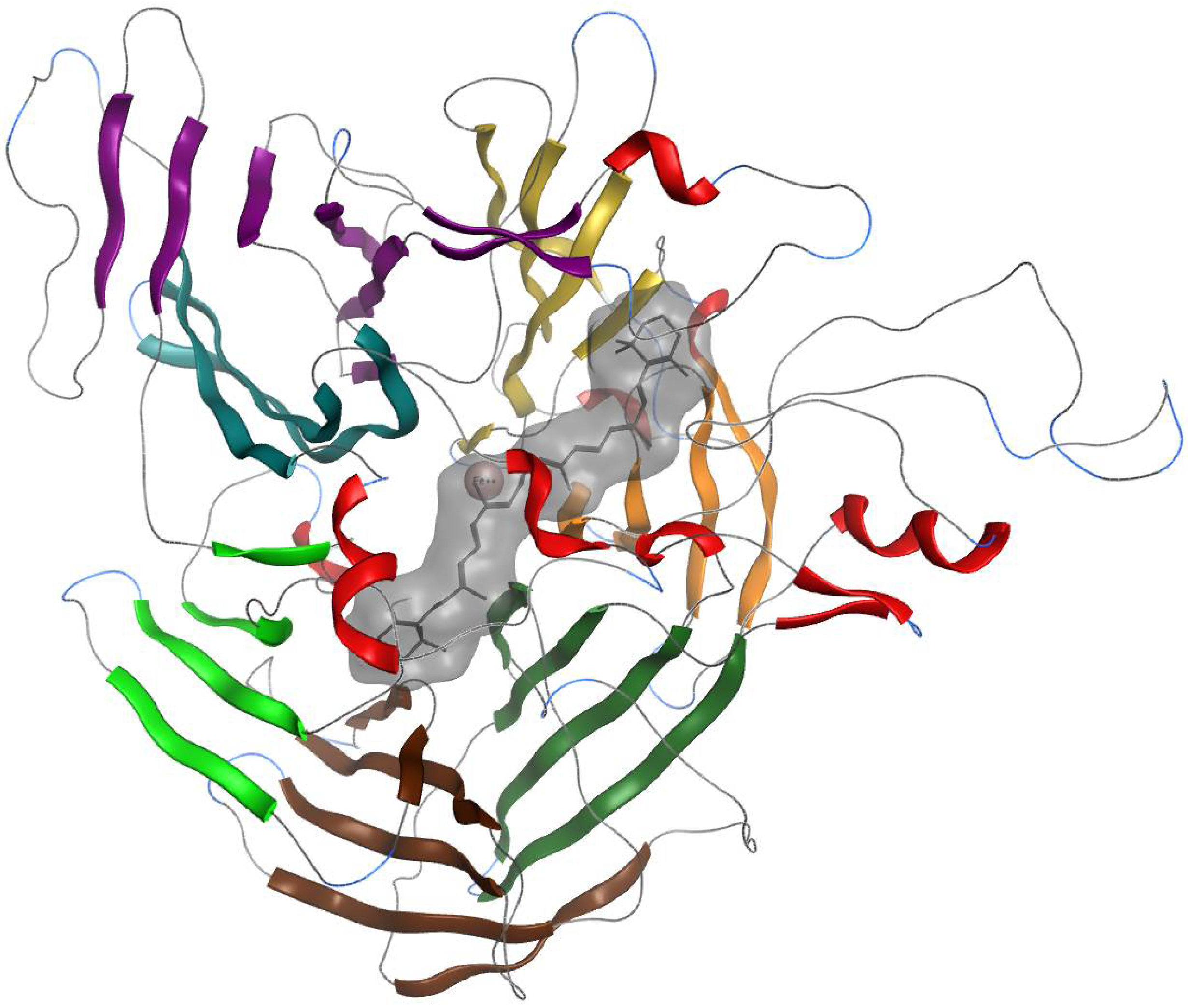
2.5. Interaction of BCO1 with Other Carotenoids
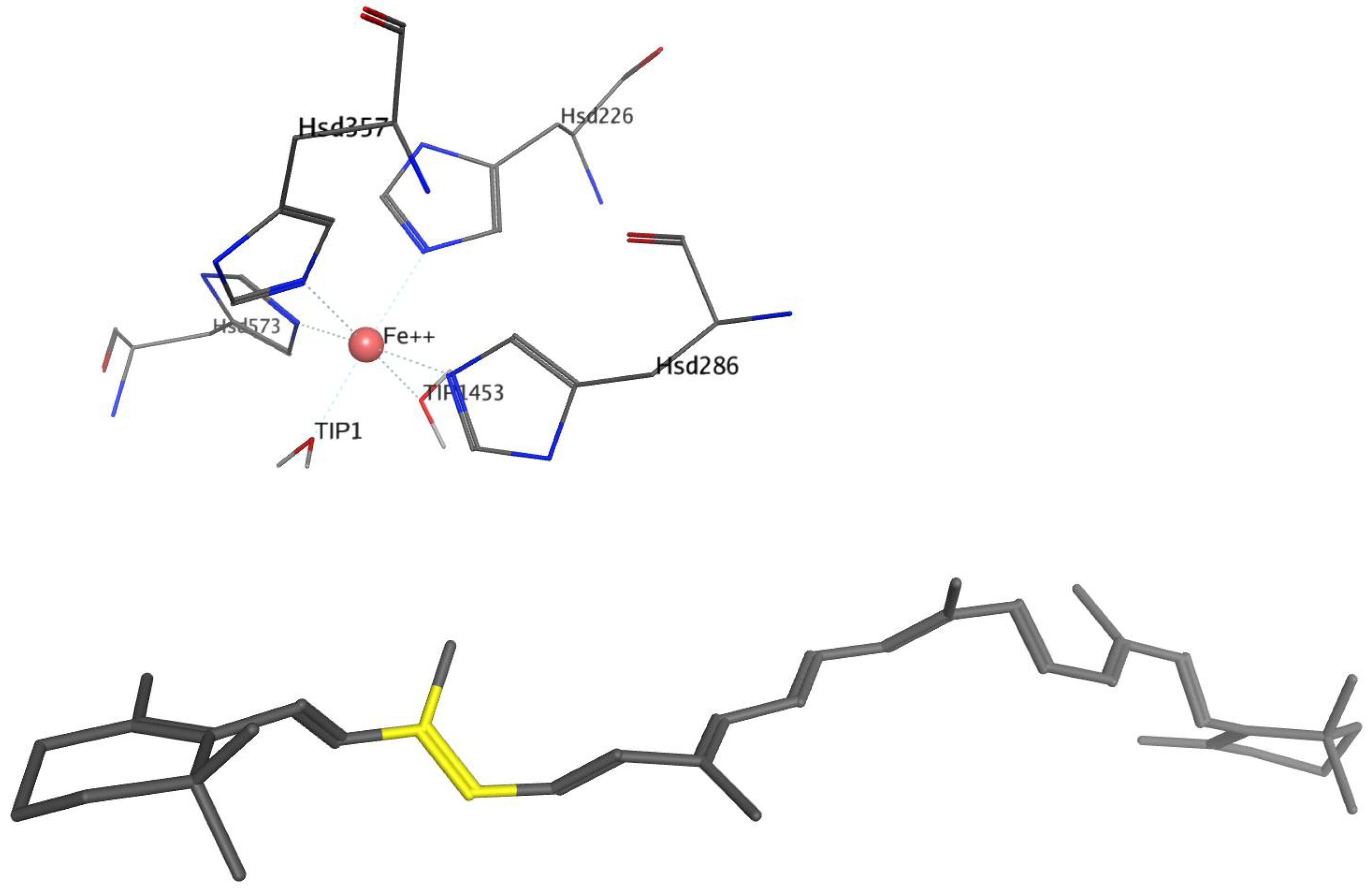
After 100 ns of simulation of the BCO1 complex with β-carotene, trajectories of 1 ns of molecular dynamics per ligand were performed. For all models subjected to 1 ns molecular dynamics, we observed that the overall structure of the enzyme–ligand complex remains the same as in the complex with β-carotene. The coordination sphere with four histidine residues and two water molecules of the catalytic center is conserved. The 15 and 15′ atoms of the carotenoids remain the closest to Fe2+ and TIP2035 water.
Table 4 describes the potential interaction energy of the different carotenoids that underwent analysis with the rest of the system (protein and solvent).
Table 5 shows the fingerprint analysis (PLIF) for the interaction of all possible ligands with the protein residues and the water molecules with which they interact at the different times of production, for 1 ns of simulation.
All carotenoids interact with Phe51, with a high interaction percentage for the linear carotenoids (15
Z)-phytofluene and (15
Z)-phytoene. Phe51 is the residue with the highest interaction percentage for the reference molecule β-carotene. For β-carotene, other residues with a high interaction percentage are Trp270 and Phe93 (
Figure 4). Both residues interact with all ligands with the exception of (15
Z)-phytofluene and (15
Z)-phytoene; furthermore, Phe93 is the residue with the highest interaction percentage for three ligands and interacts with two atoms of the ligand for eight of these carotenoids. The remaining phenylalanines that interact with β-carotene (120, 307 and 335) also interact with between ten and eleven other carotenoids.
All of these residues are aromatic, and accompany the interaction of a hydrophobic ligand with conjugated double bonds. This fact also agrees with the most important contribution being that of Van der Waals energies.
α-Carotene presents an interaction similar to that of β-carotene and the potential interaction energy between both of them differs by approximately 2 Kcal.mol
−1 (
Table 4 and
Table 5).
(3R)-β-Cryptoxanthin and (3′R)-β-cryptoxanthin interact with the same phenylalanine residues, with at least one non-aromatic, non-polar residue, and with Trp270. They differ in that (3′R)-β-cryptoxanthin interacts with a positively charged residue (Arg408) and does not interact with the solvent, whereas (3R)-β-cryptoxanthin interacts with a neutral polar residue (Cys273) and with twenty-one water molecules, suggesting that the position of the carotenoid in the pocket mainly changes the interaction with the solvent.
The interactions of (all-E)-lycopene, (5
Z)-lycopene and (5′
Z)-lycopene are very similar; furthermore, the complex–ligand interaction energies (represented in
Table 4 and
Table 5) among the three do not differ by more than 3 Kcal.mol
−1. This might suggest that the enzyme will not have specificity depending on whether the linear carotenoid is (all-E) or has a
Z configuration at the C5-bond, or on which side the isomerized carotenoid is positioned.
Unlike the other major dietary carotenoids bioavailable in humans (lutein, zeaxanthin, β-cryptoxanthin, α-carotene, β-carotene and lycopene) the cleavage of phytoene and phytofluene by CCOs has not been described and cannot be presumed due to their markedly different degree of unsaturation, hence the interest in including them in this study. Phytoene and phytofluene differ in the presence of a double bond and have similar hydrophobic interaction, but differ in the interaction with polar and charged residues. Phytofluene, which has a double bond at bond eleven, interacts with Tyr332, Tyr235, Tyr326 and His237, while phytoene, which has a single bond at bond eleven, interacts with His237 and Arg408. Phytofluene, besides having more polar interactions, has a potential interaction energy of approximately 10 Kcal.mol−1 less, this difference being more accentuated in the Van der Waals contribution.
(3R,3′S, meso)-Zeaxanthin and (3R,3′R)-lutein, two positional isomers, which differ in the position of the double bond of one of the β-rings, have very similar interaction with residues and potential interaction energy. Both interact with the same number of water molecules and with the same residues, only (3R,3′R)-lutein additionally interacts with Phe307, Trp270 and His237 and Pro455 while (3R,3′S, meso)-zeaxanthin interacts with Ala456. (3R,3′R)-Zeaxanthin, an optical isomer of (3R,3′S, meso)-zeaxanthin that differs in the orientation of one of the OH-, has a potential interaction energy with the protein of about 10 Kcal.mol−1 less, the main difference coming from the electrostatic contribution. (3R,3′R)-Zeaxanthin interacts with twelve fewer water molecules, but interacts with three neutral polar residues (Thr141, Thr424 and Tyr332), two negatively charged residues (Gln457 and Asp12) and one positively charged residue (Arg408), with which (3R,3′S, meso)-zeaxanthin does not interact. Furthermore, the residue with the highest interaction frequency for (3R,3′R)-zeaxanthin is the polar residue Thr141, with a frequency of 96%, while for (3R,3′S, meso)-zeaxanthin it is the non-polar residue Ala456, with a frequency of 85%. All this suggests to us that the position of the double bond in one of the β-rings will not significantly change the preference of the enzyme for one ligand or the other, whereas the orientation of one of the hydroxy groups may generate more significant changes with respect to the interaction.
(3S,3′S)-Astaxanthin is the ligand with the best potential interaction energy (Uab), with −124 Kcal.mol−1, followed by (3R,3′R)-zeaxanthin with −116 Kcal.mol−1. This energy difference with the rest of the carotenoids comes mainly from the electrostatic contribution. This is justified since astaxanthin has an alcohol group and a ketone group on the two β-rings. This ligand presents interaction with four non-polar residues, with three aromatic non-polar residues, with two polar residues, with one aromatic polar residue and with thirty water molecules.
Canthaxanthin has a ketone group on the two β-rings like astaxanthin, but differs from astaxanthin in that it does not have alcohol groups. This carotenoid interacts with the same aromatic residues (polar and non-polar) as (3S,3′S)-astaxanthin, but only interacts with one polar and one non-polar non-aromatic residue, and with twenty-three water molecules. It also interacts with one histidine residue. Canthaxanthin has a potential interaction energy approximately 16 Kcal.mol−1 higher than (3S,3′S)-astaxanthin, the difference coming from the electrostatic contribution.
(3S,3′S)-Violaxanthin has an alcohol group and an epoxy group present on both β-rings. This ligand interacts with fifteen residues, being the carotenoid with the highest number of interactions along with zeaxanthin. The residues with which it interacts are of all chemical natures: non-polar, aromatic non-polar, polar, aromatic polar, negatively charged and positively charged. It interacts with a single water molecule. It is the only ligand that presents a higher frequency of interaction with a charged residue, being 58% with arginine. It is also the only carotenoid that interacts with a residue through four atoms of the ligand (with threonine). With respect to the system–ligand binding energy, it is the carotenoid with the highest energy of those presenting two alcohol groups, having a very similar contribution to that of β-cryptoxanthin. Considering its structure, such changes may be attributed to the presence of the epoxide groups.
In summary, three groups of carotenoids could be arbitrarily established in terms of Uab values:
- -
Carotenes (α- and β-carotene, lycopene, phytoene and phytofluene) with Uab values in the interval ~−93 and −102 Kcal.mol−1. It should be noted that they differed clearly in structure (from none to two end rings, from three to eleven conjugated double bonds and with diverse geometrical configurations), which suggests that such differences in carotenes do not lead to energetic differences over teen units.
- -
Carotenoids with one hydroxy group (β-cryptoxanthin), two hydroxy groups (lutein, zeaxanthin), two hydroxy groups and two 5,6-epoxide groups ((3S,3′S)-violaxanthin) and two ketone groups ((3S,3′S)-astaxanthin) (all such groups in rings), with Uab values in the range ~−100 to −116 Kcal.mol−1.
- -
Carotenoids with one hydroxy and one ketone group in each terminal ring ((3S,3′S)-astaxanthin), with the lowest Uab value (~−124 Kcal.mol−1).
2.6. Interaction of BCO2 with Other Carotenoids
As in BCO1, we observe that the overall structure of the enzyme–ligand complex, for all models subjected to 1 ns molecular dynamics, remains the same as in the complex with β-carotene.
Table 6 and
Table 7 show the interaction energies and fingerprint analysis, respectively, for the same ligands as in BCO1.
Again, three groups could be arbitrarily established considering Uab values:
- -
Carotenes (α- and β-carotene, lycopene, phytoene and phytofluene) with Uab values in the interval ~−86 to −104 Kcal.mol−1.
- -
Carotenoids with one hydroxy group (β-cryptoxanthin), two hydroxy groups (lutein, zeaxanthin), two hydroxy groups and two 5,6-epoxide groups ((3S,3′S)-violaxanthin) and two ketone groups ((3S,3′S)-astaxanthin) (all such groups in rings), with Uab values in the range ~−104 to −118 Kcal.mol−1.
- -
Carotenoids with one hydroxy and one ketone group in each terminal ring ((3S,3′S)-astaxanthin), with the lowest Uab value (~−127 Kcal.mol−1).
In general, the differences in Uab values between BCO1 and BCO2 were below 5 Kcal.mol−1, except for α-carotene, (3R′)-β-cryptoxanthin, (3R,3′S, meso)-zeaxanthin, (3R,3′R)-lutein, (3S,3′S)-violaxanthin and (15Z)-phytofluene, the latter two with differences > 10 Kcal.mol−1.
For BCO2, all carotenoids interact with Phe103. Fourteen of the fifteen carotenoids interact with Phe319, Phe145 and with Phe486. Thirteen of fifteen carotenoids interact with Phe356. In addition, Phe319 is the residue with the highest interaction frequency for five of these carotenoids.
Phe145, Phe103 and Phe319 are some of the residues that interact most frequently with the reference molecule β-carotene. Unlike BCO1, in BCO2 the residues interacting with β-carotene differ significantly from those interacting with the rest of the ligands.
α-Carotene is the carotenoid with the highest potential interaction energy, which is −86.56 ± 3.89 Kcal.mol−1 with a very small electrostatic contribution. All the residues with which it interacts are aromatic. It has a similar interaction than linear carotenoids (lycopene, phytoene and phytofluene), differing from these in the contribution of the polar residue tryptophan.
With respect to (all-E)-lycopene and its isomers, we note that only (5′Z)-lycopene has an interaction similar to that of (all-E)-lycopene, while (5Z)-lycopene interacts with significantly fewer residues. On the other hand, (5Z)-lycopene is the one with a potential interaction energy very similar to (all-E)-lycopene, while (5′Z)-lycopene has a potential interaction energy approximately 8 Kcal.mol−1 higher. This differs from BCO1 in which the position of isomerization did not change the interaction.
With respect to (15Z)-phytoene and (15Z)-phytofluene, these have a similar interaction, the main difference being that phytoene interacts with two negatively charged residues (His286 and His357), whereas phytofluene does not interact with any charged residues. The potential interaction energy for both is also similar, being approximately 3 Kcal.mol−1 higher for (15Z)-phytofluene, with this difference coming from the Van der Waals contribution. This is different from that observed for BCO1 where both molecules differ in their interaction more significantly.
(3R)-β-Cryptoxanthin and (3R′)-β-cryptoxanthin have a very similar non-polar interaction, and both have a higher frequency of interaction with the same aromatic non-polar residue (Phe145). As in BCO1, they differ in polar interaction. While (3R)-β-cryptoxanthin interacts with one neutral polar residue, one positively charged residue and forty-one water molecules, (3R′)-β-cryptoxanthin interacts with three negatively charged residues and sixteen water molecules. With respect to energy, (3R′)-β-cryptoxanthin has an electrostatic contribution approximately 4 Kcal.mol−1 higher than (3R)-β-cryptoxanthin. As for BCO1, we suggest that the position of the double bond mainly changes the hydrophilic interaction.
With respect to the isomers (3R,3′R)-zeaxanthin, (3R,3′S, meso)-zeaxanthin and lutein, in BCO2, no significant differences were observed with respect to their interactions. The interaction energies do not differ among them by more than 2 Kcal.mol−1 for each component. They also show similar interactions with residues and solvent.
As in BCO1, (3S,3′S)-astaxanthin is the ligand with the best potential interaction energy, being −126.90 Kcal.mol−1, with an electrostatic contribution of −38.21 Kcal.mol−1. This carotenoid interacts with seven non-polar residues, with only one polar residue and one negatively charged residue, and with twenty-four water molecules, and presents the highest interaction frequency with one of them.
Canthaxanthin interacts with six non-polar residues, with one polar residue, with one positively charged residue, with three negatively charged residues and with forty-seven water molecules. It has a significantly higher potential interaction energy than (3S,3′S)-astaxanthin, with an approximately 16 Kcal.mol−1 higher electrostatic contribution and a 6 Kcal.mol−1 higher Van der Waals contribution.
As in BCO1, (3S,3′S)-violaxanthin is the carotenoid that interacts with the greatest number of residues (fourteen), these being of all chemical natures. It is also the carotenoid that interacts with the largest number of water molecules (sixty-two). It has a potential interaction energy of −118.04 Kcal.mol−1, with an electrostatic contribution of −33.96 Kca.mol−1. It differs from BCO1, where it is the carotenoid with the highest potential interaction energy of those with two alcohol groups.
As seen in
Table 4 and
Table 6, the differences in conformational energies at the minimum and at the current pose are always lower in absolute value than the interaction energy Uab. This ensures that the interaction energy is sufficient to compensate for the energetic cost required by the pose adopted by the ligand at the site.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














