and 0.199 for C2. Thus, the characteristic electronic properties of the −OH group are on average stronger in the C2 position, while those of the =O group are stronger in the C4 position. Stronger electronic properties are accompanied by higher ranges of their variability.
The C2 position of the uracil ring is double
ortho with respect to the two
endo N/NH atoms/groups, while the C4 position is
ortho and
para. So, two electronegative atoms in the
ortho position of the −OH group might enhance its electron-donating properties, while diminishing the electron-withdrawing by the =O group. A similar effect of
ortho N atoms on the substituent properties was observed in our recent studies on nitro and amino derivatives of pyridine, pyrimidine, pyrazine and triazine [
33].
3.2. Geometry
Analysis of geometry will be focused on the lengths of CN bonds connecting the NO
2 and NH
2 substituents and the substituted system. As shown in
Figure 5a, they vary depending on the substitution position and the tautomeric form. In the case of 5-NH
2 derivatives, the shortest CN bond occurs in the
u2 tautomer and the longest in
u6. The
u2 tautomer is also characterized by the highest electron-donating strength of the NH
2 group among 5-NH
2 derivatives (
Table 2 and
Figure 3). In the case of the
u6 tautomer in the gas phase, the NH
2 group is rotated by 90 degrees in order to form a H
2N∙∙∙HO hydrogen bond with the OH group in the
ortho position. This is accompanied by a significant extension of the CN bond, which reaches the length observed for the 5-NO
2 group in u6. In 6-NH
2 derivatives, CN bonds are shorter than in 5-NH
2, which is connected with the strong electron-donating 6-NH
2 group. A slightly longer bond relative to other tautomers occurs in
u1 and
u3. This may be due to the presence of the NH group in the
ortho position resulting in NH∙∙∙HN steric interaction.
In NO2 derivatives, shorter CN bonds are found in 5-NO2 than in 6-NO2 systems. This is in line with the electron-withdrawing strength of the 5-NO2 and 6-NO2 groups. In position 5, the shortest bond occurs in u6, where a strong NO∙∙∙HO hydrogen bond is formed, while the second shortest is in u3, in which the NO2 group has the strongest electron-accepting properties among all systems. For 6-NO2 tautomers, clearly the shortest bonds occur in u1 and u3, where the NH group is in the ortho position. This results from the attractive NO∙∙∙HN interaction.
The rotation of the NO2 group in 5-NO2 derivatives causes the elongation of CN bonds, which is related to the disturbance of the resonance effect of the NO2 group. The largest elongation occurs in the u6 5-NO2 derivative. It is caused by breaking of the NO∙∙∙HO hydrogen bond as a result of NO2 rotation. In the 6-NO2 systems, in four tautomers: u2, u4, u5 and u6 (ortho N), the NO2 rotation clearly shortens the CN bond. This is caused by the weakening of through-space repulsive interactions with the ortho endocyclic N atom. Thus, the main factor determining the CN bond lengths in the 5-NO2 derivatives is the resonance between the NO2 group and the substituted system, while in the 6-NO2 derivatives it is the ortho interaction.
The solvation effect is also reflected in the CN bond lengths.
Figure 5b shows the difference in CN bond lengths between the values obtained in the aqueous solution and the gas phase. In NH
2 derivatives, a stronger solvent effect occurs in 6-NH
2 systems, while in the case of NO
2 derivatives, in 5-NO
2 systems. This is connected with the greater variability of the substituent’s electronic properties in these systems (see, for example,
Table 3). Thus, the bond shortening is related to an increase in the characteristic electronic properties of a given substituent, due to the increase in the solvent polarity.
3.3. Intramolecular Interactions between Non-Covalently Bonded Atoms
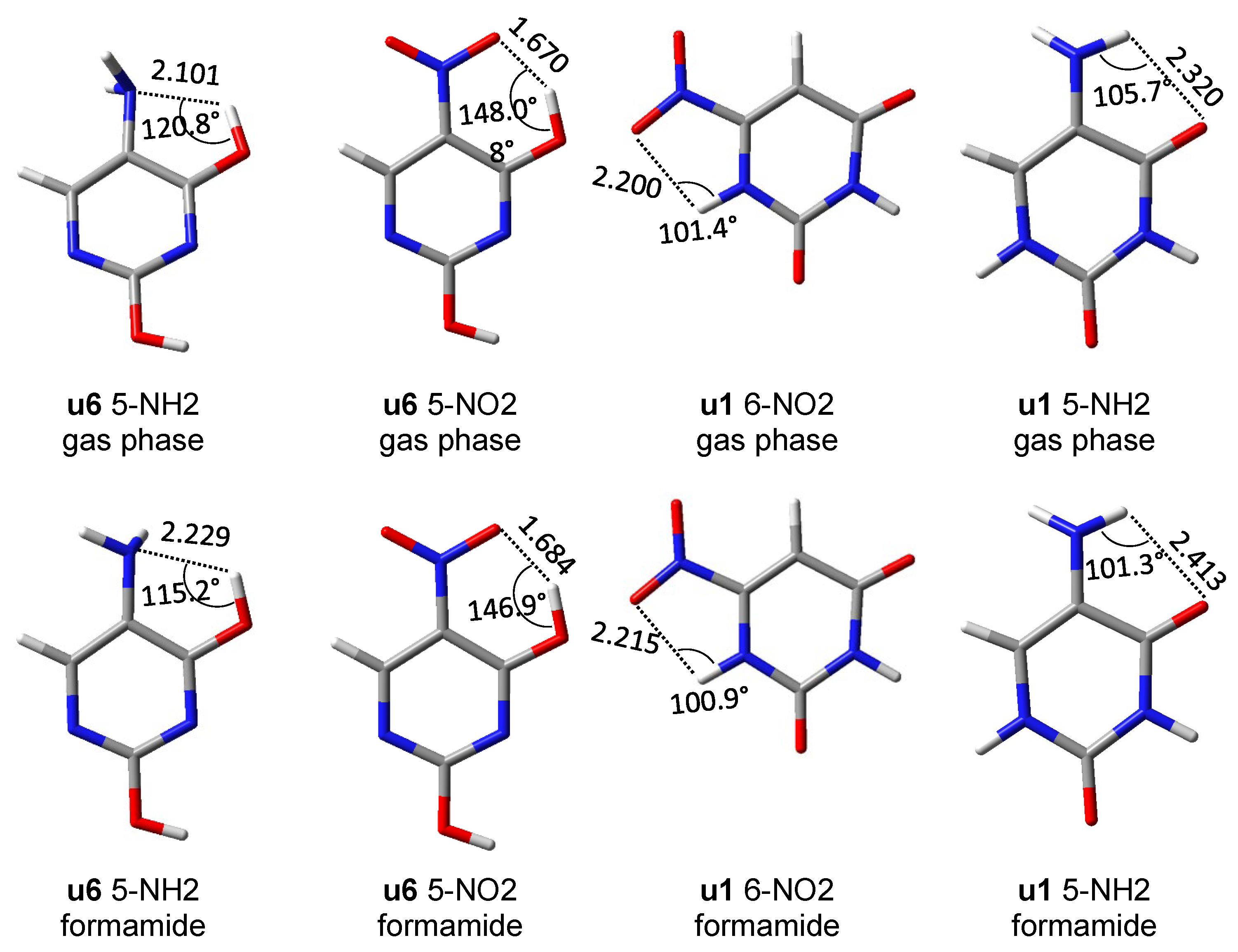
An important aspect of the interaction between the substituent and the substituted system are through-space
ortho interactions, which in some cases could already be seen by the cSAR(X) values and CN bond lengths. In order to identify these interactions, the lengths of two NH/NO bonds of the NH
2/NO
2 groups were plotted against each other (
Figure 6). Deviations from the equal length of these two bonds may indicate the existence of an asymmetric through-space interaction. Such plots also provide information about the attractive/repulsive nature of these interactions, based on the location of a point above or below the y = x line.
First of all, it should be noticed that the asymmetry in the bond lengths of the NO
2 group is about four times greater than that of the NH
2 group. Moreover, for the nitro group, the obtained results indicate greater variability of interactions, but as expected in systems with rotated groups, the lengths of both NO bonds are similar. Both repulsive and attractive interactions as well as hydrogen bonds are observed. In the latter case, the systems in which the interaction meets the Koch–Popelier criteria for hydrogen bonding [
52] are depicted as H-bonds in
Figure 6. Only one system (in the gas phase), visible in the plot,
u6 5-NO
2, fulfills the criteria. Additionally, an increase in the polarity of the solvent weakens the through-space interactions—an increase in the O∙∙∙H distance and a decrease in O∙∙∙HO angle, as shown in
Figure 7. An interesting system in which, despite the symmetry between NH bonds, there is a strong H-bond is
u6 5-NH
2. In this case, the NH
2 group rotates by 90°, and forms a H
2N∙∙∙HO hydrogen bond. Moreover, the NH
2 group in the formamide solution rotates slightly towards the coplanar conformation (76.7° dihedral angle) and the H-bond is weakened. This rotation is an interesting example of competition of attractive through-space interactions and the resonance between the group and the substituted system. In the gas phase, the H-bond has a greater influence on the structure, but in the polar solvent, due to the weakening of the H-bond, stabilization by resonance forces the group to be coplanar. The structures of
u6 5-NO
2 and
u6 5-NH
2 are shown in
Figure 7.
Based on the potential energy density at the critical point of each hydrogen bond, their energy was calculated from the Afonin equation (Equation (1)). For comparison, the hydrogen bond energy was also calculated using the rotational method [
53], i.e., the difference between
u6 and
u5 rotamers. Both methods give similar results (
Table 4), especially in the case of stronger hydrogen bonding in
u6 5-NO
2.
Figure 8 shows the energy scan along the dihedral angle between the amino group and the uracil ring plane. The global minimum corresponds to the conformer shown in
Figure 7, the minimum near scan coordinate 300 corresponds to the form rotated by 180° from the global minimum, so that NH
2∙∙∙HO bifurcated contact is present. Two maxima correspond to forms with close NH∙∙∙HO contacts (1.956 Å). Rotational barrier height is 5.08 kcal/mol, while the difference in energy between the two minima is 4.16 kcal/mol.
The NCI analysis, shown in
Figure 9, was performed to visualize all non-covalent interactions. In most cases, only weak interactions (green-shaded isosurfaces) are present. However, in systems where the asymmetry of two NH/NO bonds (
Figure 6) was high, a blue color can be noticed on the isosurfaces between the interacting atoms. This indicates a stronger attractive character of these interaction. The
u1 5-NH
2 system, which has the highest bond length asymmetry (
Figure 6) among the amino derivatives, has very slight blue features on the isosurface between NH and =O, which indicated stronger attractive interaction than in
u2–
u5 5-NH
2 systems. The intramolecular H-bond in
u6 5-NH
2, discussed earlier, appears as a mostly blue isosurface between H
2N and HO. The H-bond in the
u6 5-NO
2 system is so strong that the NCI analysis treats it as a partially covalent interaction, as the hole is pierced through the isosurface along the H∙∙∙O line. In
u1 and
u3 6-NO
2 systems, some blue accents are noticeable on the isosurface corresponding to the NO∙∙∙HN contact. Bond critical points of non-covalent interactions were found only in
u6 5-NH
2 and
u6 5-NO
2.
Interestingly, in several nitropurines, NO∙∙∙HN interactions have a bond critical point [
30]. It is possible that this interaction is on the edge of being classified as H-bonding. The reasons are probably low values of O∙∙∙HN angles (105.6° in 1H 6-nitropurine vs. 101.4° in
u1 6-NO
2 uracil), which are close to the limit of 110° proposed by Desiraju [
54], and rather high O∙∙∙H distances (2.107 Å in 1H 6-nitropurine vs. 2.200 Å in
u1 6-NO
2 uracil).
3.4. Tautomer Stability
The last section is devoted to the effects of substitution and solvation on the stability of uracil tautomers.
Table 5 presents electronic energies of each system relative to the
u1 tautomer. In all cases, this tautomer remains the most stable, irrespective of substitution and solvation. Considering the 5-NO
2 substitution, the
u6 5-NO
2 derivative is a particularly interesting case. Formation of a strong NO∙∙∙HO H-bond results in a large stabilization relative to the unsubstituted
u6 tautomer (by 12.5 kcal/mol). Consequently, among the 5-NO
2 tautomers,
u6 becomes the second most stable tautomer after
u1, despite the fact that
u6 is the least stable tautomer for unsubstituted uracil. Rotating the 5-NO
2 group by 90 degrees and breaking the hydrogen bond increases the relative energy of
u6 by 10.4 kcal/mol and in 5-NO
2 (90°),
u6 is again the least stable tautomer.
In the case of 5-NH
2 substitution, the energy difference between the
u1 and
u2 tautomers decreases compared to the unsubstituted systems, while between
u1 and others it increases. In 6-NH
2, the relative energies are smaller than for unsubstituted systems. A noteworthy increase in stability relative to
u1 is observed for
u2,
u4,
u5 and
u6 tautomers (between 5 and 6 kcal/mol), while much less for
u3 (1.1 kcal/mol). In the case 6-NO
2 tautomers, apart from
u3, the relative energies decrease slightly, but not as much as in 6-NH
2. In all cases, the relative energies between the
u1 tautomer and the second most stable one are above 5.4 kcal/mol; therefore, it is unlikely that substitution with NH
2 or NO
2 groups can significantly affect the tautomeric equilibrium. Solvation. in most cases, further increases the difference between
u1 and the other forms, as evidenced by the positive values of Δ (apart of two cases) in
Table 5. The only cases where Δ is negative are the two NH
2 derivatives of the
u3 tautomer:
u3 5-NH
2 (Δ = −2.2 kcal/mol) and
u3 6-NH
2 (Δ = −0.6 kcal/mol).
Similarly to the cSAR (X), electronic energy can be plotted against 1/ε and relations approximated with straight lines can be obtained (
Table 6). In this case, the slopes (
a) inform about the sensitivity of the energy of a given system to the solvent effect. In most cases, the
u1 and
u3 tautomers are the most sensitive, these two tautomers have an endo NH group in the 1 position of the uracil ring. The only exception is the 6-NO
2 substitution, where the
u2 and
u6 tautomers are most sensitive to the solvent effect. The
u4 and
u5 tautomers are in all but one case (H-bond forming
u6 5-NO
2) the least sensitive. In amino derivatives, the sensitivity to the solvent effect seems to be correlated with the dipole moments of the molecules, i.e., a large dipole moment is associated with a large value of
a. However, no such relation can be observed in the case of nitro derivatives.
Plotting the relative energy,
Erel, against the cSAR(X) for all systems in all solvents (
Figure 10) reveals linearly correlated groups of points for each tautomer. The linearity comes from the fact that both
Erel and cSAR change linearly with 1/ε (see
Table 3 and
Table 6). The ranges on the y and x axes for particular tautomers are a visual representation of the strength of the solvent effect on
Erel and cSAR, respectively. It is clearly visible that, in general, the greatest changes in both parameters occur for the 5-NO
2 and 6-NH
2 derivatives.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}