2.1. Syntheses
The synthesis of hybrid POMs can be achieved through different strategies. In the present study, the best synthetic procedure to get the targeted hybrid POMs has been to react first the lacunary POMs “SiW
10” and “P
2W
17” with two aminopropyltri(ethoxy)silane molecules (APTES) to give the two POM-APTES precursors (see
Figure 2) of formulas (TBA)
3H[(SiW
10O
36)(Si(CH
2)
3NH
2)
2O]·3H
2O (denoted hereafter
SiW10-APTES) and (TBA)
5H[P
2W
17O
61(Si(CH
2)
3NH
2)
2O]·6H
2O, denoted hereafter
P2W17-APTES. The syntheses of these two precursors were adapted from Mayer at al. [
28] by reaction of k
8(γ-SiW
10O
36)·12H
2O or K
10α–P
2W
17O
61·20H
2O with 3-aminopropyltriethoxy silane in presence of TBABr in H
2O/CH
3CN medium acidified by concentrated HCl (for more details see experimental section in
Supplementary Materials). Note that for each, the proton usually written as counter-cation is in fact probably an ammonium arm R-NH
3+.
The synthetic strategy to get POM-borate adducts is then to combine the amines of these POM-APTES precursors with the reactive carbonyl of the decaborate cluster [B
10H
9CO]
− (
Figure 1C) to give an amide function connecting both components. Since the boron cluster can react with water for giving a carboxylic acid and since heating the synthetic mixture above 40–50 °C led to some degradation products or to some reduction in the Dawson derivative by the hydrodecaborate cluster, reactions have been conducted at room temperature and under nitrogen atmosphere. Furthermore, the coupling reaction needs the presence of a base both to help the deprotonation of the ammonium arm(s) of the POM-APTES precursors and to trap the proton produced by the coupling reaction. A moderate and a bulky organic base, diisopropylethylamine (DIPEA), was thus used to avoid the competition with APTES for the coupling reaction with [B
10H
9CO]
−.
To quickly circumscribe the optimal conditions for the synthesis of the POM-borate adducts,
29Si,
31P and
1H NMR titrations were conducted by varying the ratios of the three reactants [B
10H
9CO]
−/POM-APTES/DIPEA (all details are given in the
Supplementary Materials).
For the [B
10H
9CO]
−/
SiW10-APTES/DIPEA system, the
29Si NMR studies in solution reveal that it is possible to modulate the coupling reaction between [B
10H
9CO]
− and POM-APTES precursors by playing on the amounts of DIPEA and of [B
10H
9CO]
−. For this tri-reactants system, the successive formation of two POM-borate species identified as mono- and di-adduct compounds was demonstrated thanks to their molecular symmetries (C
s versus C
2v). Besides, the crucial role of DIPEA in the reaction of [B
10H
9CO]
− with POM-APTES precursors was clearly evidenced. No reaction occurs when no base is used. NMR titration studies allowed establishing that the optimal quantity of base was two equivalents for one equivalent of [B
10H
9CO]
−. The
Figure 2 shows for instance the proportions of SiW
10-derivatives determined by the integration of the different peaks obtained by
29Si NMR in the system
SiW10-APTES/[B
10H
9CO]
−/DIPEA as a function of [B
10H
9CO]
−/
SiW10-APTES ratio at fixed DIPEA/[B
10H
9CO]
− ratio of 2.
Starting from
SiW10-APTES, it evidences first the formation of a mono-adduct, which predominates for ration B
10/
SiW10-APTES = 1, before being converted into a di-adduct. The NMR titrations studies allowed establishing that using proportions
SiW10-APTES/B
10H
9CO/DIPEA = 1/3/6 lead to the pure di-adduct denoted
SiW10-diB10, while using 1/1/2 ratios lead to around 80% of mono-adduct mixed with some unreacted starting POM and the di-adduct. The separation of compounds has not been possible but considering the effect of the added DIPEA amounts, we succeeded to reduce the formation of the di-adduct and thus to get the mono-adduct compound denoted
SiW10-monoB10 with a good purity by decreasing the quantity of DIPEA in the proportions
SiW10-APTES/B
10H
9CO/DIPEA = 1/1/1.5. The
Figure 3 summarizes the experimental conditions used to isolate POM-borate adducts.
Similar NMR studies were also performed in solution with the Dawson derivative
P2W17-APTES (see
Supplementary Materials). In contrast to SiW
10 derivatives, the formation of mono- and di-adduct of the Dawson derivative are not so separated as for SiW
10. Therefore, we failed to isolate the mono-adduct as pure product. Nevertheless, we can obtain quantitatively the di-adduct compound in the reaction mixture when ratios
P2W17-APTES/B
10H
9CO/DIPEA = 1/3/6 are used.
To summarize, the multistep coupling reactions have successfully been monitored by
29Si and
31P NMR, fully described in the
Supplementary Materials, revealing that intermediate products can be followed and isolated. From these results, we established the experimental conditions allowing to selectively synthesize with good yields the mono adduct of SiW
10 POM and the di-adducts of both POMs as mixed TBA
+ and DIPEAH
+ salts, namely (TBA)
3(DIPEAH)
3[(SiW
10O
36)(B
10H
9CONHC
3H
6Si)(NH
2C
3H
6Si)O]·3H
2O denoted
SiW10-monoB10, (TBA)
6.5(DIPEAH)
1.5[(SiW
10O
36)(B
10H
9CONHC
3H
6Si)
2O]·2H
2O denoted
SiW10-diB10, and (TBA)
6(DIPEAH)
4[(P
2W
17O
61)(B
10H
9CONHC
3H
6Si)
2O]·3H
2O, denoted
P2W17-diB10 (See Experimental Section in
Supplementary Materials for more details). All adducts were isolated as powders and were characterized by FT-IR, TGA, elemental analysis, MALDI-TOF and NMR techniques. It should be noted that to our knowledge,
SiW10-monoB10 is the first example of a POM-APTES monoadduct isolated so far from the direct synthesis. All studies in the literature usually reported di-adducts with such types of hybrid POMs [
29,
30,
31].
2.3. Characterizations by MALDI-TOF Mass Spectrometry
Mass spectrometry (MS) is a very efficient technique for the characterization of polyoxometalates in solution. In our case, we did not succeed in getting mass spectra with reasonable signal-to-noise ratio and exploitable data by the usual electrospray ESI-MS technique. On the contrary, Matrix-Assisted Laser Desorption/Ionization coupled to a Time-of-Flight mass spectrometer (MALDI-TOF) revealed to be an effective technique for hybrid POMs characterization, as shown for example by Mayer and coworkers on “SiW
10” and “P
2W
17“ organosilyl derivatives [
28,
32]. MALDI-TOF technique is applied on samples which are diluted in a matrix solution (DCTB in our case, DCTB = Trans-2-[3-(4-ter-Butylphenyl)-2-propenylidene] malonitrile) and then co-crystallized on a conductive target. Thanks to a laser irradiation, it allows producing singly charged species (cationic or anionic) and presenting the great advantage to strongly limit the number of peaks in comparison with ESI-MS spectra, where multiply charged species are generated. In the present study, the experiments were performed in both negative and positive modes (see
Figure S14 in the Supplementary Materials for the example of SiW
10-diB
10). According to previous works in this field, the best results were obtained in the positive mode, although the anionic character of the POM [
28,
32]. Indeed, as seen in the
Supplementary Materials for SiW
10-diB
10, the intensity reached in the negative mode appears lower, but the number of peaks is higher as there are more degradation species. Even thought our systems are polyanionic, they are more efficiently analyzed as monocationic species resulting from adducts between POMs and counter cations such as TBA
+ and H
+ in our case (H
+ coming notably from DIPEAH
+ cations or protonated amines). Furthermore, the monocationic character of the species is confirmed in all cases by the shift between peaks in the isotopic massifs.
The precursor
SiW10-APTES and the compounds
SiW10-monoB10,
SiW10-diB10 and
P2W17-diB10, were thus analyzed by this technique in the positive mode. The results are gathered in
Table S1 (see Supplementary Materials). The full spectra and a zoom on the target compounds with a spectrum simulated with IsoPro3 software are shown in
Figure 4 for SiW
10 derivatives and in
Figures S15 and S16 for P
2W
17 ones.
As shown in
Figure 4 the spectrum of the precursor
SiW10-APTES (
Figure 4a) displays a major peak centered at m/z 4087.3 and a minor peak at
m/
z 4328.3. The first peak is assigned to the monocationic species {(TBA)
3H
2[(SiW
10O
36)O(SiC
3H
6NH
2)
2](CH
3CN)
2(H
2O)
8(DCTB)
2}
+ (calculated
m/
z 4087.3), while the second peak is attributed to the species {(TBA)
4H[(SiW
10O
36)O(SiC
3H
6NH
2)
2](CH
3CN)
2(H
2O)
8(DCTB)
2}
+ (calculated
m/
z 4328.7). The two peaks correspond to the expected hybrid POM associated with some TBA
+ and H
+ cations, some solvates and two molecules of the DCTB matrix. Note that the presence of amines on the APTES part of the POM could probably favor the formation of intermolecular interactions with solvates and DCTB molecules. Such an adduct with DCTB is also observed with the precursor
P2W17-APTES (
Figure S15, Supplementary Materials) but not seen with the other POMs functionalized with B
10 clusters. The MALDI-TOF spectrum of
P2W17-APTES indeed exhibits a major peak corresponding to the expected precursor associated with one molecule of the DCTB matrix at
m/
z 6058.7 (calculated
m/
z 6057.9 for (TBA)
6H[(P
2W
17O
61)O(SiC
3H
6NH
2)
2](DCTB)}
+) and a minor peak at m/z 6300.0 (calculated
m/
z 6299.4 for (TBA)
7[(P
2W
17O
61)O(SiC
3H
6NH
2)
2](DCTB)}
+). The attribution of the peaks is definitely confirmed thanks to the fitting of the isotopic distribution massifs. The latter are mainly due to the isotopic distribution of the 10 or 17 tungsten atoms of the POMs, which appears consistent with the experimental spectrum (see
Figure 4a and
Figure S15, respectively).
The spectrum of
SiW10-monoB10 depicted in
Figure 4b shows only one experimental peak at
m/
z 3843.2 which is perfectly consistent with the calculated mass for the monocationic product {(TBA)
4H
3[(SiW
10O
36)O(SiC
3H
6NH
2)(SiC
3H
6NHCOB
10H
9)](CH
3CN)(H
2O)
3}
+ (calculated
m/
z 3843.1). It evidences the formation of the expected adduct
SiW10-monoB10 and thus indirectly the grafting of one (B
10H
9CO)
− cluster to SiW
10-APTES. The simulated spectrum agrees well with the experimental data, which supports this assumption although the presence of one B
10 cluster does not modify significantly the isotopic massif.
The MALDI-TOF spectrum of
SiW10-diB10 shown in
Figure 4c displays a major peak centered at
m/
z 4085.8, which fully agrees with the expected di-grafted compound {(TBA)
4H
5[(SiW
10O
36)O(SiC
3H
6NHCOB
10H
9)
2](CH
3CN)
2(H
2O)
6}
+ (
m/
z calculated 4084.4) and a minor peak at
m/
z = 4330.2 consistent with the species {(TBA)
5H
4[(SiW
10O
36)O(SiC
3H
6NHCOB
10H
9)
2](CH
3CN)
3(H
2O)
4}
+ (
m/
z calculated 4330.8). This result confirms the formation of the expected di-grafted compound.
Finally, the case of
P2W17-diB10, appears more complicated, certainly due to a higher charge of the hybrid POM (10-) and a larger surface, which both favor intermolecular interactions with solvent molecules and cations. For technical reasons, the MALDI-TOF spectrum shown in
Figure S16 (see Supplementary Materials) was recorded in linear mode, which does not favor the high resolution in contrast with other compounds. The spectrum displays an intense and broad experimental peak centered at
m/
z 6048.1, while four smaller peaks are found, respectively, at
m/
z 6289.6, 6431.7, 6672.5 and 6813.8. All these peaks are consistent with di-grafted species of general formula {(TBA)
xH
y[(P
2W
17O
61)O(SiC
3H
6NHCOB
10H
9)
2](CH
3CN)
z(H
2O)
t}
+ (x + y = 11, z = 0–3 and t = 5–6). Regarding the main peak, the latter appears much broader than expected for only one species. Moreover, the resolution of the isotopic massif is lost. In fact, the experimental spectrum likely corresponds to a spectra superimposition of monocationic species of general formula {(TBA)
5H
6[(P
2W
17O
61)O(SiC
3H
6NHCOB
10H
9)
2](CH
3CN)
x(H
2O)
y}
+ with x ranging from 1 to 5 and y from 0 to 8 (
m/
z in the range 6035.70 to 6076.75). Some simulated spectra are given in
Figure S16 in Supplementary Materials.
2.4. NMR Studies in Solution
In the absence of crystallographic data, the three obtained hybrid systems have been thoroughly characterized by multinuclear NMR spectroscopy in order to verify their structures in solution.
1H,
11B,
13C,
15N,
29Si,
31P, and
183W NMR spectra were recorded in CD
3CN at room temperature. The data are gathered in
Table S2 (Supplementary Materials), while selected spectra are given in
Figure 5 and
Figure 6 and in
Figures S17–S32 (Supplementary Materials).
As shown in
Figure 5a and in
Figures S17–S20 (Supplementary Materials),
11B{
1H} NMR spectrum of [B
10H
9CO]
− undergoes a significant change upon coupling with
SiW10-APTES or
P2W17-APTES. In particular, the signal at −44.4 ppm specific for the equatorial boron atom bearing the substituent CO in [B
10H
9CO]
− (B2 atom, see
Figure 1c) is strongly shifted to ca. −25 ppm in the spectra of
SiW10-monoB10,
SiW10-diB10 and
P2W17-APTES in agreement with the grafting of the cluster on the POM.
Concomitantly, the
1H NMR spectrum of the mono adduct
SiW10-monoB10, exhibits a splitting of the signals for the three methylene groups –CH
2- of the APTES linker, denoted a, b, c (see
Figure 5b), because of the lowering of the symmetry of
SiW10-APTES from C
2v to C
s. In addition, a new peak at 6.12 ppm assigned to an amide function is observed. For the remaining amine function, a broad signal is observed at 7.4 ppm (d), but together with two other broad signals at 5.70 and 6.33 ppm (d’ and d’’), attributed to the amine function in a frozen configuration in which the interaction with B
10 cluster generates two inequivalent protons as depicted in
Figure 7a (DFT optimized structure). These assumptions are confirmed by
1H-
15N HMBC (Heteronuclear Multiple Bond Correlation) NMR spectrum (
Figure S25) revealing two
15N signals at −251 ppm (amide) correlated to the proton signal at 6.12 ppm and at −272 ppm (free amine) correlated to the two protons peaks at 5.70 and 6.33 ppm.
Grafting a second [B
10H
9CO]
− group on the
SiW10-APTES platform allows recovering the C
2v symmetry and thus one set of peaks was observed for the linker and especially the protons “a”, in addition to an amide peak at 5.94 ppm (
Figure S24, Supplementary Materials). The signals of the amine at 7.4, 5.7, and 6.3 ppm disappear in agreement with the reaction of [B
10H
9CO]
− groups with this function. Similarly, the
1H NMR spectrum of
P2W17-diB10 compared to that of
P2W17-APTES (
Figure S27, Supplementary Materials) evidences the appearance of a sharp peak at 5.94 ppm assigned to an amide function, while the signal of the free amine at 7.03 ppm in the precursor P
2W
17-APTES disappears.
To further confirm our assignments of the signal of the amide function,
1H-
1H ROESY (Rotating frame Overhause Effect SpectroscopY) and
13C NMR experiments were performed on
SiW10-diB10 and
P2W17-diB10 (
Figures S28–S32, Supplementary Materials). Cross REO peaks involving the amide proton (5.94 ppm in both compounds) and some equatorial B-H protons of the B
10 cluster at 0.4 ppm and the protons “c” of the APTES chains can be seen in
Figures S28 and S29 (Supplementary Materials). This demonstrates the spatial proximity between these protons that interact between each other through dipolar contacts.
13C NMR spectra of
SiW10-monoB10,
SiW10-diB10, and P
2W
17-diB
10 (
Figures S30–S32) notably exhibits a signal at 203 ppm assigned to a carbon atom from an amide group, which is confirmed by 2D
1H-
13C HMBC NMR spectrum of
SiW10-monoB10 evidencing a correlation between this
13C signal at 203 ppm and the
1H amide signal at 6.12 ppm. Besides, in both cases of
SiW10-monoB10 and
SiW10-diB10 this
13C signal appears as a poorly resolved quadruplet with a coupling constant of 95 Hz consistent with a
1J
13C-11B coupling.
Therefore, 1H, 1H-15N HMBC, 1H-1H ROESY, 13C and 1H-13C HMBC NMR experiments unambiguously confirm the formation of an amide group in our three adducts by reaction of the amines of POM-APTES precursors with the carbonyl group of the cluster [B10H9CO]−. The modification of the 11B{1H} NMR spectra of the boron cluster after its reaction with the POM-APTES precursors further confirms such results.
29Si,
31P and
183W NMR probe the POM part in compounds
SiW10-APTES,
SiW10-monoB10, SiW10-diB10, P2W17-APTES and
P2W17-diB10 (see
Figure 6 and
Figures S21–S23 in Supplementary Materials). The unsymmetrical environment in the mono adduct
SiW10-monoB10 is clearly confirmed through the appearance of two peaks for Si of the different linker arms at −61.9 and −63.3 ppm, while only one signal was observed at −62.3 ppm for the symmetrical di adduct
SiW10-diB10 with only a small shift from the initial SiW
10-APTES precursor (−62.5 ppm). In addition, for all the compounds, a single peak is observed for the Si atom in the central cavity of the SiW
10 POM moiety which is almost not affected by the grafting of the boron clusters and the resulting changes of symmetry of the adducts (
Figure S21, Supplementary Materials). In case of
P2W17-APTES and
P2W17-diB10, both compounds exhibit only one signal assigned to the two equivalent Si atoms of the APTES linker (
Figure S22, Supplementary Materials).
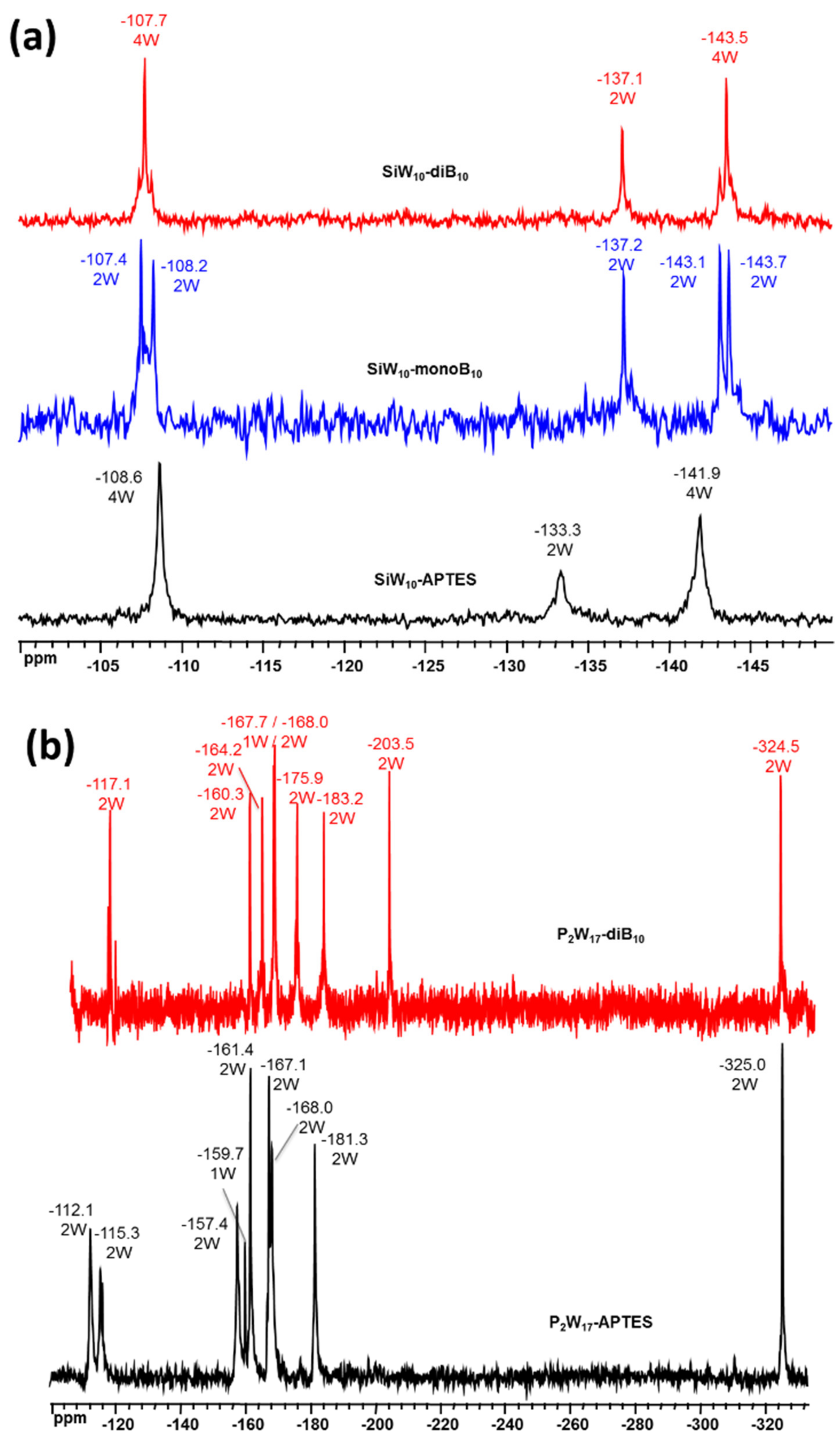
The
183W NMR spectra of precursors and adducts are given in
Figure 6. For
SiW10-monoB10 the
183W NMR spectrum displays five peaks of intensities 2:2:2:2:2 in agreement with the expected low C
s symmetry, while three resonances are observed for the di adduct
SiW10-diB10 and the initial precursor
SiW10-APTES of intensities (4:2:4) consistent with their C
2v symmetry (
Figure 6a).
Figure 6b shows the
183W NMR spectrum of P
2W
17-diB
10 which differs significantly from its precursor. Both compounds exhibit nine NMR lines of integration 2:2:2:1:2:2:2:2:2 in agreement with the expected C
s symmetry, but their positions are slightly changed. This is due to the modification of the P
2W
17 moiety induced by the grafting of the two [B
10H
9CO]
− clusters. Additionally,
31P NMR spectra of
P2W17-APTES and
P2W17-diB10 display two signals (
Figure S23, Supplementary Materials), wherein one of them showed a common chemical shift, while the second exhibited a small shift from −13.4 ppm in
P2W17-APTES to −13.6 ppm in
P2W17-diB10.In conclusion, these experiments focused on the POM part fully agree in terms of molecular symmetries with the formation of the expected mono- or di-adducts with B10 clusters.
2.5. Computational Studies
The molecular geometries of
SiW10-APTES,
SiW10-monoB10, and
SiW10-diB10, as well as those of P
2W
17-APTES, P
2W
17-monoB
10 and P
2W
17-diB
10 were fully optimized at a DFT level including implicit solvent effects (see
Figure 7 and
Supplementary Materials for computational details). We considered the most relevant plausible conformers. Firstly, regarding
SiW10-APTES, it exhibits two main conformers as defined by the orientation of the two amine organic arms, which we called them open and closed forms. The small difference in their relative energy, less than 1 kcal·mol
−1 in favor of the closed form (represented in
Figure 1a), forecasted that further substitution would easily overcome any initial geometric preference in the reactants. Indeed, upon B
10 incorporation a much more complex situation arises. For
SiW10-monoB10 we characterized five conformers, two arising from the closed reactant and three species from the open reactant form. In the most stable conformer (
Figure 7a), which arise from the closed form, the decaborate moiety interacts favorably with the amine hydrogens (d’ and d’’) of the unreacted arm through strong dihydrogen contacts. In the most stable open form (
Figure 7b), although interaction between arms is almost neglected, the H amide atom develops other interactions. Overall, the most stable conformer given in
Figure 7a is 11 kcal·mol
−1 below the second one (
Figure 7b). All five conformers lie in a narrow 20 kcal·mol
−1 range. For the double substituted
SiW10-diB10, since the additional repulsion arising from the negatively charged B
10 groups, we could only characterize two forms: an open (not shown) and a closed one (two views on
Figure 7c,d). The energy difference between both species was computed to be just only 5 kcal·mol
−1. We highlight, as dashed lines in
Figure 7, those hydrogen atoms of the organic arm and the B
10 moiety that lie close in three-dimensional space.
Due to the monovacant character of P
2W
17, the Si-O-Si angle of the APTES moiety grafted to the POM strongly differs from that observed for the divacant SiW
10 POM (see
Figure 1). The topology of the two arms, and thus the connectivity of the two POM-APTES derivatives, strongly differs. Therefore, as seen in
Figure 1, closed form is not possible for P
2W
17-APTES. Only one geometry could thus be considered. Then, for the mono-substituted P
2W
17-monoB
10, two conformers were characterized, one open and one folded, the folded one being more stable by only 2.2 kcal·mol
−1. For the di-substituted Dawson derivative, only one open shaped product could be characterized (see
Figure 7e,f).
Hydrogen atoms of the decaborate moieties possess a hydride character. Consequently, they can establish hydrogen-hydrogen contacts with protic solvent or with functional groups like amines or amides. In the present structures, many H-H dihydrogen contacts between the amine organic arms from APTES moiety and hydrogen atoms from decaborate clusters were observed. For instance, for
SiW10-monoB10 (
Figure 7a), the hydrogen atoms d’ and d’’ from the «free» amino group are found 2.21 and 1.99 Å far from an H atom belonging to the B
10 cluster, which are quite short distances. This fact agrees with the couplings observed in the NMR experiments.
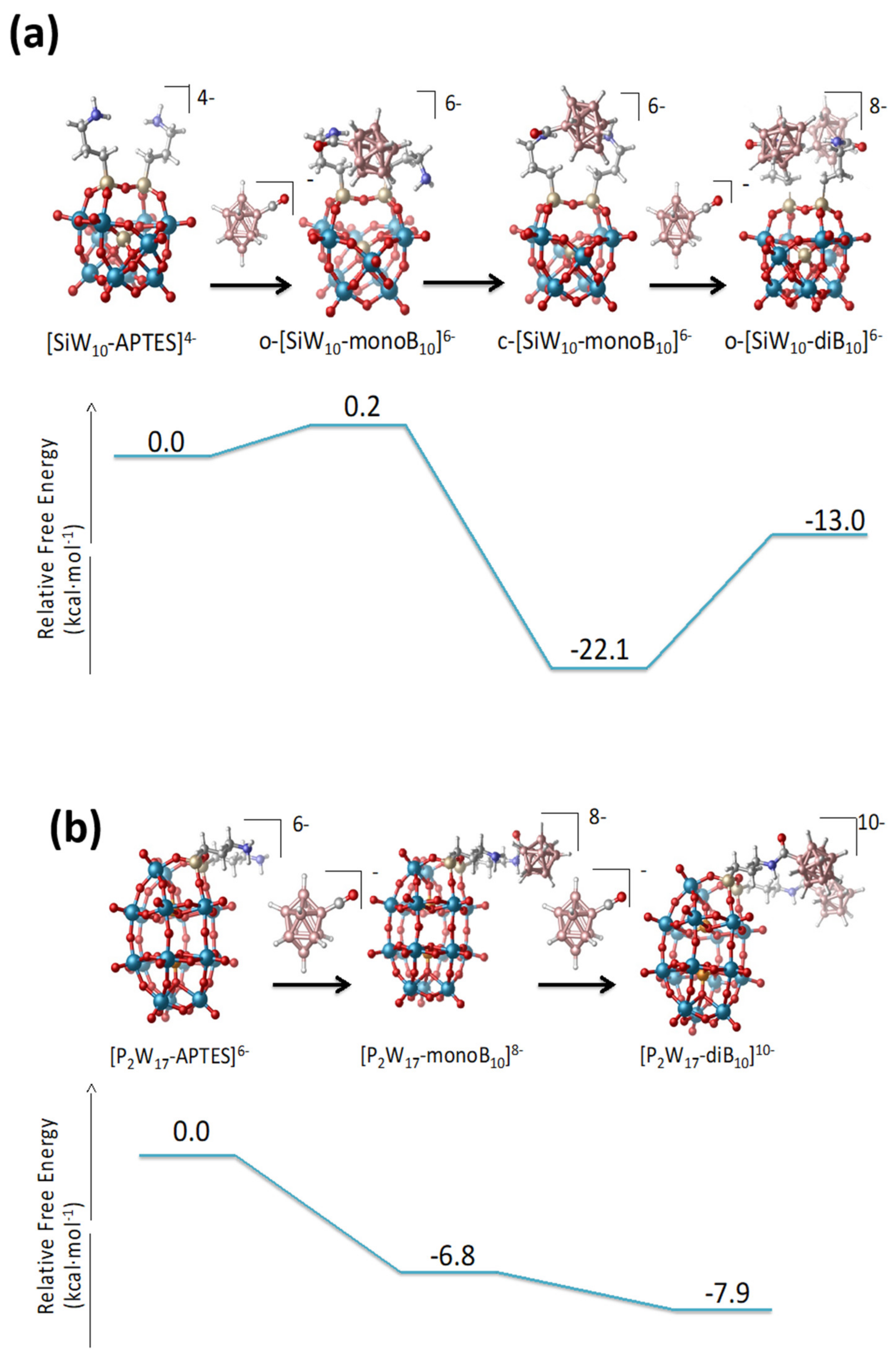
The thermodynamics of the formation of the mono- and di-adducts of the Keggin and the Dawson species is computed exergonic in all cases as seen in
Figure 8. For
SiW10-APTES, both the formation of the mono- and bi-derivative were computed exergonic, 22.1 kcal·mol
−1 for the
SiW10-monoB10, and 13.0 kcal·mol
−1 for the
SiW10-diB10. The formation of
SiW10-monoB10 is clearly favored and the strong dihydrogen contacts between amino group and the grafted B
10 cluster undoubtedly strongly stabilize such a species compared to the di-adduct
SiW10-diB10. For P
2W
17-APTES, also the two substitutions are favorable, 6.8 kcal·mol
−1 for the first, and 7.9 kcal·mol
−1 for the second.
The computed reaction free energies for the
SiW10-APTES and P
2W
17-APTES derivatives are fully consistent with the experimental findings. For the keggin derivatives, the strong stabilization of the mono-adduct allows isolating both mono and di-adduct thanks to the formation of strong H-H contacts. In contrast, for the Dawson derivatives, the small difference of energies between mono- and bi-adducts (1.1 eV only) does not permit isolating the mono-adduct. Besides, an excess of [B
10H
9CO]
− (3 equivalents/POM instead of 2) is needed to get the pure di-adduct compound to avoid the formation of a mixture between mono and di-grafted adducts. A similar situation was previously obtained with Anderson-type derivatives since the mono and di-adduct of B
10 with [MnMo
6(Tris)
2]
3− are only separated by 6 eV and it was not possible to get mono-adduct [
22]. This result highlights the role of the topology of the POM-APTES compounds and their faculty to stabilize species thanks to intramolecular interactions.
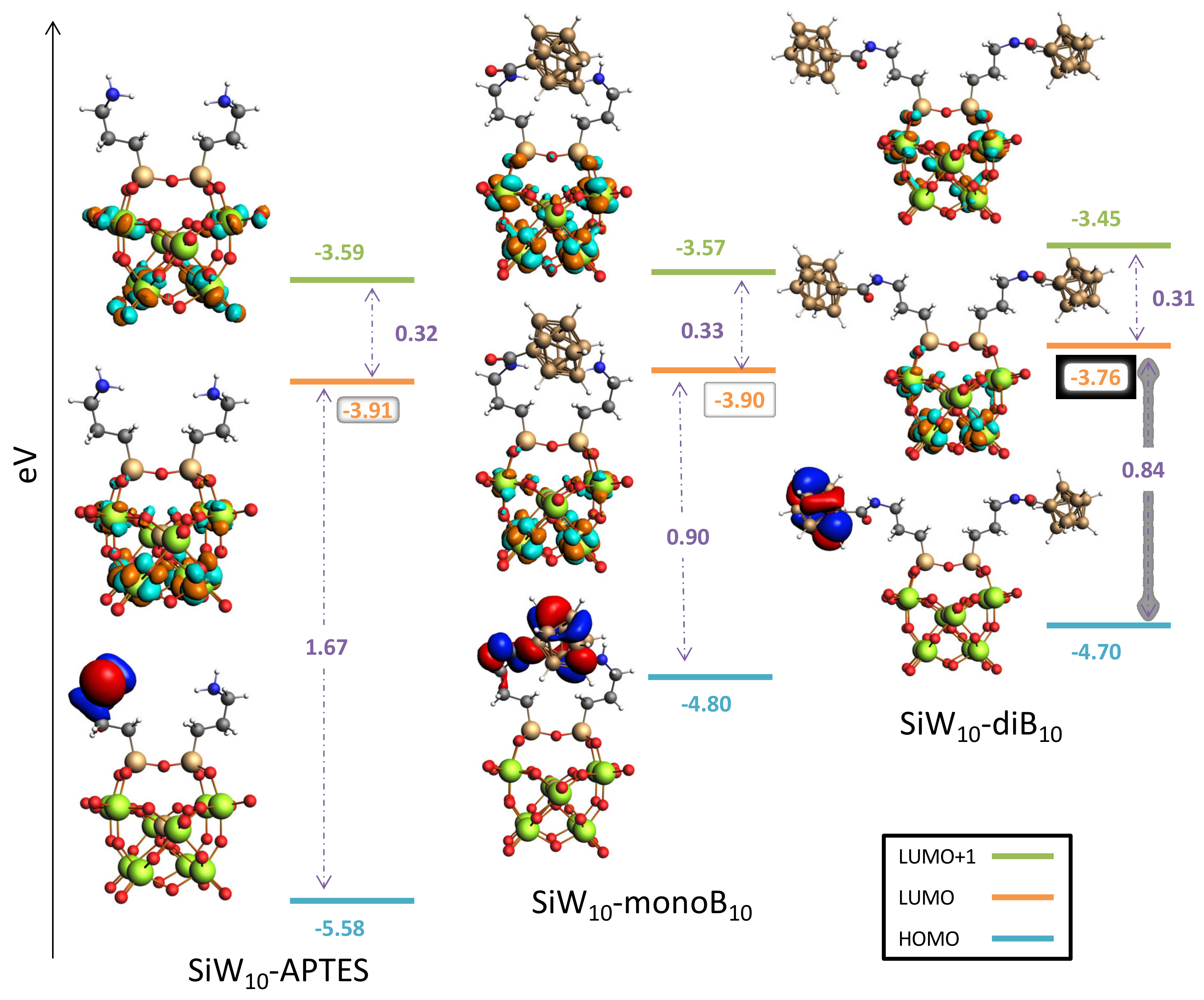
Finally, DFT studies provided the frontier orbitals for each compound (see
Figure 9 and
Figure S33 in Supplementary Materials). The results obtained for Keggin and Dawson derivatives exhibit the same feature. For POM-APTES the HOMO is located on one (for
SiW10-APTES) or two (
P2W17-APTES) amines of the APTES part, while the LUMO are localized on the W atoms of the POM part. By grafting the B
10 clusters, the LUMO levels are slightly affected. LUMO remains localized on W atoms and only minor changes in energy are observed.
Conversely, the HOMO levels are drastically modified by the introduction of B10 clusters. Electrons of the HOMO orbitals are now mainly localized on one grafted B10 cluster. Interestingly, for SiW10-monoB10 the HOMO is delocalized between one B10 cluster and the amine of the second arm, which strongly interacts with the B10 through H-H contacts. The HOMO energy level increases in all cases within the range 0.78 to 0.92 eV. The HOMO-LUMO gaps, are thus significantly reduced upon the B10 grafting. Indeed, for SiW10-APTES, the gap decreases from 1.67 to 0.90 eV for the first substitution, and to 0.94 eV for the second. For P2W17-APTES, the gap evolves from 1.56 to 0.77 eV for the first substitution, and to 0.76 eV for the second.
2.6. Electrochemical Properties
The electronic spectra of compounds
SiW10-monoB10,
SiW10-diB10 and
P2W17-diB10 recorded in CH
3CN containing 0.1 mol.L
−1 TBAClO
4 (TBAP, tetrabutylammonium perchlorate) at room temperature and 2.10
−4 mol.L
−1 concentration are depicted in
Figures S35 and S36 (Supplementary Materials).
The electronic spectra of precursors
SiW10-APTES and
P2W17-APTES display absorption bands in ultraviolet region corresponding to transition between p-orbitals of the oxo ligands and d-type orbitals centered on tungsten [
33,
34], while the cluster [B
10H
9CO]
− exhibits weak absorption band between 300 and 200 nm notably assigned to π−π* transitions [
35]. Considering that the main contribution of the spectra comes from the LMCT band involving the W atoms and that the LUMO band centered on tungsten atoms are only slightly modified upon grafting of B
10 cluster, no drastic changes are expected in the POM-B
10 adducts. Indeed, the electronic spectra of
SiW10-monoB10 and
P2W17-diB10 match well with the sum of the spectra of
SiW10-APTES or
P2W17-APTES and one or two times that of [B
10H
9CO]
−, respectively. The spectrum of
SiW10-diB10 slightly differs from the sum of the component’s spectra probably due to a larger variation of LUMO energy level from
SiW10-APTES to
SiW10-diB10 and additional constraints due to the vicinity of the two boron clusters.
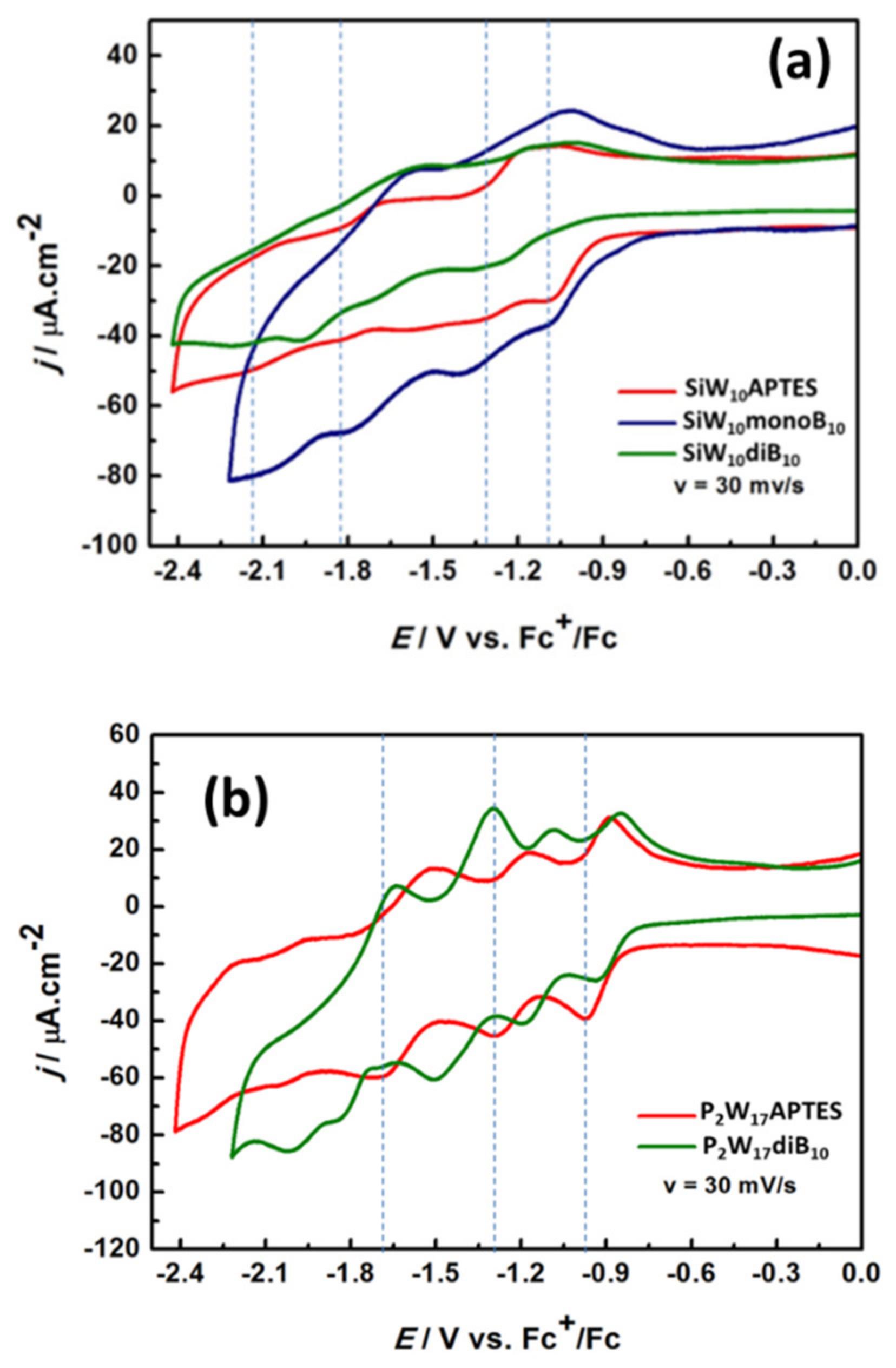
As depicted in
Figure 10a and
Figure S37, the CV of
SiW10-APTES is poorly resolved and it is difficult to identify confidently all the reduction processes corresponding to the successive reduction in W
VI centers into W
V, well-known to be monoelectronic in non-aqueous solvents [
36]. These waves seem nevertheless reversible with processes better resolved in oxidation. Besides, an irreversible process attributed to the oxidation of amine function of the APTES linker is also observed in oxidation around +0.452 V vs. Fc
+/Fc (see
Supplementary Materials). As for their parent precursor, CVs of
SiW10-monoB10 and
SiW10-diB10 display poorly resolved reversible electronic transfers, which appear shifted towards more negative potentials and one irreversible oxidation process around +0.452 V vs. Fc
+/Fc assigned to the oxidation of the remaining amine function and/or of the B
10 cluster (see
Figure S39, Supplementary Materials). This behavior agrees with the increase in the charge from 4- in
SiW10-APTES to 6- in
SiW10-monoB10 and 8- in
SiW10-diB10 and the electron donating character of the boron cluster [
37] but does not evidence a strong electronic effect of the boron cluster on the POMs electronic properties.
The CVs of
P2W17-APTES and
P2W17-diB10 are given in
Figure 10b and in
Figure S38 (Supplementary Materials) and appears much more resolved than those of SiW
10 derivatives. The CV of
P2W17-APTES displays four reversible electronic transfers with cathodic peak potentials assigned to successive mono- or bi-electronic reductions of W
VI centers into W(+V) [
38] and two irreversible oxidation processes at E
pa = +0.452 and +0.759 V, assigned to the oxidation of the terminal amine groups of the APTES linkers. Conversely to the di-adduct compound
SiW10-diB10, the CVs of the Dawson derivative
P2W17-diB10 give four reversible reduction processes significantly shifted towards the more positive potential compared to
P2W17-APTES and one irreversible reduction process at E
pc = −2.030 V vs. Fc
+/Fc, which was not observed in the precursor. The opposite effect was expected. This effect probably results from a combination of a charge effect, the presence of protons (in DIPEAH+ cations) and of an electronic effect of boron cluster on P
2W
17 moiety but at this stage it is difficult to have a clear explanation of the contribution of all these effects which can be antagonist.
Although reduction waves in the Dawson derivatives are not very well resolved, it can be observed that the di-substituted species (green line in
Figure 10) is reduced at lower potentials than
P2W17-APTES, in agreement with the fact that the LUMO and LUMO+1 raise in energy upon B
10 attachment. Also, the successive reduction waves seem just shifted left, which would conform with the almost constant difference in the LUMO and LUMO+1 energies along the series.
2.7. Electrocatalytic Properties for the Reduction in Protons into Hydrogen (HER)
Many POMs are known to catalyze protons reduction into hydrogen in aqueous or in non-aqueous conditions [
7,
39,
40]. We verified by UV-Vis spectroscopy that [B
10H
9CO]
− and its adducts with POMs are stable in CH
3CN in the presence of excess acetic acid (20 equivalents). In these conditions, it was interesting to study the reactivity of these compounds in regard to the electro-catalytic reduction in protons into hydrogen. The experiments were performed in CH
3CN + 0.1 M TBAP by using acetic acid as a source of protons, and as a weak acid in such a medium (pKa = 22.3) [
41].
Figure 11 and
Figure S40 (Supplementary Materials) show the evolution of CVs upon stepwise addition of acetic acid up to 20 equivalents of acid/POM for all P
2W
17 and SiW
10 derivatives, respectively. For all the compounds, the addition of acetic acid, gives a new irreversible reduction wave, which grows gradually with the amount of acid, expressed as γ = [acid]/[POM]. As shown in
Figure 11 and
Figure S40, at a given potential of −2.2 V vs Fc
+/Fc, a linear dependence of the catalytic current versus γ is obtained, a behavior featuring the electro-catalytic reduction of protons. However, the effect of the addition of acetic acid in the solution appears stronger for P
2W
17 derivatives than for SiW
10 based compounds.
To evidence the electrocatalytic process, linear voltammetry of
P2W17-diB10 in the presence of 20 equivalents of acetic acid was performed and compared to similar experiments performed without catalyst or on platinum electrode (
Figure 11d). We notice that in the presence of the catalyst
P2W17-diB10, the current density is almost doubled and there is a 250-mV overvoltage decrease compared to the solution without catalyst. Indeed, the proton reduction with respect to platinum starts at −1.400 V vs. Fc+/Fc, while it starts at −1.750 V vs. Fc
+/Fc with catalyst and at −2.000 V vs. Fc
+/Fc without catalyst. Finally, the formation of hydrogen is unambiguously demonstrated by gas chromatography analysis during electrolysis performed at −2.200 V vs Fc+/Fc during 4.5 h (
Figure S40, Supplementary Materials).
To compare the efficiency of all compounds, the catalytic efficiency (CAT) can be estimated using Equation (1):
Table 1 summarizes the
CAT values measured for our products at −2.2 V vs Fc+/Fc. Interestingly, the two precursors
SiW10-APTES and
P2W17-APTES exhibit similar efficiency. Also, the efficiency of
SiW10-monoB10 and
SiW10-diB10 adducts are lower than that of
SiW10-APTES, while it is the opposite for
P2W17-diB10, which appears much more efficient than its parent precursor, in agreement with cyclic voltammetry experiments. Indeed, a less negative reduction potential of the POM part should facilitate the electro-catalytic reduction in protons.
In terms of mechanism, three key steps have to be considered: the protonation, the reduction in the catalyst and the transfer of electron towards the protons to give dihydrogen. For protonation step, since catalysis is observed in all compounds, it must occur on the most basic sites, either on the oxo groups of the POM moiety, on the free amine groups in
SiW10-APTES and
P2W17-APTES or on boron clusters for
SiW10-diB10 and
P2W17-diB10. DFT calculations evidence that the most nucleophilic sites are found on the oxo ligands of the POM parts which are consequently the preferential sites for protonation (see
Figure 12 and
Figure S34 in Supplementary Materials).
For the reduction step, as seen in
Figure S41c,d in Supplementary Materials, during electrolysis, the P
2W
17 derivatives turned to blue as expected for the reduction in such species before returning back colorless when the current is stopped indicating that the reduced POM probably transfers electrons to protons to produce hydrogen. We understand well that if this reduction occurs at higher potential, it should favor the process.
P2W17-diB10 is thus logically the most efficient compound.
To sum up, even if the decaborate cluster is probably not directly involved in the HER process, it plays two indirect roles: (1) the covalent grafting on POMs increases the electronic density on the POM which should facilitate the protonation step, and (2) the covalent grafting can modifies the reduction potential of the POM moieties in POM-borate adducts, which favors the reduction step of the POM species when shifted towards more positive potentials as observed in P2W17-diB10.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}