

Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities
 ,
,
Abstract
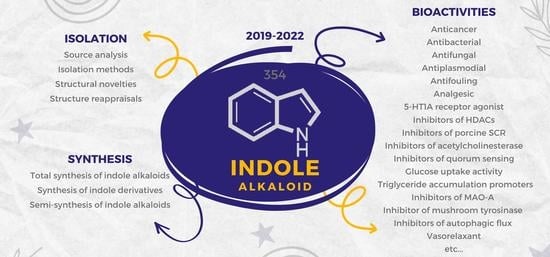
1. Introduction
2. Isolation of Novel Indole Alkaloids
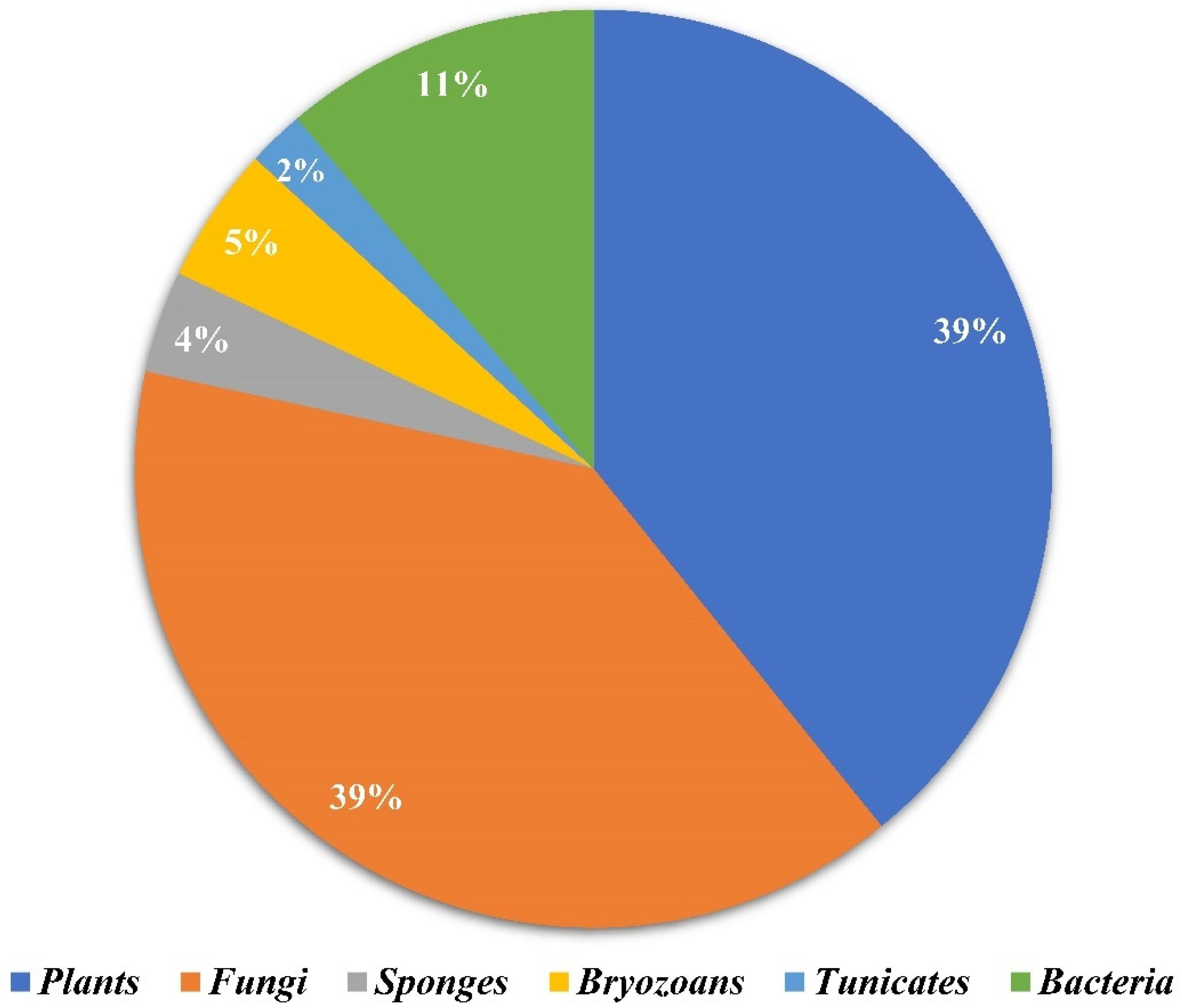
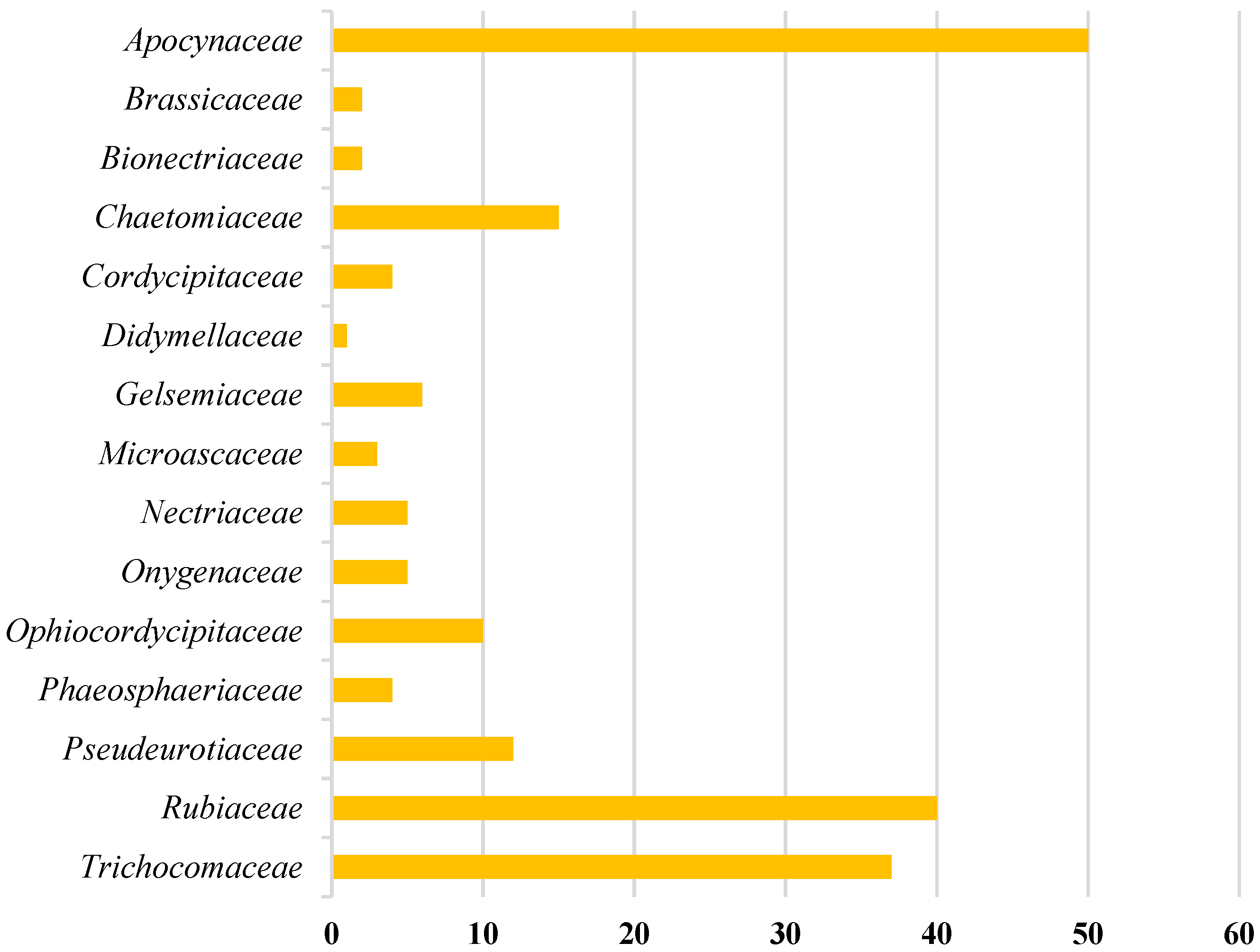
2.1. Source Analysis
2.2. Isolation Methods
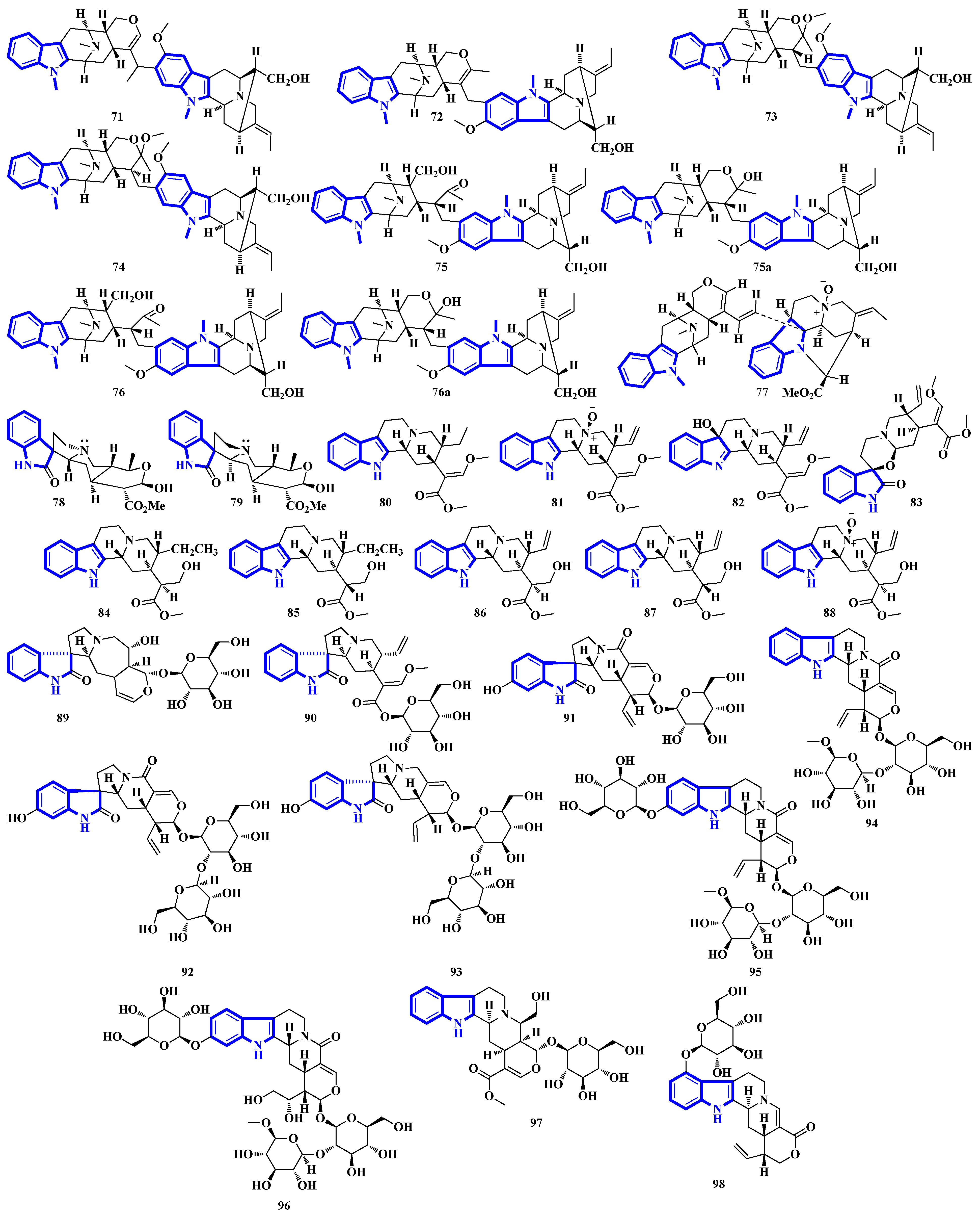
2.3. Structural Novelties
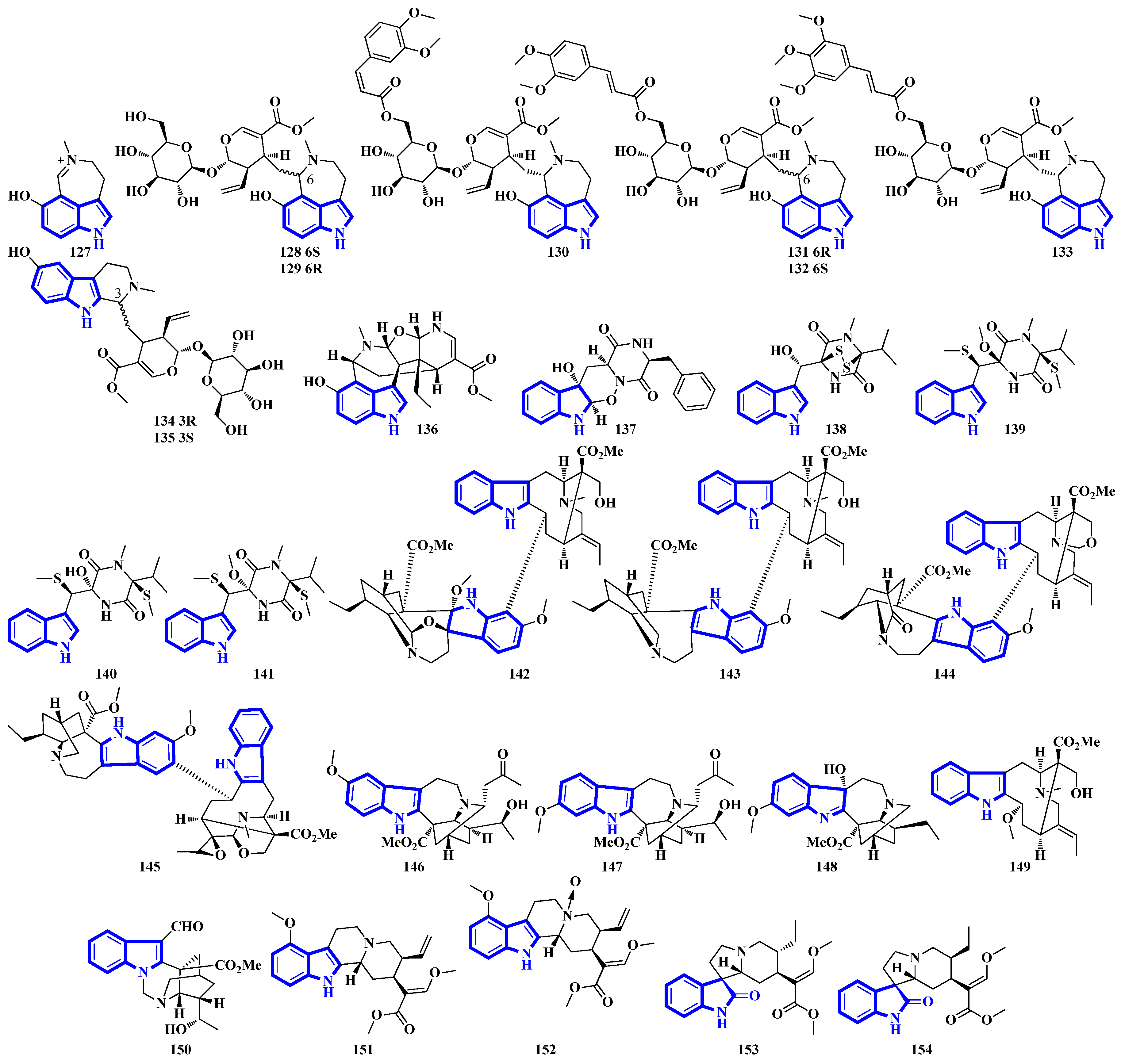
2.4. Structure Reappraisals
3. Synthesis of Indole Alkaloids
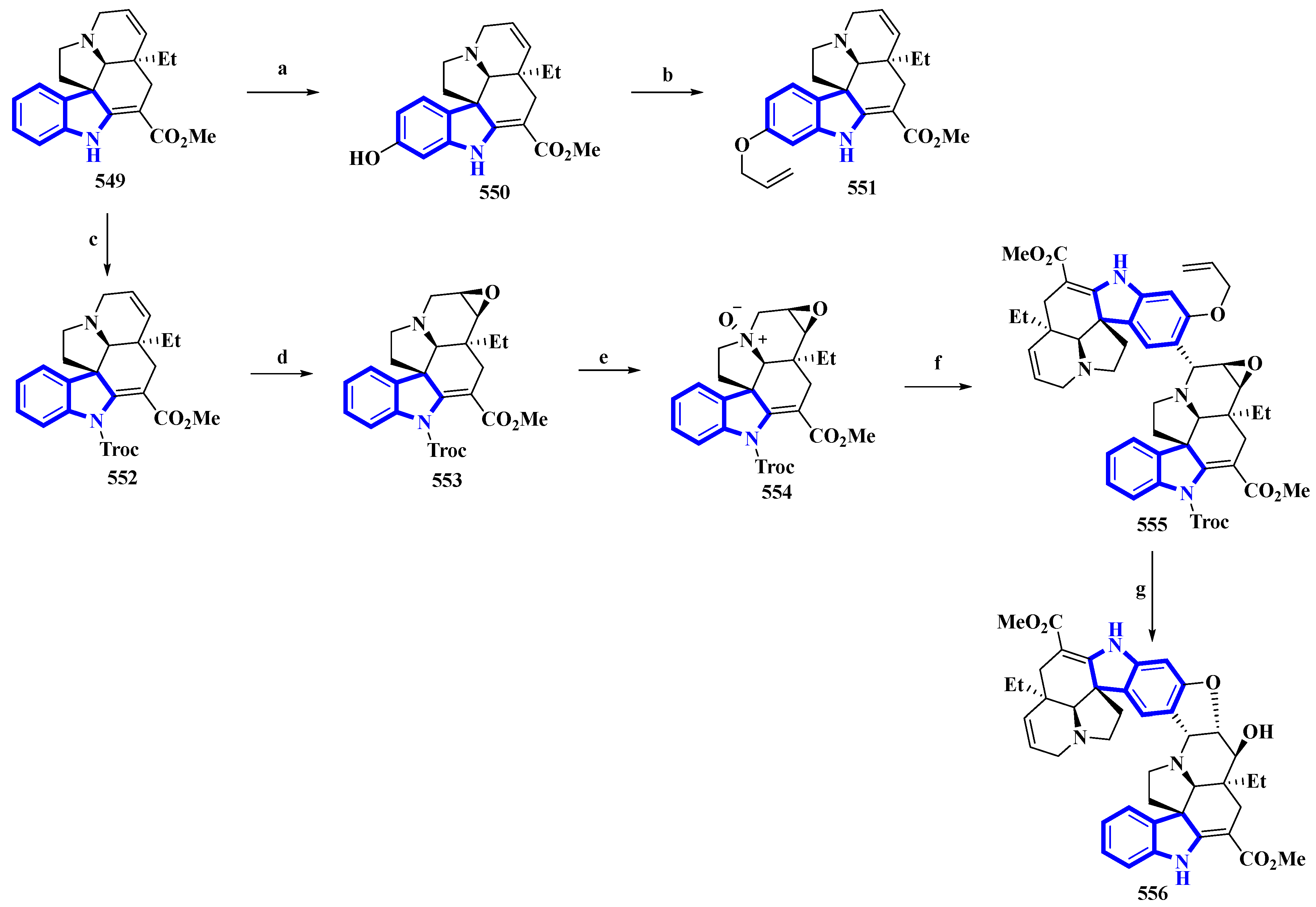
3.1. Total Synthesis of Indole Alkaloids
3.2. Synthesis of Indole Derivatives
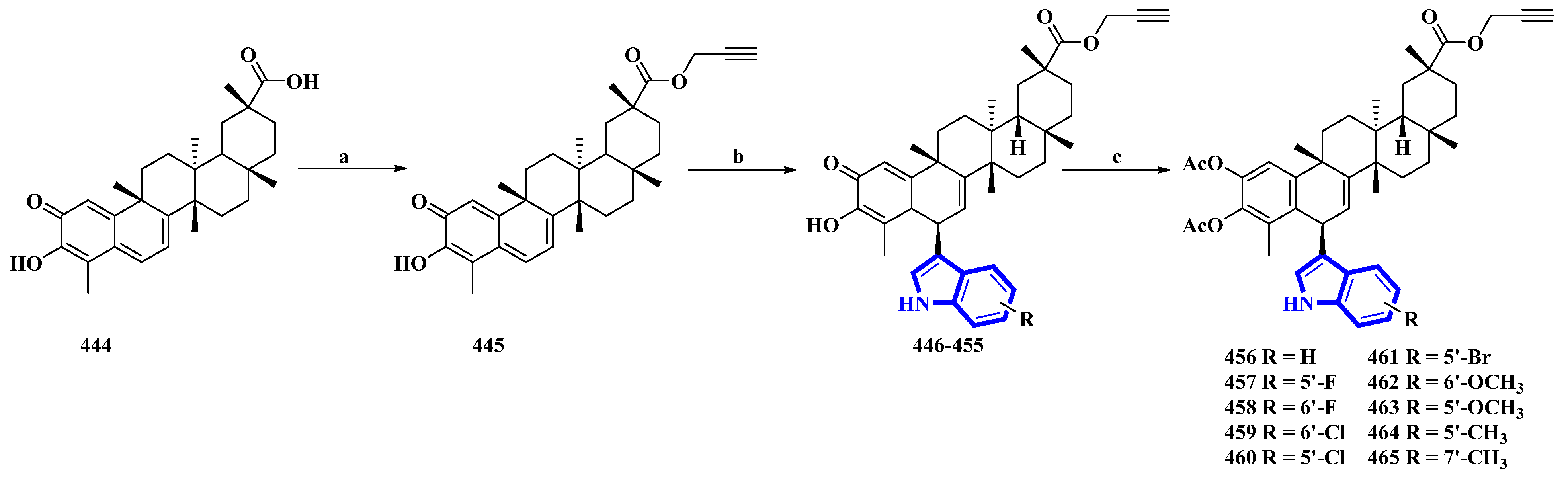
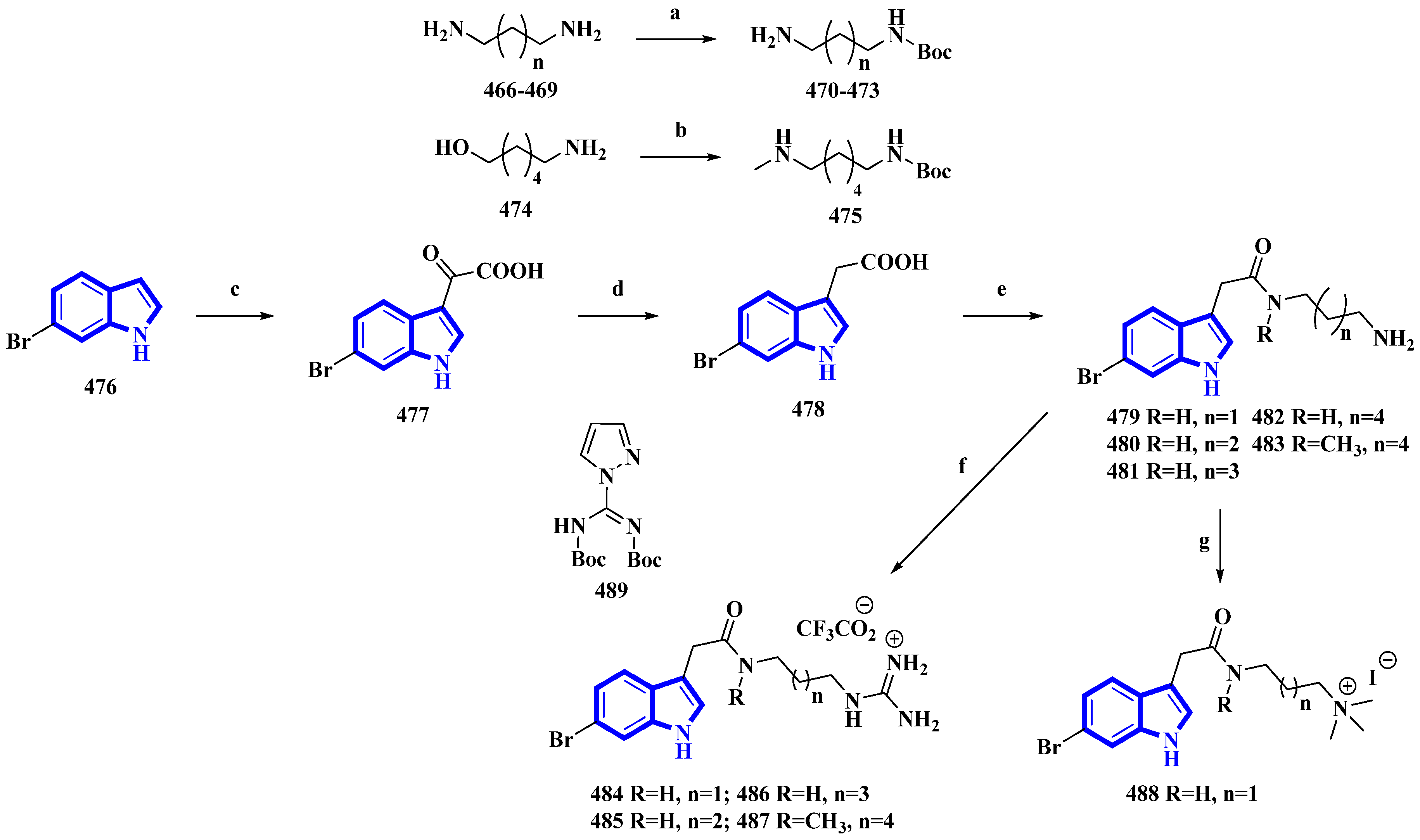
3.3. Semi-Synthesis of Indole Alkaloids

{kind=link}
{kind=link}
{kind=link}
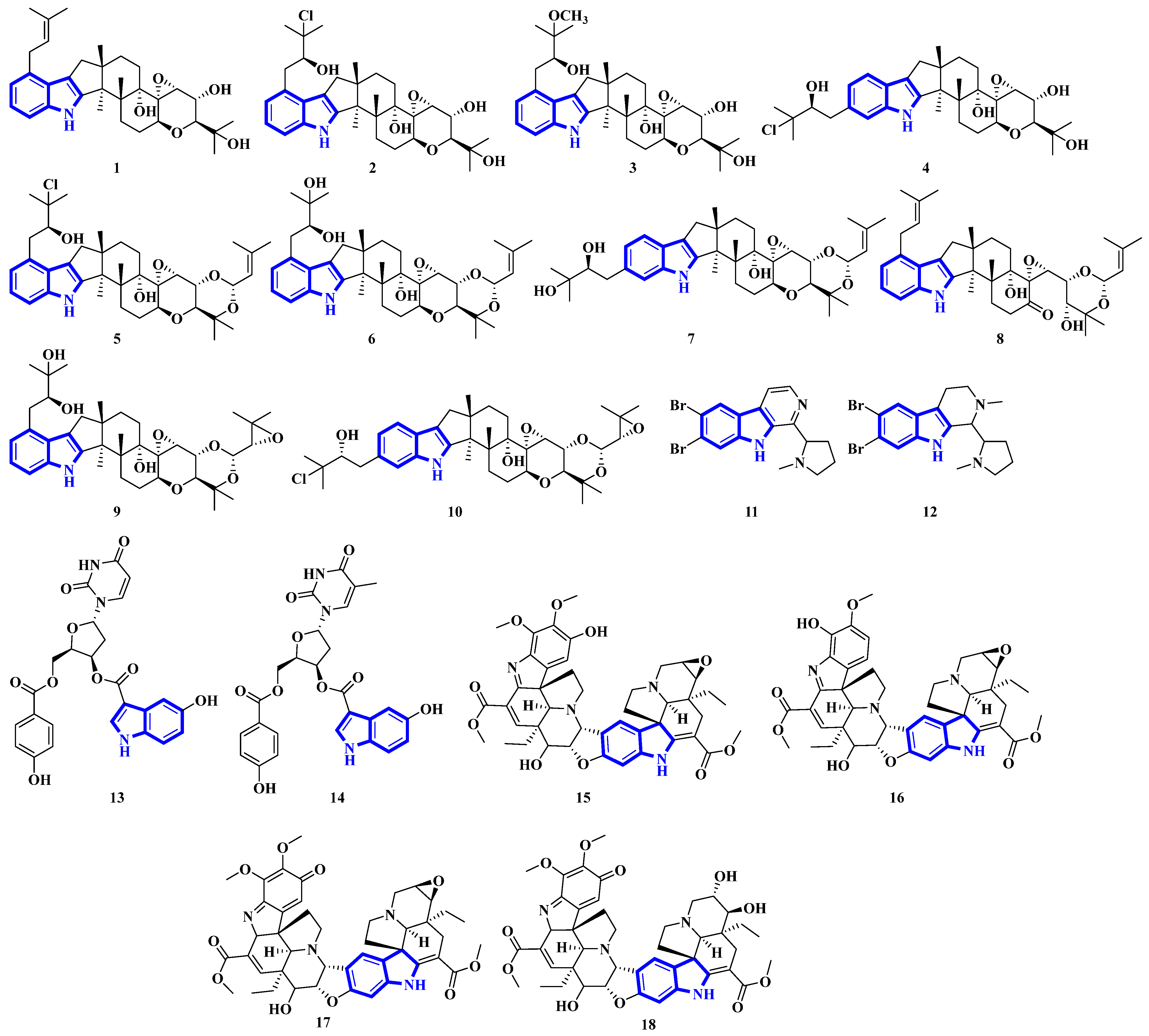{kind=link}
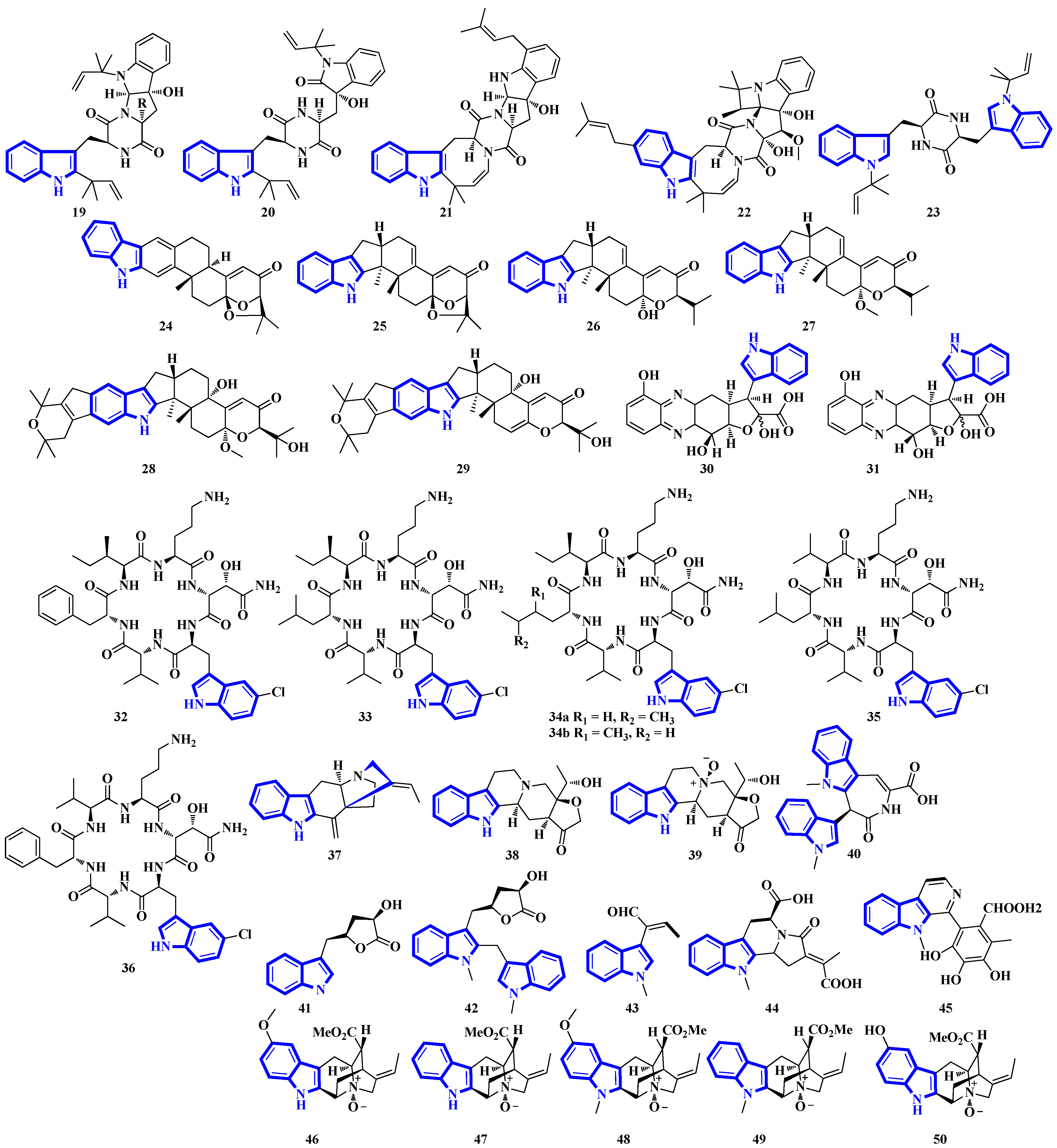{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
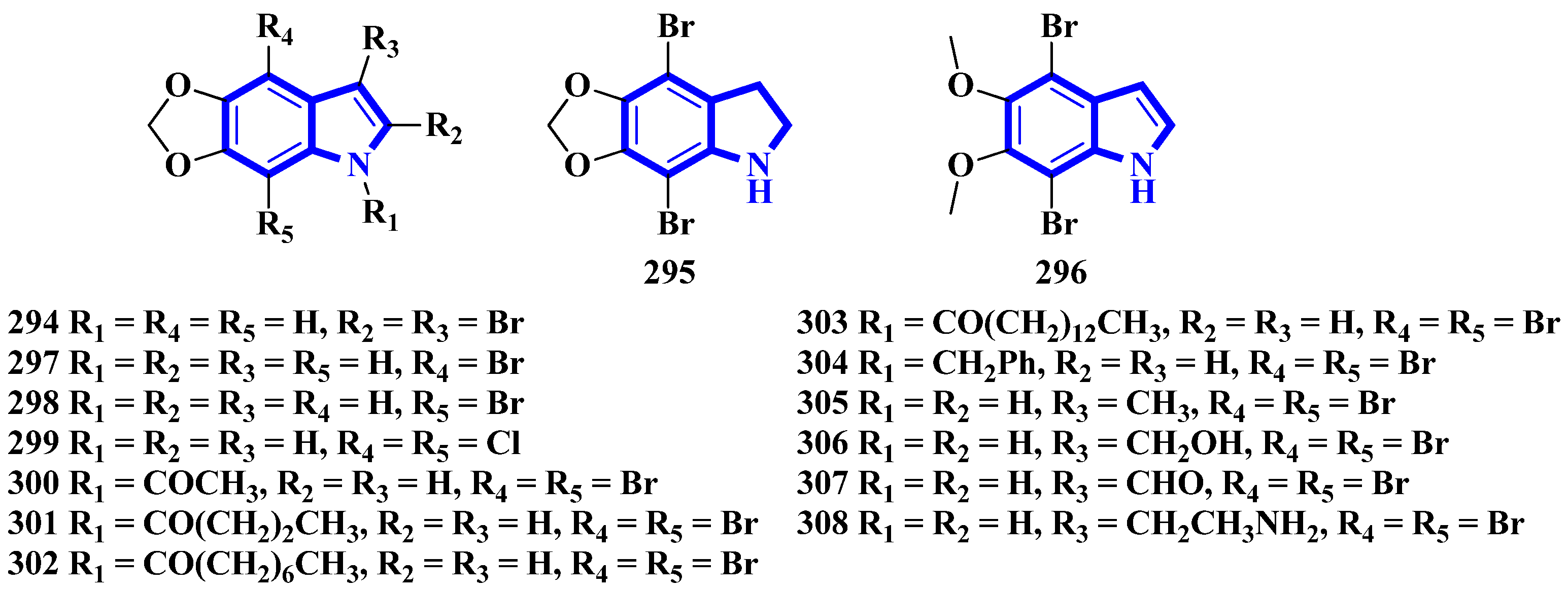{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cytotoxicity (GI50) µM [Cell Line] | Ref. |
|---|---|---|
| 1 | 38 [Huh7], 32 [LN229], 19 [HCT116], 16 [MGC803], 16 [A549], 21 [MDA231] | [20] |
| 15 | 1.1 [KB/S], 4.1 [KB/VJ300 b], 4.7 [PC-3], 4.8 [MCF7], 3.6 [MDA-MB-231], 4.3 [HCT 116], 2.2 [HT-29h] | [22] |
| 16 | 0.1 [KB/S], 1.3 [KB/VJ300 b], 7.9 [PC-3], 9.6 LNCaP, 0.6 [MCF7], 1.4 [MDA-MB-231], 1.1 [HCT 116], 0.3 [HT-29h] | [22] |
| 17 | 0.1 [KB/S], 8.6 [KB/VJ300], 1.5 [KB/VJ300 b], 0.7 [PC-3], 0.3 [MCF7], 0.4 [MDA-MB-231], 0.2 [HCT 116], 0.2 [HT-29h] | [22] |
| 18 | 0.2 [KB/S], 2.6 [LNCaP], 3.5 [MCF7], 5.4 [MDA-MB-231], 3.1 [HCT 116], 2.3 [HT-29h] | [22] |
| 24 | 3.6 [L5178Y], 8.7 [A2780b], 40 [J82], 29 [HEK-293] | [24] |
| 25 | 5.3 [L5178Y], 12 [A2780b], 42 [J82], 22 [HEK-293] | [24] |
| 26 | 5.3 [L5178Y], 28 [HEK-293] | [24] |
| 27 | 12 [A2780b], 55 [J82] | [24] |
| 28 | 32 [A2780b], 100 [J82] | [24] |
| 29 | 8.1 [L5178Y], 7.8 [A2780b], 32 [J82], 37 [HEK-293] | [24] |
| 37 | 6.2 [HT-29] | [27] |
| 51–54a | 4.1 × 10−4 [U87], 7.5 × 10−4 [SKOV3], 5.4 [MDA-MB-231 and HCT116] | [30] |
| 55 | 7.3 [HeLa] | [31] |
| 56 | 6.4 [HeLa] | [31] |
| 71 | 0.6 [KB/S], 6.7 [KB/VJ300], 0.2 [KB/VJ300 b], 8.2 [PC-3], 6.2 [LNCaP], 4.5 [MCF7], 4.5 [MDA-MB-231], 0.3 [HT-29], 7 [A549], 7.9 [MRC-5] | [35] |
| 72 | 2.8 [KB/S], 5.9 [KB/VJ300], 6.1 [PC-3], 2 [MCF7], 4.8 [MDA-MB-231], 0.07 [HT-29], 4.2 [HCT 116], 9 [A549], 9.7 [CCD-18Co] | [35] |
| 73 | 1.1 [KB/S], 3.9 [KB/VJ300B], 3.7 [LNCaP], 5.3 [MCF7], 4.8 [MDA-MB-231], 0.07 [HT-29] | [35] |
| 74 | 2.4 [KB/S], 2.9 [KB/VJ300], 3.1 [PC-3], 1.3 [MCF7], 1.4 [MDA-MB-231], 0.03 [HT-29], 2.9 [HCT 116], 6.8 [A549], 4.3 [CCD-18Co] | [35] |
| 75 | 2.5 [KB/S], 5.5 [KB/VJ300], 8.8 [KB/VJ300 b], 7.7 [MCF7], 4.1 [MDA-MB-231], 0.03 [HT-29], 1.5 [HCT 116], 3.9 [A549], 4.1 [CCD-18Co] | [35] |
| 76 | 1.7 [KB/S], 3.6 [KB/VJ300], 4.2 [KB/VJ300 b], 8.5 [MCF7], 0.02 [HT-29], 2.1 [HCT 116], 3 [A549], 5.1 [CCD-18Co] | [35] |
| 117 | 81 [HeLa] | [43] |
| 118 | 42 [HeLa] | [43] |
| 119 | 84 [Huh7], 92 [HeLa] | [43] |
| 123 | 2.45 [A549], 0.96 [Huh7], 0.93 [HeLa] | [43] |
| 145 | 7.5 [SW480] | [48] |
| 157 | 18.7 [HepG2], 28.7 [A-549] | [50] |
| 160 | 17 [U251], 4.5 [U87MG] | [51] |
| 161 | 36 [U251], 43 [U87MG] | [51] |
| 162 | 12 [U251], 2.3 [U87MG] | [51] |
| 164 | 27 [U251], 8.9 [U87MG] | [51] |
| 166 | 36 [U251], 29 [U87MG] | [51] |
| 167 | 19 [U251], 17 [U87MG] | [51] |
| 168 | 12 [U251], 7.4 [U87MG] | [51] |
| 170 | 13 [U251], 11 [U87MG] | [51] |
| 172 | 15 [U251], 19 [U87MG] | [51] |
| 187 | 6.8 [SK-MEL-28], 9.8 [SW480], 6.3 [HepG2], 9.8 [T47D] | [55] |
| 191 | 8.3 [SK-MEL-28], 7.4 [HepG2] | [55] |
| 192 | 6.7 [SK-MEL-28], 7.8 [SW480], 2.5 [HepG2], 8.7 [T47D] | [55] |
| 193 | 9.5 [T47D] | [55] |
| 211 | 5 [KB/S], 7.6 [KB/VJ300], 6.1 [PC-3], 5.9 [MDA-MB-231], 0.2 [HT-29], 4.7 [HCT 116], 2.1 [A549] | [60] |
| 217 | 44.0 [A549], 40.4 [HT-29], 37.3 [HepG2] | [63] |
| 218 | 8.2 [A549], 4.7 [A427], 8.0 [HCT116], 6.7 [HT-29], 6.2 MCF-7, 7.1 [HeLa], 9.1 [HepG2], 44.1 [LO2] | [63] |
| 219 | 13.8 [A427], 38.5 [HCT116], 20.3 [HeLa] | [63] |
| 220 | 18.9 [HeLa], 46.3 [LO2] | [63] |
| 221 | 7.9 [A549], 6.6 [A427], 10.6 [HCT116], 8.2 [HT-29], 7.2 MCF-7, 8.4 [HeLa], 9.6 [HepG2], 35.5 [LO2] | [63] |
| 222 | 9.5 [A549], 4.8 [A427], 12.2 [HCT116], 8.9 [HT-29], 6.5 MCF-7, 8.6 [HeLa], 9.1 [HepG2], 36.3 [LO2] | [63] |
| 223 | 6.9 [A549], 7.5 [A427], 11.2 [HCT116], 8.8 [HT-29], 6.8 MCF-7, 10.1 [HeLa], 10.7 [HepG2], 41.8 [LO2] | [63] |
| 224 | 21.5 [A549], 26.1 [HCT116], 41.5 [HeLa] | [63] |
| 225 | 23.3 [A549], 30.9 [HCT116], 43.8 [HT-29], 44.4 [HepG2] | [63] |
| 226 | 15.9 [A549], 21.2 [HCT116] | [63] |
| 227 | 40.4 [A549], 45.3 [HCT116] | [63] |
| 239 | 1.7 [HeLa], 1.6 [A549], 1.8 [HepG2], 1.5 [SMMC7721] | [70] |
| 242 | 7.9 [HeLa], 7.8 [A549], 8.1 [HepG2], 6.7 [SMMC7721] | [70] |
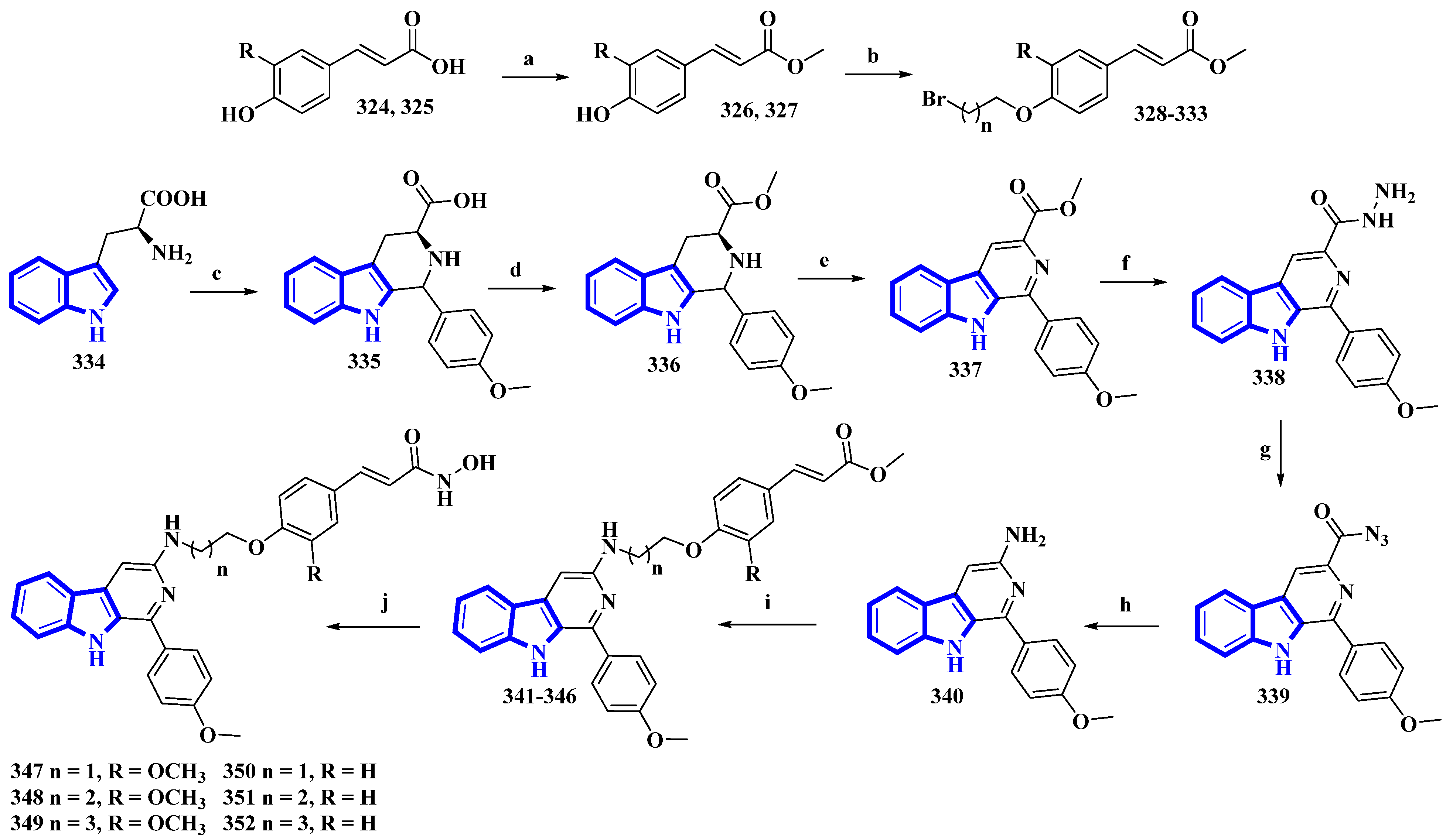
| 347 | 4 [SMMC-7721], 4.8 [HepG2], 1.8 [Bel7402], 2.8 [Huh7] | [88] |
| 348 | 2.9 [SMMC-7721], 1.4 [HepG2], 1.0 [Bel7402], 1.1 [Huh7] | [88] |
| 349 | 7.1 [SMMC-7721], 5.3 [HepG2], 4.6 [Bel7402], 6.2 [Huh7] | [88] |
| 350 | 6.3 [SMMC-7721], 5.8 [HepG2], 5.2 [Bel7402] | [88] |
| 351 | 4.1 [SMMC-7721], 3.4 [HepG2], 4.0 [Bel7402] | [88] |
| 352 | 9.5 [HepG2], 8.9 [Bel7402] | [88] |
| 535 | 0.51 [MV4-11] | [96] |
| 536 | 0.42 [MV4-11] | [96] |
| 537 | 0.41 [MV4-11] | [96] |
| 538 | 0.36 [MV4-11] | [96] |
| 539 | 0.56 [MV4-11] | [96] |
| 540 | 0.32 [MV4-11] | [96] |
| 541 | 0.59 [MV4-11] | [96] |
| 542 | 0.43 [MV4-11] | [96] |
| 543 | 0.44 [MV4-11] | [96] |
| 544 | 0.36 [MV4-11] | [96] |
| 545 | 0.96 [MV4-11] | [96] |
| 546 | 0.70 [MV4-11] | [96] |
| 547 | 0.39 [MV4-11] | [96] |
| 548 | 0.51 [MV4-11] | [96] |
| Antibacterial (MIC; µg/mL) | ||
| 1 | 25 [B. cereus], 12.5 [S. aureus c] | [20] |
| 2 | 50 [S. aureus c] | [20] |
| 8 | 12.5 [B. cereus], 25 [S. aureusc], 25 [M. lysodeikticus], 25 [B. paratyphosum], 25 [B. subtilis], 25 [E. aerogenes], 25 [S. typhi], 25 [P. vulgaris] | [20] |
| 32 | 4 [B. subtilis], 16 [S. typhimurium], 8 [M. luteus], 8 [M.phlei] | [26] |
| 33 | 32 [B. subtilis], 32 [S. typhimurium], 32 [M. luteus], 32 [M. phlei] | [26] |
| 40 | 8 [X. o. pv. oryzae], 32 [R. solanacearum], 32 [X. o. pv. oryzicola], 128 [P. s. pv. lachrymans] | [28] |
| 42 | 32 [X. o. pv. oryzae] | [28] |
| 44 | 32 [X. o. pv. oryzae], 128 [R. solanacearum], 64 [X. o. pv. oryzicola] | [28] |
| 45 | 64 [X. o. pv. oryzae] | [28] |
| 64 | 25 [H. influenzae ATCC 4] | [33] |
| 65 | 50 [H. influenzae ATCC 4] | [33] |
| 66 | 25 [H. influenzae ATCC 4] | [33] |
| 231 | 25 [M. smegmatis], 25 [M. abscessus], 25 [M. bovis] | [65] |
| Antifungal (MIC; µg/mL) | ||
| 1 | 6.25 [A. fragariae], 25 [C. cassiicola], 25 [A. alternata], 6.25 [B. cinereal Pers], 25 [C. personata], 6.25 [V. dahliaekleb], 25 [S. sclerotiorum] | [20] |
| 2 | 25 [A. fragariae] | [20] |
| 3 | 25 [A. fragariae] | [20] |
| 4 | 50 [A. fragariae] | [20] |
| 5 | 6.25 [A. fragariae] | [20] |
| 6 | 25 [A. fragariae] | [20] |
| 7 | 6.25 [A. fragariae] | [20] |
| 9 | 50 [A. fragariae] | [20] |
| 10 | 25 [A. fragariae] | [20] |
| 229 | 50 [F. oxysporum] | [64] |
| Antiplasmodial (IC50 µM) | ||
| 63 | 6.1 d [P. falciparum] | [32] |
| 110 | 8.7 [P. falciparum FcB1] | [42] |
| 111 | 9.5 [P. falciparum FcB1] | [42] |
| 112 | 2.6 [P. falciparum FcB1] | [42] |
| 113 | 5.2 [P. falciparum FcB1] | [42] |
| 114 | 3.0 [P. falciparum FcB1] | [42] |
| 210 | 1.05 [P. falciparum FcB1] | [59] |
| 234 | 15.1 [P. falciparum 3D7] | [67] |
| Antiviral (EC50 µM) | ||
| 109 | 70 [HSV-2] | [41] |
| 137 | 5.7 [HCV] | [46] |
| 196 | 7.5 [ZIKV] | [56] |
| 197 | 38 [ZIKV] | [56] |
| 198 | 50 [ZIKV] | [56] |
| 249 | 9.4 [H1N1], 6.7 [RSV] | [72] |
| 250 | 3.7 [H1N1], 2.8 [RSV] | [72] |
| Antifouling activity (EC50 µM) | ||
| 486 | 2.2 [A. improvisus] | [93] |
| 487 | 0.7 [A. improvisus] | [93] |
| Inhibition of protein phosphatases (IC50 µM) | ||
| 67 | 14 [PTP1B], 38 [PTPsigma] | [34] |
| 68 | 27 [PTP1B] | [34] |
| 70 | 23 [PTP1B], 35 [TCPTP] | [34] |
| Inhibition of DC secretion of IL-12p40 (% inhibition at 10 µg/mL) | ||
| 177 | 38% | [53] |
| 185 | 36% | [53] |
| Promotion of DC secretion of IL-10 (% promotion at 10 µg/mL) | ||
| 176 | 19% | [53] |
| Promotion of hBM-MSC secretion of adiponectin (EC50 µM) | ||
| 212 | 9.86 | [61] |
| 213 | 6.20 | [61] |
| Inhibition of LPS-induced B-cell proliferation (IC50 µM) | ||
| 237 | 0.38 | [69] |
| 238 | 47.37 | [69] |
| Analgesic activity | ||
| 205 | 64.7% (1 mg/kg) | [58] |
| 206 | 50% (5 mg/kg) | [58] |
| 207 | 67.6% (0.04 mg/kg), 76.1% (0.2 mg/kg) | [58] |
| 208 | 55% (5 mg/kg) | [58] |
| 209 | 53% (5 mg/kg) | [58] |
| 5-HT1A receptor agonist (EC50 µM) | ||
| 83 | 10 | [37] |
| 84 | 2.2 | [37] |
| 86 | 0.1 | [37] |
| 87 | 54 | [37] |
| Inhibition of HDACs (EC50 nM) | ||
| 347 | 32 [HDAC1], 125 [HDAC3], 17 [HDAC6] | [88] |
| 348 | 27 [HDAC1], 148 [HDAC3], 13 [HDAC6] | [88] |
| 349 | 61 [HDAC1] | [88] |
| 350 | 75 [HDAC1] | [88] |
| 351 | 57 [HDAC1] | [88] |
| 352 | 125 [HDAC1] | [88] |
| Inhibition of porcine SCR (EC50 µM) | ||
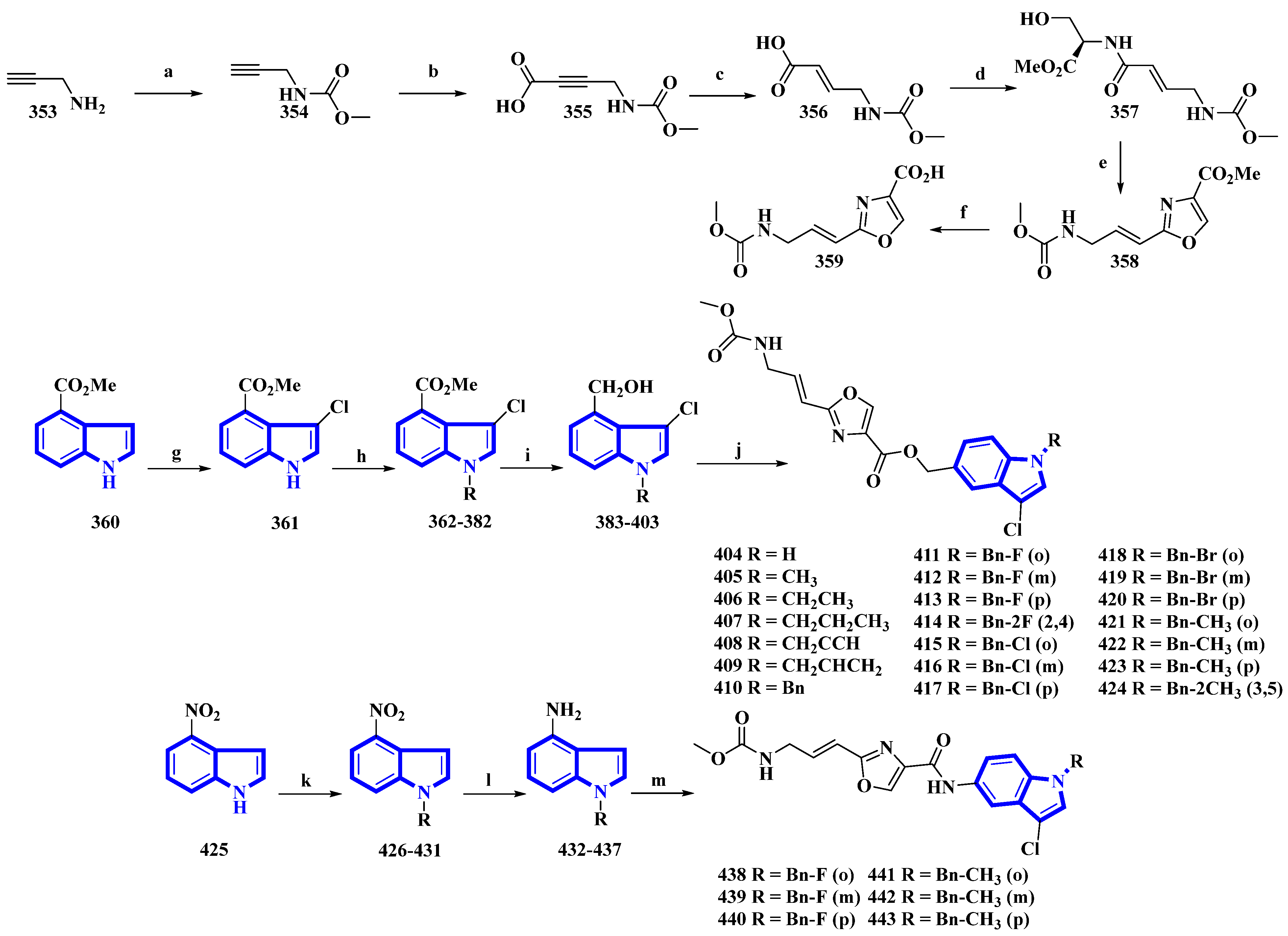
| 411 | 10 | [90] |
| 438 | 1.34 | [90] |
| 439 | 1.75 | [90] |
| 440 | 1.46 | [90] |
| 442 | 0.70 | [90] |
| Inhibition of acetylcholinesterase (IC50 µM) | ||
| 98 | 10.5 | [38] |
| Inhibition of quorum sensing (MIC µg/well) | ||
| 104 | 32 [C. violaceum CV026] | [39] |
| 105 | 32 [C. violaceum CV026] | [39] |
| Glucose uptake activity in L6 myoblasts at 50 µM (mmol/L) | ||
| 203 | 5 | [57] |
| 204 | 5 | [57] |
| Triglyceride accumulation promotion in 3T3-L1 cells (EC50 µM) | ||
| 126 | 1.03 | [44] |
| Inhibition of MAO-A (IC50 µM) | ||
| 127 | 0.9 [MAO-A] | [45] |
| Inhibition of misc. enzymes (inhibition % at 10 µM) | ||
| 127 | 31 [MAO-B], 21 [BChE] | [45] |
| Inhibition of mushroom tyrosinase (IC50 µM) | ||
| 141 | 33.2 | [47] |
| Inhibition of autophagic flux (EC50 µM) | ||
| 145 | 20.2 | [48] |
| Vasorelaxant activity (EC50 µM) | ||
| 158 | 2.8 | [50] |
| 159 | 2.4 | [50] |
| Potentiation of imipenem activity (4 ug/mL of imipenem; MIC µg/mL) | ||
| 173 | 8–16 [S. aureus c] | [52] |
| 174 | 2–4 [S. aureus c] | [52] |
| Inhibition of RANKL-induced multinuclear osteoclasts (IC50 µM) | ||
| 233 | 10.5 [RAW264] | [66] |
| 294 | 50 e [RAW264] | [66] |
| 295 | 25.6 [RAW264] | [66] |
| 296 | 6.3 [RAW264] | [66] |
| 297 | 16.8 [RAW264] | [66] |
| 298 | 35.4 [RAW264] | [66] |
| 299 | 40.0 [RAW264] | [66] |
| 300 | 11.7 [RAW264] | [66] |
| 301 | 7.9 [RAW264] | [66] |
| 302 | 11.1 [RAW264] | [66] |
| 305 | 8.1 [RAW264] | [66] |
| 306 | 10.0 [RAW264] | [66] |
| 307 | 13.9 [RAW264] | [66] |
| 308 | 5.9 [RAW264] | [66] |
4. Bioactivities of Novel Indole Alkaloids
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| LSD | Lysergic acid diethylamide |
| DCs | Dendritic cells |
| HDAC | Histone deacetylase |
| MIAs | Monoindole alkaloids |
| CANPA | Computer-Assisted Natural Products Anticipation |
| ETP | Epipolythiodioxopiperazine |
| HMDA | Hydroxy-4-methyldecanoic acid |
| EDB | Edible |
| LCPA | Long-chain polyamine |
| DFT | Density-functional theory |
| THF | Tetrahydrofuran |
| TBAF | Tetra-n-butylammonium fluoride |
| RANKL | Receptor activator of nuclear factor kappa-Β ligand |
| DCM | Dichloromethane |
| TiPSOTf | Triisopropylsilyl trifluoromethanesulfonate |
| HOBt | Hydroxybenzotriazole |
| DIPEA | N,N-Diisopropylethylamine |
| HATU | Hexafluorophosphate azabenzotriazoletetramethyl uronium |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| TrocCl | 2,2,2-Trichloroethoxycarbonyl chloride |
| TFAA | Trifluoroacetic anhydride |
| MAO | Monoamine oxidase |
| SCR | Succinate-cytochrome c reductase |
| LPS | Lipopolysaccharide |
| IL | Interleukin |
| PTP | Protein tyrosine phosphatase |
| HSV-2 | Herpes simplex virus 2 |
| hBM-MSC | Human bone marrow mesenchymal stem cells |
| TFA | Trifluoroacetic acid |
| Et3N | Triethylamine |
| Et2O | Diethyl ether |
| NBS | N-Bromosuccinimide |
| AcOH | Acetic acid |
| Boc2O | Di-tert-butyl dicarbonate |
| Ac2O | Acetic anhydride |
| DMAP | 4-Dimethylaminopyridine |
| TMSOTf | Trimethylsilyl trifluoromethanesulfonate |
| DMF | Dimethylformamide |
| n-BuLi | n-Butyl lithium |
| EtOAc | Ethyl acetate |
| i-BuOCOCl | Isobutyl chloroformate |
| DAST | Diethylamino sulfur trifluoride |
| DBU | 1,8-Diazabicyclo [5.4.0]undec-7-ene |
| DIBAL-H | Diisobutylaluminium hydride |
| EDCl | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| MsCl | Methanesulfonyl chloride |
| NCS | N-Chlorosuccinimide |
| TCDI | Thiocarbonyldiimidazole |
| m-CPBA | Meta-chloroperoxybenzoic acid |
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| 5-HT1A | 5-hydroxytryptamine 1A |
| ZIKV | Zika virus |
| TCPTP | T-cell protein tyrosine phosphatase |
| RSV | Respiratory syncytial virus |
| ATP | Adenosine triphosphate |
References
- Lahlou, M. The success of natural products in drug discovery. Pharmacol. Pharm. 2013, 4, 17–31. [Google Scholar] [CrossRef]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200. [Google Scholar] [CrossRef] [PubMed]
- John, J.E. Natural products-based drug discovery: Some bottlenecks and considerations. Curr. Sci. 2009, 96, 753–754. [Google Scholar]
- Eddershaw, P.J.; Beresford, A.P.; Bayliss, M.K. ADME/PK as part of a rational approach to drug discovery. Drug Discov. Today 2000, 5, 409–414. [Google Scholar] [CrossRef]
- Appendino, G.; Fontana, G.; Pollastro, F. Natural Products Drug Discovery. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 205–236. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Mohr, J.T.; Krout, M.R.; Stoltz, B.M. Natural products as inspiration for the development of asymmetric catalysis. Nature 2008, 455, 323–332. [Google Scholar] [CrossRef]
- Grigalunas, M.; Burhop, A.; Christoforow, A.; Waldmann, H. Pseudo-natural products and natural product-inspired methods in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 2020, 56, 111–118. [Google Scholar]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef]
- Hunter, P. Harnessing nature’s wisdom. Turning to nature for inspiration and avoiding her follies. EMBO Rep. 2008, 9, 838–840. [Google Scholar] [CrossRef]
- Hanson, J.R. Natural Products: The Secondary Metabolites; Royal Society of Chemistry: Cambridge, UK, 2003; Volume 17. [Google Scholar]
- Harden, R.N. Chronic neuropathic pain: Mechanisms, diagnosis, and treatment. Neurologist 2005, 11, 111–122. [Google Scholar]
- Esu, E.B.; Effa, E.E.; Opie, O.N.; Meremikwu, M.M. Artemether for severe malaria. Cochrane Database Syst. Rev. 2019, 6, CD010678. [Google Scholar] [PubMed]
- Kaur, R.; Arora, S. Alkaloids-important therapeutic secondary metabolites of plant origin. J. Crit. Rev. 2015, 2, 1–8. [Google Scholar]
- Seigler, D.S. Plant Secondary Metabolism; Springer: New York, NY, USA, 2001; p. 628. [Google Scholar]
- Kiesecker, C.; Zitron, E.; Luck, S.; Bloehs, R.; Scholz, E.P.; Kathofer, S.; Thomas, D.; Kreye, V.A.; Katus, H.A.; Schoels, W.; et al. Class Ia anti-arrhythmic drug ajmaline blocks HERG potassium channels: Mode of action. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 423–435. [Google Scholar] [CrossRef]
- Moore, P.W.; Rasimas, J.J.; Donovan, J.W. Physostigmine is the antidote for anticholinergic syndrome. J. Med. Toxicol. 2015, 11, 159–160. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. (Eds.) Chapter 8—Anticancer drugs targeting tubulin and microtubules. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; pp. 229–249. [Google Scholar]
- Koenig, X.; Hilber, K. The anti-addiction drug ibogaine and the heart: A delicate relation. Molecules 2015, 20, 2208–2228. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Hai, P.; Zhang, S.B.; Xiao, J.F.; Gao, Y.; Ma, B.J.; Fu, H.Y.; Chen, Y.M.; Yang, X.L. Prenylated indole diterpene alkaloids from a mine-soil-derived Tolypocladium sp. J. Nat. Prod. 2019, 82, 221–231. [Google Scholar] [CrossRef]
- Tabudravu, J.N.; Pellissier, L.; Smith, A.J.; Subko, K.; Autreau, C.; Feussner, K.; Hardy, D.; Butler, D.; Kidd, R.; Milton, E.J.; et al. LC-HRMS-database screening metrics for rapid prioritization of samples to accelerate the discovery of structurally new natural products. J. Nat. Prod. 2019, 82, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sim, D.S.; Navanesan, S.; Sim, K.S.; Gurusamy, S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Conolodinines A-D, Aspidosperma-Aspidosperma bisindole alkaloids with antiproliferative activity from Tabernaemontana corymbosa. J. Nat. Prod. 2019, 82, 850–858. [Google Scholar] [CrossRef]
- Yu, X.; Müller, W.E.; Guo, Z.; Lin, W.; Zou, K.; Liu, Z.; Proksch, P. Indole alkaloids from the coprophilous fungus Aphanoascus fulvescens. Fitoterapia 2019, 136, 104168. [Google Scholar] [CrossRef]
- Ariantari, N.P.; Ancheeva, E.; Wang, C.; Mandi, A.; Knedel, T.O.; Kurtan, T.; Chaidir, C.; Muller, W.E.G.; Kassack, M.U.; Janiak, C.; et al. Indole diterpenoids from an endophytic Penicillium sp. J. Nat. Prod. 2019, 82, 1412–1423. [Google Scholar] [CrossRef]
- Wang, X.; Abbas, M.; Zhang, Y.; Elshahawi, S.I.; Ponomareva, L.V.; Cui, Z.; Van Lanen, S.G.; Sajid, I.; Voss, S.R.; Shaaban, K.A.; et al. Divergent fused phenazine-based metabolites from a Himalayan Streptomyces. J. Nat. Prod. 2019, 82, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Mudalungu, C.M.; von Torne, W.J.; Voigt, K.; Ruckert, C.; Schmitz, S.; Sekurova, O.N.; Zotchev, S.B.; Sussmuth, R.D. Noursamycins, chlorinated cyclohexapeptides identified from molecular networking of Streptomyces noursei NTR-SR4. J. Nat. Prod. 2019, 82, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Wong, S.P.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. A cytotoxic indole characterized by incorporation of a unique carbon-nitrogen skeleton and two pentacyclic corynanthean alkaloids incorporating a substituted tetrahydrofuranone ring from Kopsia arborea. J. Nat. Prod. 2019, 82, 1902–1907. [Google Scholar] [CrossRef]
- Yan, W.; Zhao, S.S.; Ye, Y.H.; Zhang, Y.Y.; Zhang, Y.; Xu, J.Y.; Yin, S.M.; Tan, R.X. Generation of indoles with agrochemical significance through biotransformation by Chaetomium globosum. J. Nat. Prod. 2019, 82, 2132–2137. [Google Scholar] [CrossRef]
- Ramos, A.E.F.; Pavesi, C.; Litaudon, M.; Dumontet, V.; Poupon, E.; Champy, P.; Genta-Jouve, G.; Beniddir, M.A. CANPA: Computer-assisted natural products anticipation. Anal. Chem. 2019, 91, 11247–11252. [Google Scholar] [CrossRef]
- Kim, M.C.; Cullum, R.; Machado, H.; Smith, A.J.; Yang, I.; Rodvold, J.J.; Fenical, W. Photopiperazines A-D, photosensitive interconverting diketopiperazines with significant and selective activity against U87 glioblastoma cells, from a rare, marine-derived Actinomycete of the family Streptomycetaceae. J. Nat. Prod. 2019, 82, 2262–2267. [Google Scholar] [CrossRef]
- Li, H.; Xu, D.; Sun, W.; Yang, B.; Li, F.; Liu, M.; Wang, J.; Xue, Y.; Hu, Z.; Zhang, Y. HPLC-DAD-directed isolation of linearly fused prenylated indole alkaloids from a soil-derived Aspergillus versicolor. J. Nat. Prod. 2019, 82, 2181–2188. [Google Scholar] [CrossRef]
- Knestrick, M.A.; Wilson, N.G.; Roth, A.; Adams, J.H.; Baker, B.J. Friomaramide, a highly modified linear hexapeptide from an Antarctic sponge, inhibits Plasmodium falciparum liver-stage development. J. Nat. Prod. 2019, 82, 2354–2358. [Google Scholar] [CrossRef]
- Bankeu, J.J.K.; Kagho, D.U.K.; Fongang, Y.S.F.; Toghueo, R.M.K.; Mba’ning, B.M.; Feuya, G.R.T.; Fekam, F.B.; Tchouankeu, J.C.; Ngouela, S.A.; Sewald, N.; et al. Constituents from Nauclea latifolia with anti-haemophilus influenzae type b inhibitory activities. J. Nat. Prod. 2019, 82, 2580–2585. [Google Scholar] [CrossRef]
- Zhou, L.M.; Kong, F.D.; Fan, P.; Ma, Q.Y.; Xie, Q.Y.; Li, J.H.; Zheng, H.Z.; Zheng, Z.H.; Yuan, J.Z.; Dai, H.F.; et al. Indole-diterpenoids with protein tyrosine phosphatase inhibitory activities from the marine-derived fungus Penicillium sp. KFD28. J. Nat. Prod. 2019, 82, 2638–2644. [Google Scholar] [CrossRef]
- Yeap, J.S.; Saad, H.M.; Tan, C.H.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Macroline-sarpagine bisindole alkaloids with antiproliferative activity from Alstonia penangiana. J. Nat. Prod. 2019, 82, 3121–3132. [Google Scholar] [CrossRef]
- Salim, F.; Yunus, Y.M.; Anouar, E.H.; Awang, K.; Langat, M.; Cordell, G.A.; Ahmad, R. Absolute configuration of alkaloids from Uncaria longiflora through experimental and computational approaches. J. Nat. Prod. 2019, 82, 2933–2940. [Google Scholar] [CrossRef]
- Liang, J.H.; Luan, Z.L.; Tian, X.G.; Zhao, W.Y.; Wang, Y.L.; Sun, C.P.; Huo, X.K.; Deng, S.; Zhang, B.J.; Zhang, Z.J.; et al. Uncarialins A-I, monoterpenoid indole alkaloids from Uncaria rhynchophylla as natural agonists of the 5-HT1A receptor. J. Nat. Prod. 2019, 82, 3302–3310. [Google Scholar] [CrossRef]
- Guo, Q.; Si, X.; Shi, Y.; Yang, H.; Liu, X.; Liang, H.; Tu, P.; Zhang, Q. Glucoconjugated monoterpene indole alkaloids from Uncaria rhynchophylla. J. Nat. Prod. 2019, 82, 3288–3301. [Google Scholar] [CrossRef]
- Kong, F.D.; Zhang, S.L.; Zhou, S.Q.; Ma, Q.Y.; Xie, Q.Y.; Chen, J.P.; Li, J.H.; Zhou, L.M.; Yuan, J.Z.; Hu, Z.; et al. Quinazoline-containing indole alkaloids from the marine-derived fungus Aspergillus sp. HNMF114. J. Nat. Prod. 2019, 82, 3456–3463. [Google Scholar] [CrossRef]
- Rodriguez, J.P.G.; Bernardi, D.I.; Gubiani, J.R.; Magalhaes de Oliveira, J.; Morais-Urano, R.P.; Bertonha, A.F.; Bandeira, K.F.; Bulla, J.I.Q.; Sette, L.D.; Ferreira, A.G.; et al. Water-soluble glutamic acid derivatives produced in culture by Penicillium solitum IS1-A from King George Island, Maritime Antarctica. J. Nat. Prod. 2020, 83, 55–65. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, K.; Wang, W.; Zhang, G.; Zhu, T.; Che, Q.; Gu, Q.; Li, D. Amphiepicoccins A-J: Epipolythiodioxopiperazines from the fish-gill-derived Fungus Epicoccum nigrum HDN17-88. J. Nat. Prod. 2020, 83, 524–531. [Google Scholar] [CrossRef]
- Alcover, C.F.; Bernadat, G.; Kabran, F.A.; Le Pogam, P.; Leblanc, K.; Ramos, A.E.F.; Gallard, J.F.; Mouray, E.; Grellier, P.; Poupon, E.; et al. Molecular networking reveals serpentinine-related bisindole alkaloids from Picralima nitida, a previously well-investigated species. J. Nat. Prod. 2020, 83, 1207–1216. [Google Scholar] [CrossRef]
- Wang, M.H.; Zhang, X.Y.; Tan, X.M.; Niu, S.B.; Sun, B.D.; Yu, M.; Ding, G.; Zou, Z.M. Chetocochliodins A-I, epipoly(thiodioxopiperazines) from Chaetomium cochliodes. J. Nat. Prod. 2020, 83, 805–813. [Google Scholar] [CrossRef]
- Li, C.J.; Chen, P.N.; Li, H.J.; Mahmud, T.; Wu, D.L.; Xu, J.; Lan, W.J. Potential antidiabetic fumiquinazoline alkaloids from the marine-derived fungus Scedosporium apiospermum F41-1. J. Nat. Prod. 2020, 83, 1082–1091. [Google Scholar] [CrossRef]
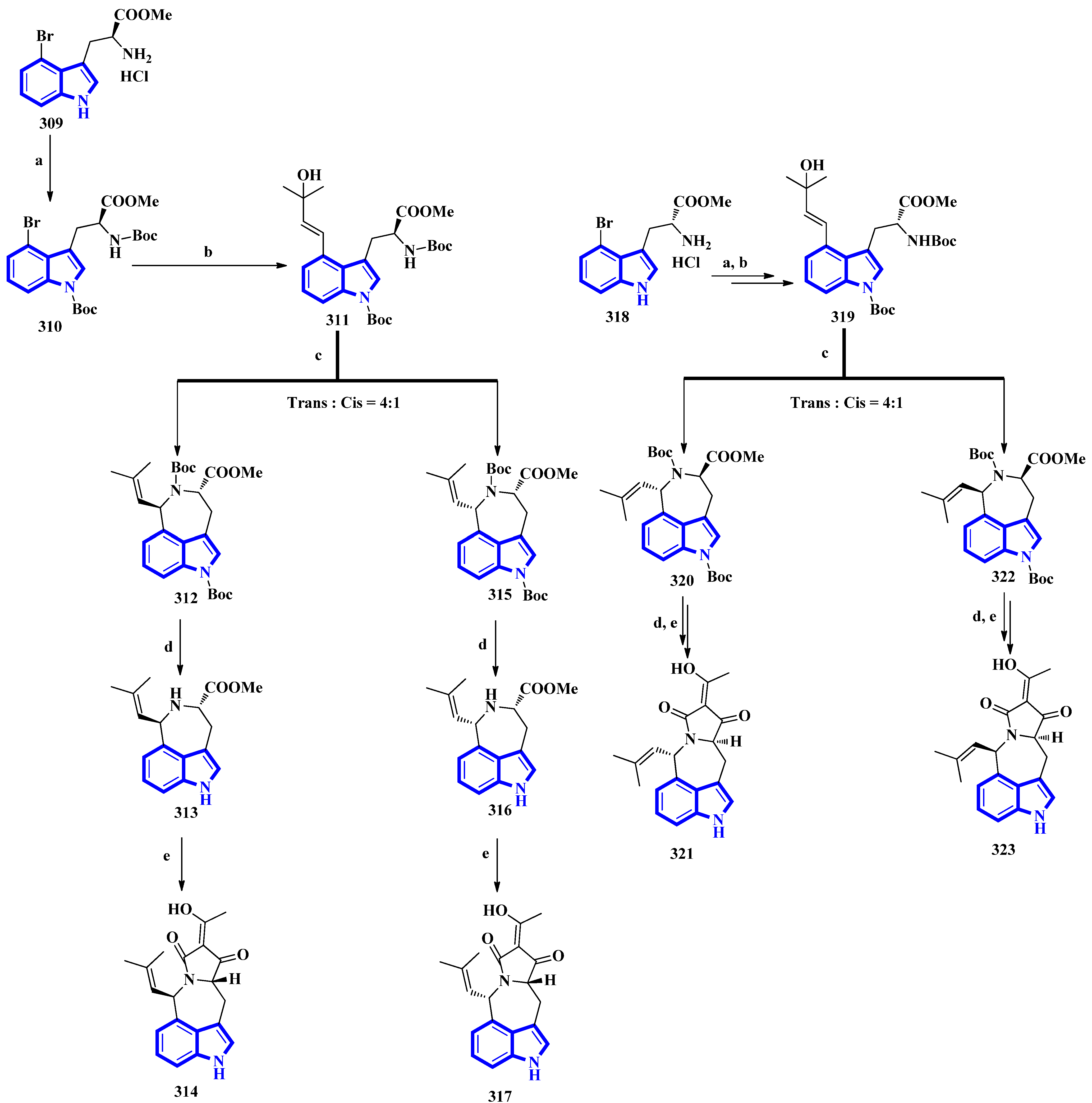
- Klein-Junior, L.C.; Cretton, S.; Heyden, Y.V.; Gasper, A.L.; Nejad-Ebrahimi, S.; Christen, P.; Henriques, A.T. Bioactive azepine-indole alkaloids from Psychotria nemorosa. J. Nat. Prod. 2020, 83, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, Y.; Hao, X.; Tan, J.; Li, F.; Qiao, X.; Chen, S.; Xiao, C.; Chen, M.; Peng, Z.; et al. Raistrickindole A, an anti-HCV oxazinoindole alkaloid from Penicillium raistrickii IMB17-034. J. Nat. Prod. 2019, 82, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.J.; Huo, G.M.; Zhang, Q.; Li, D.; Wang, D.C.; Qi, J.Z.; Han, W.B.; Gao, J.M. Phaeosphaones: Tyrosinase inhibitory thiodiketopiperazines from an endophytic Phaeosphaeria fuckelii. J. Nat. Prod. 2020, 83, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ding, X.; Yuan, Y.X.; Guo, L.L.; Hao, X.J. Cytotoxic monoterpenoid indole alkaloids from Tabernaemontana corymbosa as potent autophagy inhibitors by the attenuation of lysosomal acidification. J. Nat. Prod. 2020, 83, 1432–1439. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Raja, H.A.; Graf, T.N.; Augustinovic, M.; Wallace, E.D.; Hematian, S.; Kellogg, J.J.; Todd, D.A.; Cech, N.B.; Oberlies, N.H. The chemistry of Kratom [Mitragyna speciosa]: Updated characterization data and methods to elucidate indole and oxindole alkaloids. J. Nat. Prod. 2020, 83, 2165–2177. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.W.; Li, Y.; Wei, C.J.; Xie, J.; Yuan, M.F.; Zhang, D.M.; Ye, W.C.; Zhang, X.Q. Structurally diverse indole alkaloids with vasorelaxant activity from Melodinus hemsleyanus. J. Nat. Prod. 2020, 83, 2313–2319. [Google Scholar] [CrossRef]
- Qin, L.; Yi, W.; Lian, X.Y.; Zhang, Z. Bioactive alkaloids from the Actinomycete Actinoalloteichus sp. ZZ1866. J. Nat. Prod. 2020, 83, 2686–2695. [Google Scholar] [CrossRef]
- Perez-Bonilla, M.; Oves-Costales, D.; Gonzalez, I.; de la Cruz, M.; Martin, J.; Vicente, F.; Genilloud, O.; Reyes, F. Krisynomycins, imipenem potentiators against methicillin-resistant Staphylococcus aureus, Produced by Streptomyces canus. J. Nat. Prod. 2020, 83, 2597–2606. [Google Scholar] [CrossRef]
- Di, X.; Wang, S.; Oskarsson, J.T.; Rouger, C.; Tasdemir, D.; Hardardottir, I.; Freysdottir, J.; Wang, X.; Molinski, T.F.; Omarsdottir, S. Bromotryptamine and imidazole alkaloids with anti-inflammatory activity from the Bryozoan Flustra foliacea. J. Nat. Prod. 2020, 83, 2854–2866. [Google Scholar] [CrossRef]
- Kleks, G.; Holland, D.C.; Kennedy, E.K.; Avery, V.M.; Carroll, A.R. Antiplasmodial alkaloids from the Australian Bryozoan Amathia lamourouxi. J. Nat. Prod. 2020, 83, 3435–3444. [Google Scholar] [CrossRef]
- Yi, W.F.; Ding, X.; Chen, Y.Z.; Adelakun, T.A.; Zhang, Y.; Hao, X.J. Tabernaesines A-I, cytotoxic Aspidosperma-Aspidosperma-type bisindole alkaloids from Tabernaemontana pachysiphon. J. Nat. Prod. 2020, 83, 3215–3222. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.W.; Liu, X.J.; Yuan, J.; Li, H.J.; Mahmud, T.; Hong, M.J.; Yu, J.C.; Lan, W.J. L-Tryptophan induces a marine-derived Fusarium sp. to produce indole alkaloids with activity against the Zika virus. J. Nat. Prod. 2020, 83, 3372–3380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Hu, K.; Wang, F.; Tang, J.W.; Zhang, L.; Sun, H.D.; Cai, X.H.; Puno, P.T. 3-Hydroxy-4-methyldecanoic acid-containing cyclotetradepsipeptides from an endolichenic Beauveria sp. J. Nat. Prod. 2021, 84, 1244–1253. [Google Scholar] [CrossRef]
- Jin, P.; Zhan, G.; Zheng, G.; Liu, J.; Peng, X.; Huang, L.; Gao, B.; Yuan, X.; Yao, G. Gelstriamine A a triamino monoterpene indole alkaloid with a caged 6/5/7/6/6/5 scaffold and analgesic alkaloids from Gelsemium elegans stems. J. Nat. Prod. 2021, 84, 1326–1334. [Google Scholar] [CrossRef]
- Otogo N’Nang, E.; Le Pogam, P.; Ndong Mba, T.; Sima Obiang, C.; Mouray, E.; Grellier, P.; Kumulungui, B.; Champy, P.; Beniddir, M.A. Targeted isolation of hemitheion from Mostuea brunonis, a proposed biosynthetic intermediate of theionbrunonines. J. Nat. Prod. 2021, 84, 1409–1413. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Yeap, J.S.; Lim, S.H.; Low, Y.Y.; Sim, K.S.; Kam, T.S. The bisindole alkaloids Angustilongines M and A from Alstonia penangiana induce mitochondrial apoptosis and G0/G1 cell cycle arrest in HT-29 cells through promotion of tubulin polymerization. J. Nat. Prod. 2021, 84, 1524–1533. [Google Scholar] [CrossRef]
- Kwon, O.; Ahn, S.; Jeon, J.; Park, I.G.; Won, T.H.; Sim, C.J.; Park, H.; Oh, D.; Oh, K.; Noh, M. Psammocindoles A–C: Isolation, synthesis, and bioactivity of indole-γ-lactams from the sponge Psammocinia vermis. Org. Lett. 2021, 23, 4667–4671. [Google Scholar] [CrossRef]
- Wang, F.; Bi, J.; He, L.; Chen, J.; Zhang, Q.; Hou, X.; Xu, H. The indole alkaloids from the roots of Isatidis Radix. Fitoterapia 2021, 153, 104950. [Google Scholar] [CrossRef]
- Duan, F.F.; Liu, L.; Gao, Y.; Peng, X.G.; Meng, X.G.; Ruan, H.L. [11]-Chaetoglobosins from Pseudeurotium bakeri induce G2/M cell cycle arrest and apoptosis in human cancer cells. J. Nat. Prod. 2021, 84, 1904–1914. [Google Scholar] [CrossRef]
- Jiang, C.X.; Yu, B.; Miao, Y.M.; Ren, H.; Xu, Q.; Zhao, C.; Tian, L.L.; Yu, Z.Q.; Zhou, P.P.; Wang, X.; et al. Indole alkaloids from a soil-derived Clonostachys rosea. J. Nat. Prod. 2021, 84, 2468–2474. [Google Scholar] [CrossRef]
- Fouotsa, H.; Le Pogam, P.; Mkounga, P.; Lannang, A.M.; Bernadat, G.; Vanheuverzwijn, J.; Zhou, Z.; Leblanc, K.; Rharrabti, S.; Nkengfack, A.E.; et al. Voatriafricanines A and B, trimeric Vobasine-Aspidosperma-Aspidosperma alkaloids from Voacanga africana. J. Nat. Prod. 2021, 84, 2755–2761. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, Y.; Nakashima, Y.; Kato, H.; Hitora, Y.; Maki, K.; Inada, N.; Murakami, S.; Inazumi, T.; Ise, Y.; Sugimoto, Y.; et al. Amakusamine from a Psammocinia sp. Sponge: Isolation, synthesis, and SAR study on the inhibition of RANKL-induced formation of multinuclear osteoclasts. J. Nat. Prod. 2021, 84, 2738–2743. [Google Scholar] [CrossRef] [PubMed]
- Holland, D.C.; Prebble, D.W.; Er, S.; Hayton, J.B.; Robertson, L.P.; Avery, V.M.; Domanskyi, A.; Kiefel, M.J.; Hooper, J.N.A.; Carroll, A.R. α-Synuclein aggregation inhibitory prunolides and a dibrominated β-carboline sulfamate from the ascidian Synoicum prunum. J. Nat. Prod. 2022, 85, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Vergis, J.; Mahmud, T. EDB gene cluster-dependent indole production is responsible for the ability of Pseudomonas fluorescens NZI7 to repel grazing by Caenorhabditis elegans. J. Nat. Prod. 2022, 85, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.B.; Ai, H.L.; Duan, K.T.; Feng, T.; Liu, J.K. Ophiorrhines F and G, key biogenetic intermediates of ophiorrhine alkaloids from Ophiorrhiza japonica and their immunosuppressant activities. J. Nat. Prod. 2022, 85, 453–457. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Q.; Liu, J.; Wu, J.; Jia, H.; Liu, D.; Gu, Y.; Liu, J.; Huang, J.; Fan, A.; et al. Sclerotiamides C-H, notoamides from a marine gorgonian-derived fungus with cytotoxic activities. J. Nat. Prod. 2022, 85, 1067–1078. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Li, X.; Han, X.; Wang, Z.; Zhang, W.; Dou, B.; Lu, Z.; Li, P.; Li, G. Neopetrosins A-D and haliclorensin D, indole-C-mannopyranosides and a diamine alkaloid isolated from the south china sea marine sponge Neopetrosia chaliniformis. J. Nat. Prod. 2022, 85, 1626–1633. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Li, L.; Li, Y.Q.; Luo, J.H.; Li, W.; Li, L.F.; Zheng, C.J.; Cao, F. Oxalierpenes A and B, unusual indole-diterpenoid derivatives with antiviral activity from a marine-derived strain of the fungus Penicillium oxalicum. J. Nat. Prod. 2022, 85, 1880–1885. [Google Scholar] [CrossRef]
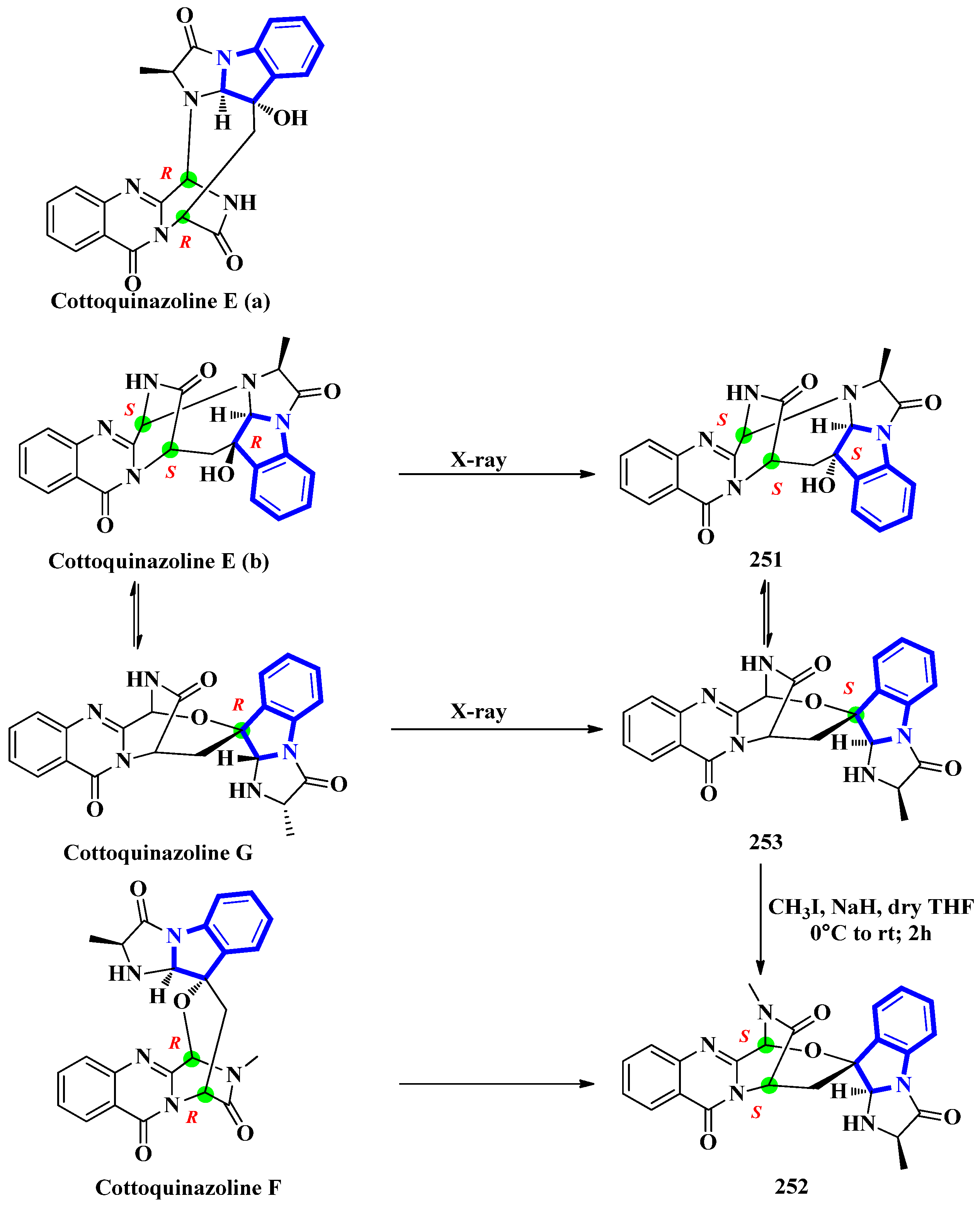
- Shan, W.G.; Wang, S.L.; Lang, H.Y.; Chen, S.M.; Ying, Y.M.; Zhan, Z.J. Cottoquinazolines E and F from Neosartorya fischeri NRRL 181. Helv. Chim. Acta 2015, 98, 552–556. [Google Scholar] [CrossRef]
- Lin, S.; Yu, H.; Yang, B.; Li, F.; Chen, X.; Li, H.; Zhang, S.; Wang, J.; Hu, Y.; Hu, Z.; et al. Reisolation and configurational reinvestigation of cottoquinazolines E-G from an arthropod-derived strain of the fungus Neosartorya fischeri. J. Nat. Prod. 2020, 83, 169–173. [Google Scholar] [CrossRef]
- Ovenden, S.P.; Capon, R.J. Echinosulfonic acids A-C and echinosulfone A: Novel bromoindole sulfonic acids and a sulfone from a southern australian marine sponge, echinodictyum. J. Nat. Prod. 1999, 62, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Rubnov, S.; Chevallier, C.; Thoison, O.; Debitus, C.; Laprevote, O.; Guénard, D.; Sévenet, T. Echinosulfonic acid D: An ESI MS n evaluation of a new cytotoxic alkaloid from the new-Caledonian sponge Psammoclemma sp. Nat. Prod. Res. 2005, 19, 75–79. [Google Scholar] [CrossRef] [PubMed]
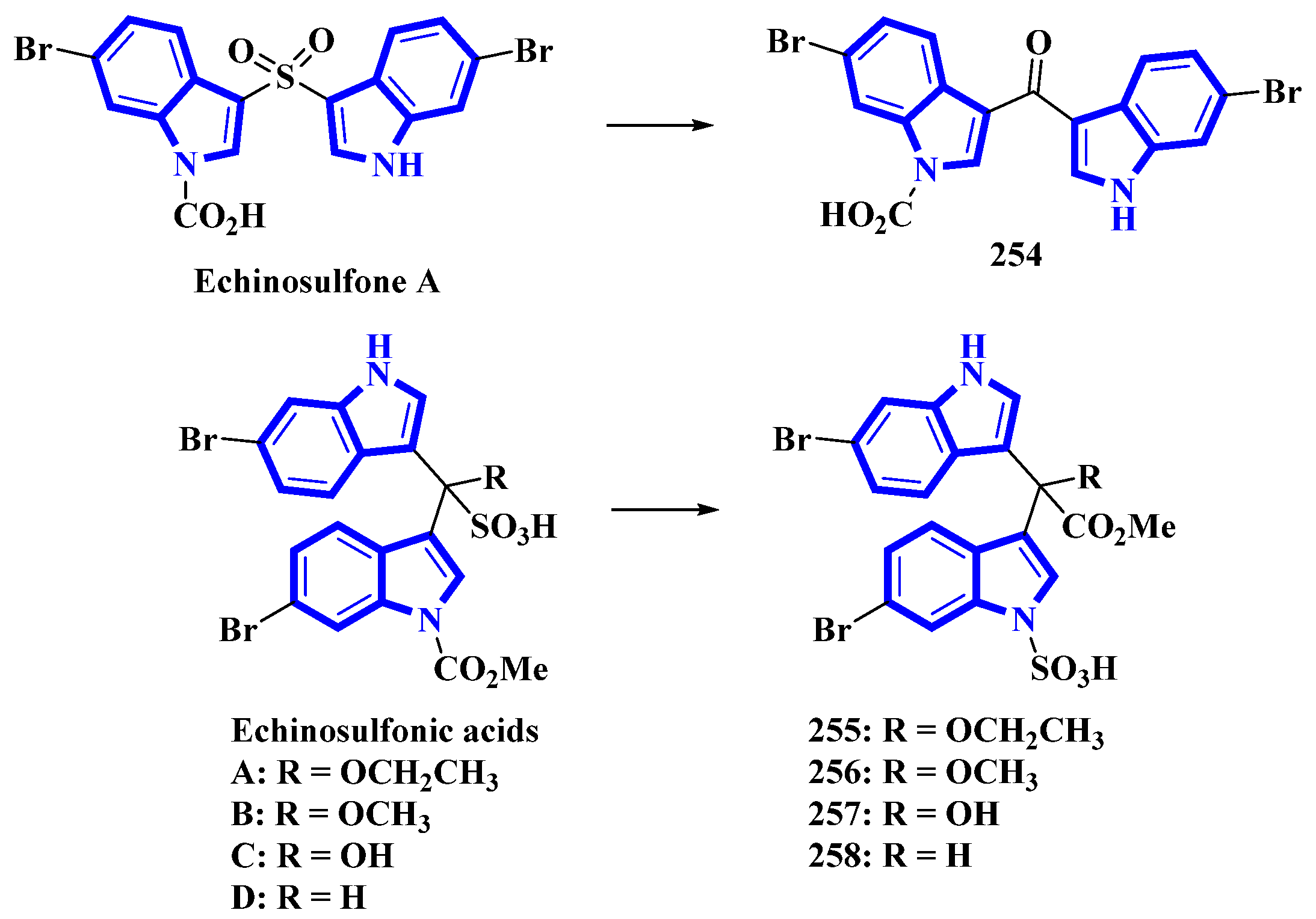
- Sala, S.; Nealon, G.L.; Sobolev, A.N.; Fromont, J.; Gomez, O.; Flematti, G.R. Structure reassignment of echinosulfone A and the echinosulfonic acids A-D supported by single-crystal X-ray diffraction and density functional theory analysis. J. Nat. Prod. 2020, 83, 105–110. [Google Scholar] [CrossRef]
- Matsunaga, S.; Jimbo, M.; Gill, M.B.; Lash-Van Wyhe, L.L.; Murata, M.; Nonomura, K.I.; Swanson, G.T.; Sakai, R. Isolation, amino acid sequence and biological activities of novel long-chain polyamine-associated peptide toxins from the sponge Axinyssa aculeata. ChemBioChem 2011, 12, 2191–2200. [Google Scholar] [CrossRef]
- Irie, R.; Miyako, K.; Matsunaga, S.; Sakai, R.; Oikawa, M. Structure revision of protoaculeine B, a post-translationally modified N-terminal residue in the peptide toxin aculeine B. J. Nat. Prod. 2021, 84, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
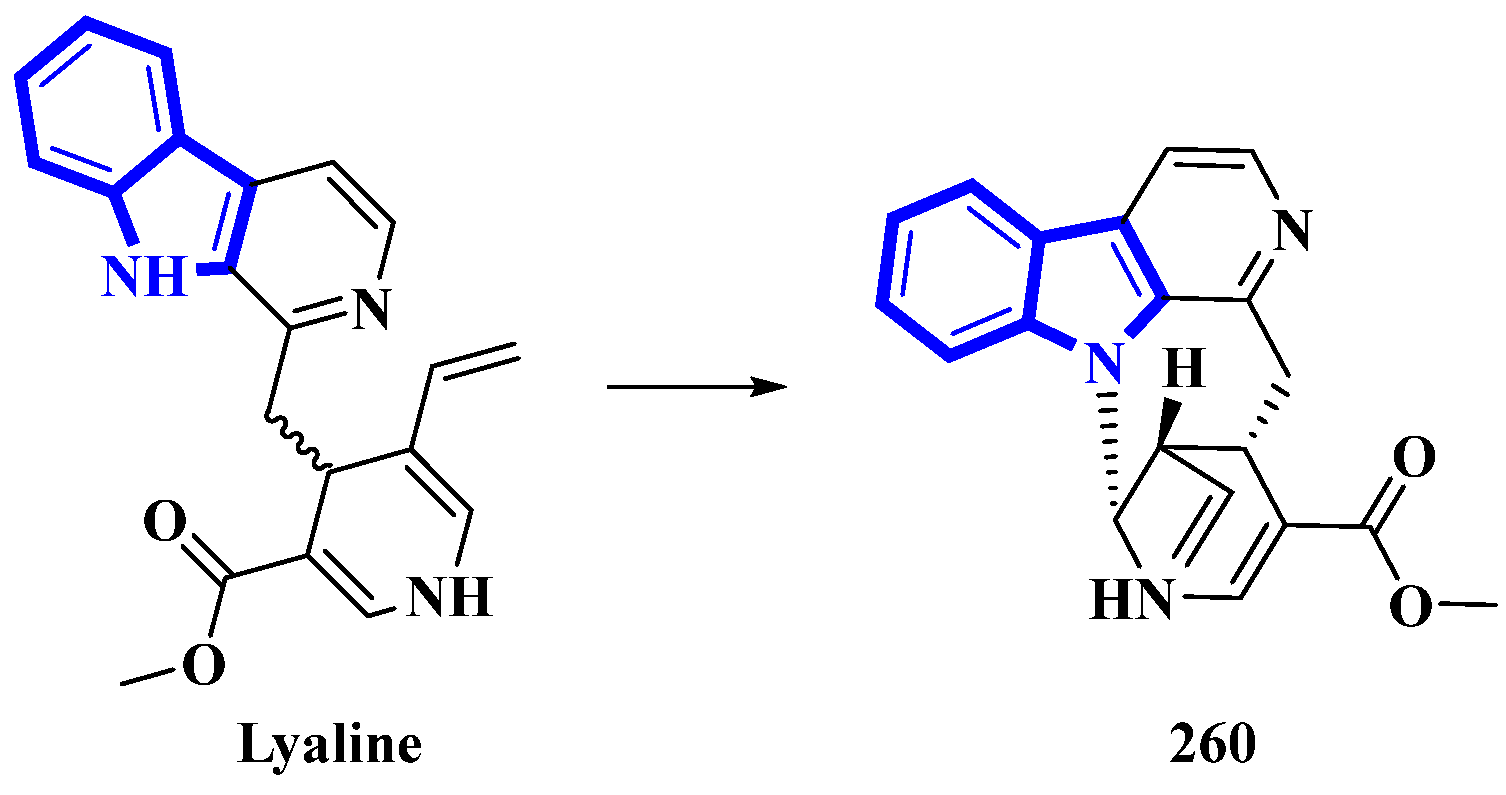
- Levesque, J.; Pousset, J.; Cavé, A. Lyaline and lyadine-new alkaloids from Pauridiantha-lyalli brem(rubiaceae). Comptes Rendus Hebd. Seances L Acad. Des Sci. Ser. C 1974, 278, 959–961. [Google Scholar]
- Bennasar, M.L.; Roca, T.; Monerris, M. Total synthesis of the proposed structures of indole alkaloids lyaline and lyadine. J. Org. Chem. 2004, 69, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Jagora, A.; Gallard, J.F.; Beniddir, M.A.; Le Pogam, P. A reappraisal of the structure of lyaline as the first naturally occurring nacycline monoterpene indole alkaloid. J. Nat. Prod. 2021, 84, 2617–2622. [Google Scholar] [CrossRef]
- Kam, T.S.; Pang, H.S.; Choo, Y.M.; Komiyama, K. Biologically active ibogan and vallesamine derivatives from Tabernaemontana divaricata. Chem. Biodivers. 2004, 1, 646–656. [Google Scholar] [CrossRef]
- Tarselli, M.A.; Raehal, K.M.; Brasher, A.K.; Streicher, J.M.; Groer, C.E.; Cameron, M.D.; Bohn, L.M.; Micalizio, G.C. Synthesis of conolidine, a potent non-opioid analgesic for tonic and persistent pain. Nat. Chem. 2011, 3, 449–453. [Google Scholar] [CrossRef]
- Chen, G.; Wang, C.; Zou, L.; Zhu, J.; Li, Y.; Qi, C. Six-step total synthesis of(+/-)-conolidine. J. Nat. Prod. 2019, 82, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Genta-Jouve, G.; Zheng, Y.; Zhang, Q.; Chen, C.; Zhou, Q.; Wang, J.; Zhu, H.; Zhang, Y. Griseofamines A and B: Two indole-tetramic acid alkaloids with 6/5/6/5 and 6/5/7/5 ring systems from Penicillium griseofulvum. Org. Lett. 2018, 20, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Ma, C.; Zhang, G.; Pan, X.; Liu, Z. Asymmetric total synthesis of griseofamine B and its three stereoisomers. J. Nat. Prod. 2022, 85, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Li, Y.; Zhu, R.; Qian, J.; Liu, J.; Gao, W.; Meng, C.; Miao, J.; Xiong, B.; Qiu, X.; et al. Hydroxamic acid derivatives of β-carboline/hydroxycinnamic acid hybrids inducing apoptosis and autophagy through the PI3K/Akt/mTOR pathways. J. Nat. Prod. 2019, 82, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Botelho, J.C.; Guzman, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Xiong, M.Q.; Chen, T.; Wang, Y.X.; Zhu, X.L.; Yang, G.F. Design and synthesis of potent inhibitors of bc1 complex based on natural product neopeltolide. Bioorg. Med. Chem. Lett. 2020, 30, 127324. [Google Scholar] [CrossRef]
- Venkatesha, S.H.; Moudgil, K.D. Celastrol suppresses experimental autoimmune encephalomyelitis via MAPK/SGK1-regulated mediators of autoimmune pathology. Inflamm. Res. 2019, 68, 285–296. [Google Scholar] [CrossRef]
- He, Q.W.; Feng, J.H.; Hu, X.L.; Long, H.; Huang, X.F.; Jiang, Z.Z.; Zhang, X.Q.; Ye, W.C.; Wang, H. Synthesis and biological evaluation of celastrol derivatives as potential immunosuppressive agents. J. Nat. Prod. 2020, 83, 2578–2586. [Google Scholar] [CrossRef]
- Labriere, C.; Elumalai, V.; Staffansson, J.; Cervin, G.; Le Norcy, T.; Denardou, H.; Rehel, K.; Moodie, L.W.K.; Hellio, C.; Pavia, H.; et al. Phidianidine A and synthetic analogues as naturally inspired marine antifoulants. J. Nat. Prod. 2020, 83, 3413–3423. [Google Scholar] [CrossRef]
- Wang, L.; Mei, X.; Wang, C.; Zhu, W. Biomimetic semi-synthesis of fradcarbazole A and its analogues. Tetrahedron 2015, 71, 7990–7997. [Google Scholar] [CrossRef]
- Fu, P.; Zhuang, Y.; Wang, Y.; Liu, P.; Qi, X.; Gu, K.; Zhang, D.; Zhu, W. New indolocarbazoles from a mutant strain of the marine-derived actinomycete Streptomyces fradiae 007M135. Org. Lett. 2012, 14, 6194–6197. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, Y.; Zuo, M.; Liu, W.; Wang, L.; Zhu, W. Semisynthetic derivatives of fradcarbazole A and their cytotoxicity against acute myeloid leukemia cell lines. J. Nat. Prod. 2019, 82, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Li, Y.; Wang, Y.Y.; Cai, X.H.; Liu, Y.P.; Luo, X.D. Cytotoxic indole alkaloids from Melodinus tenuicaudatus. J. Nat. Prod. 2010, 73, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Walia, M.; Teijaro, C.N.; Gardner, A.; Tran, T.; Kang, J.; Zhao, S.; O’Connor, S.E.; Courdavault, V.; Andrade, R.B. Synthesis of(-)-melodinine K: A case study of efficiency in natural product synthesis. J. Nat. Prod. 2020, 83, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Andrade, R.B. Domino Michael/Mannich/N-alkylation route to the tetrahydrocarbazole framework of aspidosperma alkaloids: Concise total syntheses of (-)-aspidospermidine, (-)-tabersonine, and (-)-vincadifformine. J. Am. Chem. Soc. 2013, 135, 13334–13337. [Google Scholar] [CrossRef]
- Fricke, J.; Sherwood, A.M.; Halberstadt, A.L.; Kargbo, R.B.; Hoffmeister, D. Chemoenzymatic synthesis of 5-methylpsilocybin: A tryptamine with potential psychedelic activity. J. Nat. Prod. 2021, 84, 1403–1408. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, M.A. Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [Google Scholar] [CrossRef]
- Yue, J.; Lopez, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Wang, F.; Ma, H.; Liu, Z.; Huang, W.; Xu, X.; Zhang, X. α-Mangostin inhibits DMBA/TPA-induced skin cancer through inhibiting inflammation and promoting autophagy and apoptosis by regulating PI3K/Akt/mTOR signaling pathway in mice. Biomed. Pharmacother. 2017, 92, 672–680. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Zhang, R.; Wu, Q.-Y.; Song, Y.-J.; Wang, Y.-X.; Yang, J.-F.; Yang, G.-F. Natural product neopeltolide as a cytochrome bc 1 complex inhibitor: Mechanism of action and structural modification. J. Agric. Food Chem. 2019, 67, 2774–2781. [Google Scholar] [CrossRef]



























| Compound Serial | Source | Isolation Method | Reference |
|---|---|---|---|
| 1 to 10 | Tolypocladium | Extracted with DCM/CH3OH (1:1); residue suspended in water and extracted with EtOAc | [20] |
| 11 to 14 | Didemnidae, Eudistoma, Pseudodistoma | Extracted with CH3OH, followed by DCM | [21] |
| 15 to 18 | Tabernaemontana corymbosa | Extracted with EtOH, concentrated, and partitioned with 3% tartaric acid | [22] |
| 19 to 23 | Aphanoascus fulvescens | Extracted with EtOAc | [23] |
| 24 to 29 | Penicillium | Extracted with EtOAc; residue extracted with hexane/CH3OH (90%) | [24] |
| 30 to 31 | Streptomyces sp. PU-10A | Extracted with CH3OH; mixed with 3% (w/v) XAD-16 resin, filtered, washed with water, and extracted with CH3OH | [25] |
| 32 to 36 | Streptomyces noursei | Extracted with CH3OH; the aq. layer was freeze-dried | [26] |
| 37 to 39 | Kopsia arborea | Extracted with CH3OH, concentrated, and partitioned with 3% tartaric acid | [27] |
| 40 to 45 | Chaetomium globosum | Extracted with EtOAc | [28] |
| 46 to 50 | Alstonia balansae | Basified with NH4OH (6 M) and extracted with DCM; extracted using HCl (0.1 M); basified with NH4OH (pH = 10) and extracted with DCM | [29] |
| 51 to 54 | Actinomycete AJS-327 | Extracted with EtOAc | [30] |
| 55 to 62 | Aspergillus versicolor | Extracted with 95% EtOH; residue suspended in water and extracted with EtOAc (1:1 v/v) | [31] |
| 63 | Inflatella | Extracted with DCM: CH3OH (1:1), followed by CH3OH: H2O (1:1) | [32] |
| 64 to 66 | Nauclea latifolia | Extracted with CH3OH; residue suspended in 5% H2SO4 and extracted with DCM; solution neutralized with NH3 and extracted with DCM | [33] |
| 67 to70 | Penicillium | Extracted with EtOAc | [34] |
| 71 to 77 | Alstonia penangiana | Extracted with CH3OH, concentrated, and partitioned with 3% tartaric acid | [35] |
| 78 to 79 | Uncaria longiflora var. pteropoda | Extracted with CH3OH, followed by trituration using hexane, CHCl3, EtOAc, and CH3OH; CHCl3 extract was acidified (5% HCl) and filtered; the filtrate was basified with 37% NH4OH | [36] |
| 80 to 88 | Uncaria rhynchophylla | Extracted with 95% EtOH and 75% EtOH; residue suspended in water and extracted with DCM and n-BuOH | [37] |
| 89 to 98 | Uncaria rhynchophylla | Extracted with 95% EtOH, 75% EtOH, and 50% EtOH; residue dissolved in 4% HCl and filtered; filtrate basified with NH3. H2O to pH 9–10 and partitioned sequentially with CHCl3 and EtOAc; remaining alkaline soln. was neutralized with HCl and subjected to microporous resin HP-20 | [38] |
| 99 to 105 | Aspergillus | Extracted with EtOAc | [39] |
| 106 to 108 | Penicillium solitum | Extracted with EtOAc; residue dissolved in H2O:CH3OH (5:95; v/v) and partitioned with hexane; aq. fraction was extracted by a 1:1:1 mixture of XAD 2, 4, and 7; water-soluble organic material was desorbed with CH3OH and CH3OH/acetone (1:1; v/v) | [40] |
| 109 | Epicoccum nigrum | Supernatant extracted with EtOAc; mycelia were macerated and extracted with acetone | [41] |
| 110 to 114 | Picralima nitida | Extracted with EtOH | [42] |
| 115 to 123 | Chaetomium cochliodes | Extracted with DCM/CH3OH (1:1); residue dispersed in water and extracted with DCM and EtOAc | [43] |
| 124 to 126 | Scedosporium apiospermum | Extracted with CH3OH | [44] |
| 127 to 136 | Psychotria nemorosa | Extracted with CH3OH; suspended in 1 M HCl and partitioned with DCM; aq. phase was basified using NH4OH (pH = 9.5) and partitioned with DCM | [45] |
| 137 | Penicillium raistrickii | Extracted with EtOAc | [46] |
| 138 to 141 | Phaeosphaeria fuckelii | Extracted with EtOAc | [47] |
| 142 to 150 | Tabernaemontana corymbosa | Extracted with CH3OH; solution acidified with saturated tartaric acid (pH = 2–3) and extracted with EtOAc; aq. phase basified using saturated NaOH and extracted with DCM | [48] |
| 151 to 154 | Mitragyna speciosa, Havil. Rubiaceae | Extracted with CHCl3:CH3OH (1:1) and 10% KOH by maceration; residue dissolved in 1 M HCl: hexane (1:1); aq. phase basified using conc. NH4OH (pH = 9) and extracted with CHCl3 | [49] |
| 155 to 159 | Melodinus hemsleyanus | Extracted with 95% EtOH; suspended in water and acidified using 5% HCl (pH = 2–3); partitioned with CHCl3; aq. phase basified using 10% NH3 soln. (pH = 9–10) and extracted with CHCl3 | [50] |
| 160 to 172 | Actinoalloteichus | Extracted with EtOAc | [51] |
| 173 to 174 | Streptomyces canus | Extracted with acetone | [52] |
| 175 to 185 | Flustra foliacea | Extracted with CH3OH, DCM; residue suspended in water and partitioned with EtOAc; organic fraction suspended in 90% CH3OH and partitioned with hexane; CH3OH fraction suspended in 60% CH3OH and partitioned with DCM | [53] |
| 186 | Amathia lamourouxi | Extracted with CH3OH | [54] |
| 187 to 195 | Tabernaemontana pachysiphon | Extracted with CH3OH, and acidified using saturated tartaric acid (pH = 2–3); extracted with EtOAc; aq. phase basified using saturated NaOH (pH = 9–10) and extracted with DCM | [55] |
| 196 to 200 | Fusarium | Extracted with EtOAc | [56] |
| 201 to 204 | Beauveria | Extracted with EtOAc | [57] |
| 205 to 209 | Gelsemium elegans | Extracted with 95% EtOH; suspended in water, acidified using 0.3 M HCl (pH = 3), and partitioned with CHCl3; aq. phase basified using NH3 soln. (pH = 10) and extracted with CHCl3; aq. extract partitioned with n-BuOH | [58] |
| 210 | Mostueabrunonis | Alkalinized with NH4OH (25%) and extracted with EtOAc; extracted with 1% HCl, and basified using NH4OH (pH = 10); extracted with EtOAc | [59] |
| 211 | Alstonia penangiana | Extracted with CH3OH, concentrated, and partitioned with 3% tartaric acid | [60] |
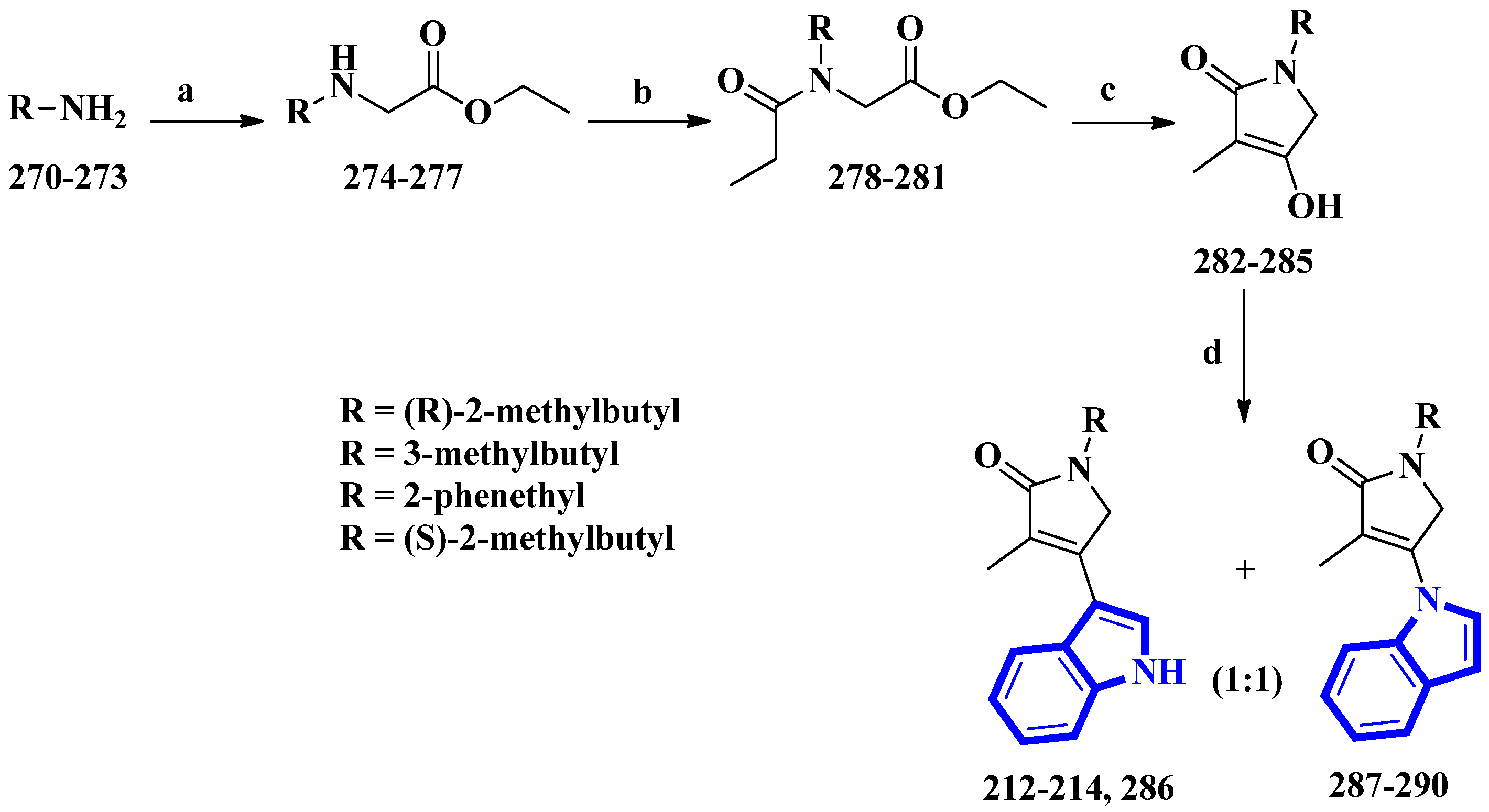
| 212 to 214 | Psammocinia vermis | Macerated and extracted with CH3OH and DCM; partitioned between H2O and n-BuOH; latter fraction was repartitioned between H2O-CH3OH (15:85) and hexane | [61] |
| 215 to 216 | Isatis indigotica | Submerged in 2.5% ammonia for 6 h and extracted with 80% and 70% ethanol; concentrated and extracted with ethyl acetate | [62] |
| 217 to 228 | Pseudeurotium bakeri | Extracted with EtOAc | [63] |
| 229 to 230 | Clonostachys rosea | Macerated culture extracted with acetone/DCM/CH3OH; residue partitioned with petroleum ether, EtOAc, and n-BuOH | [64] |
| 231 to 232 | Voacanga africana | Extracted with CH3OH; suspended in CH3OH: H2O (10%) and basified using NH3 (pH = 12); extracted with DCM | [65] |
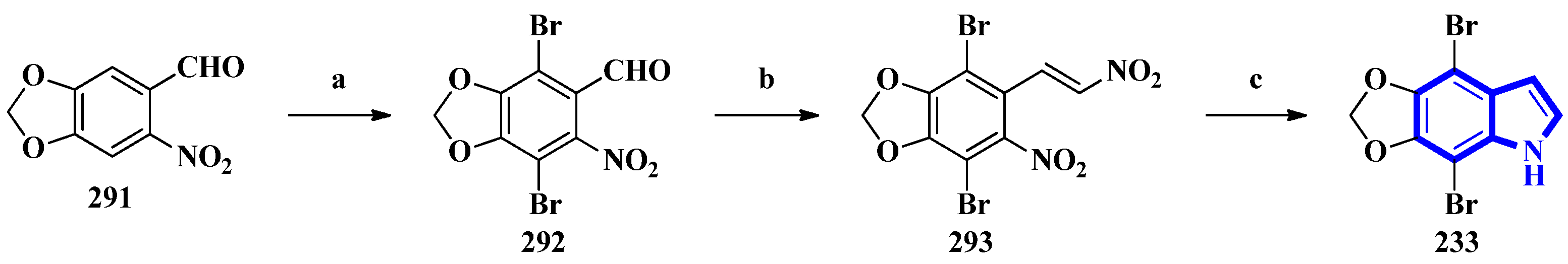
| 233 | Psammocinia | Extracted with EtOH and CH3OH; residual solution extracted with EtOH; fraction partitioned between hexane and 90% CH3OH: H2O | [66] |
| 234 | Synoicum prunum | Extracted with CH3OH | [67] |
| 235 to 236 | Escherichia coli | Extracted with EtOAc | [68] |
| 237 to 238 | Ophiorrhiza japonica | Extracted with 90% CH3OH; residue dissolved in 0.5% aq. HCl and extracted with EtOAc; aq. phase basified using 10% NH4OH (pH = 8–9) and extracted with EtOAc | [69] |
| 239 to 244 | Aspergillus sclerotiorum | Extracted with EtOAc; residue dissolved in CH3OH: H2O (9:1) and partitioned with cyclohexane | [70] |
| 245 to 248 | Neopetrosia chaliniformis | Extracted with CH3OH | [71] |
| 249 to 250 | Penicillium oxalicum | Extracted with EtOAc | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umer, S.M.; Solangi, M.; Khan, K.M.; Saleem, R.S.Z. Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities. Molecules 2022, 27, 7586. https://doi.org/10.3390/molecules27217586
Umer SM, Solangi M, Khan KM, Saleem RSZ. Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities. Molecules. 2022; 27(21):7586. https://doi.org/10.3390/molecules27217586
Chicago/Turabian StyleUmer, Syed Muhammad, Mehwish Solangi, Khalid Mohammed Khan, and Rahman Shah Zaib Saleem. 2022. "Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities" Molecules 27, no. 21: 7586. https://doi.org/10.3390/molecules27217586
APA StyleUmer, S. M., Solangi, M., Khan, K. M., & Saleem, R. S. Z. (2022). Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities. Molecules, 27(21), 7586. https://doi.org/10.3390/molecules27217586