

Coumarin-Based Dual Inhibitors of Human Carbonic Anhydrases and Monoamine Oxidases Featuring Amino Acyl and (Pseudo)-Dipeptidyl Appendages: In Vitro and Computational Studies

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Chemistry
3. Biochemical Assays and SAR Analysis
3.1. CA Inhibition Results
- Due to the complete lack of activity towards hCA I and II showing a Ki ˃ 10,000 nM, the test compounds fulfill the primary goal of selectivity towards membrane-anchored hCAs vs. the cytosolic enzymes;
- All molecules, apart from 1 and 11, present the structural requisites that are compatible with a potent (one-/two-digit nanomolar Ki values) hCA XII inhibition;
- hCA IX inhibitory activity is maintained in the low nanomolar range for the smaller amino acyl-coumarin series (compounds 1, 2, 6–8), whereas the extension of the (pseudo)-peptidyl chain results in up to an eleven-fold reduction of Ki values when compared to the most effective inhibitor 6 (excluding compounds 18 and 20, which is similar in potency to 2 and 7, respectively);
- In the amino acyl coumarin series, hCA inhibitory potency decreases from compounds with small N-protecting groups to analogs bearing more extended and lipophilic moieties;
- The general inhibitory profile discloses a significant selectivity towards hCA XII compared to the other isoform that is relevant to cancer; the most remarkable SI values are found in the (pseudo)-dipeptidyl coumarin series of derivatives (except 20), which all share the urea structural motif;
- The replacement of AMC for AMMC in similar structures (pairs 6/7 and 17/20) gives rise to unexpected effects, with a slight reduction of activity against both isoforms for the amino acyl-coumarin derivatives, whereas for the (pseudo)-dipeptidyl coumarin analogs the same replacement was found to cause a six-fold increase in hCA IX inhibition and a similar reduction in inhibitory activity towards hCA XII;
- The external amino acid also affects activity as observed with the (pseudo)-dipeptidyl AMC conjugates. The inhibition potency towards CA IX increases in the order βAla, Gly, Ala, and Phe, suggesting that, in the active site, a larger space is available for additional hydrophobic interactions involving the side chain; Ala and βAla, on the other hand, are the preferred residues for potent hCA XII inhibitory activity, which may be explained by suitable conformational rearrangements into the isoform binding site.
3.2. MAO Inhibition Results
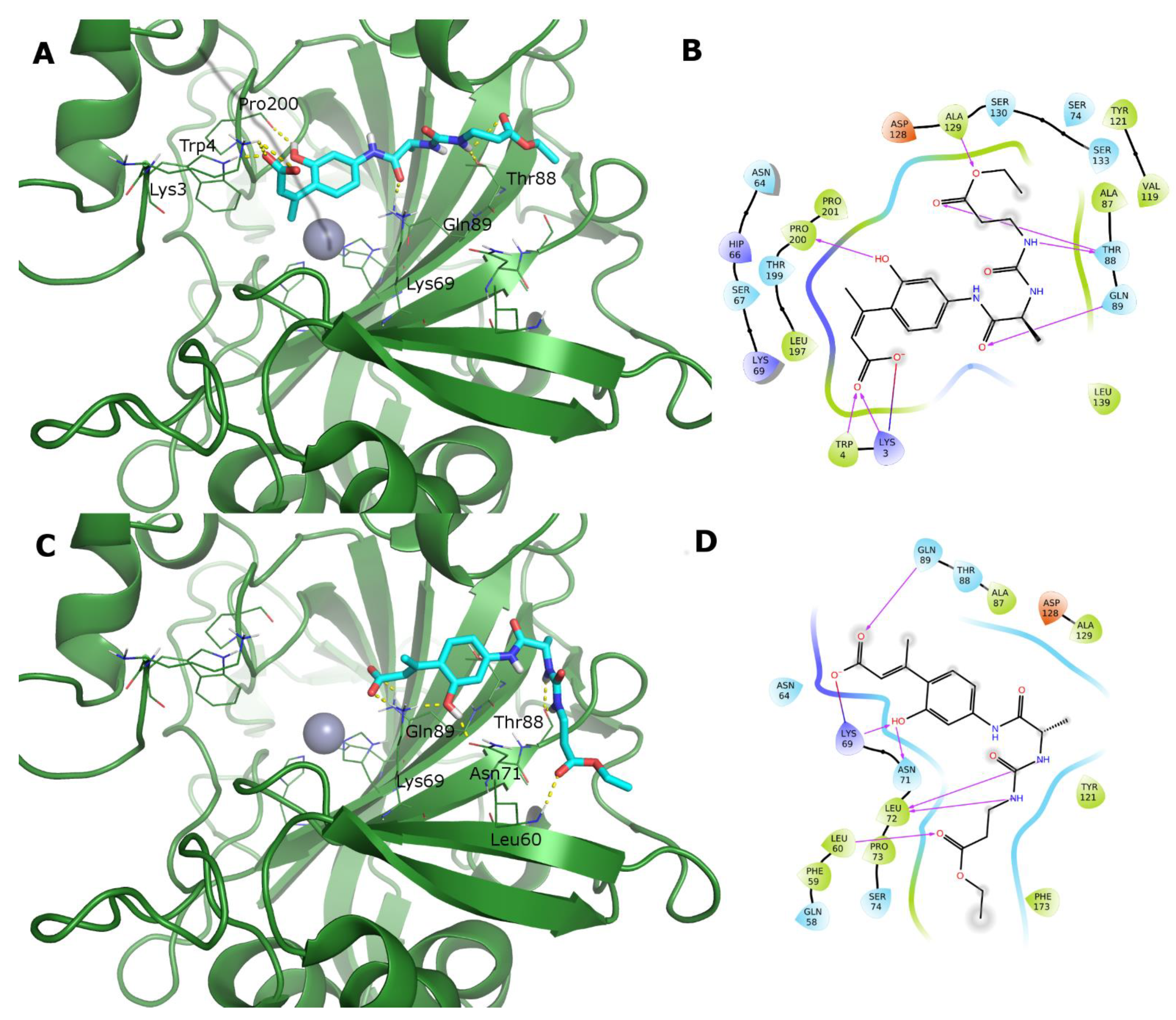
3.3. Computational Studies
4. Conclusions
5. Experimental Section
5.1. Chemical Synthesis
5.1.1. General Procedure for the Synthesis of Z/Fmoc-Xaa-AMC/AMMC Derivatives (1–3, 8, and 9)
5.1.2. General Protocol for the N-Deprotection of Fmoc-Xaa-AMC/AMMC Derivatives (2, 3, 9): Preparation of 4, 5, and 10
5.1.3. General Procedure for the Synthesis of Ac-Ala-AMC/AMMC (6 and 7)
5.1.4. Synthesis of Z-Ala-tLeu-AMMC (11)
5.1.5. General Protocol for the Preparation of Active Carbamates (12–15)
5.1.6. General Procedure for the Synthesis of (Pseudo)-Dipeptidyl Coumarins (16–20)
5.2. Biological Methods
5.2.1. hCA Inhibition Assay
5.2.2. hMAO Inhibition Studies
5.3. Computational Studies
5.3.1. Ligand Preparation
5.3.2. hCA Structure Preparation and Docking
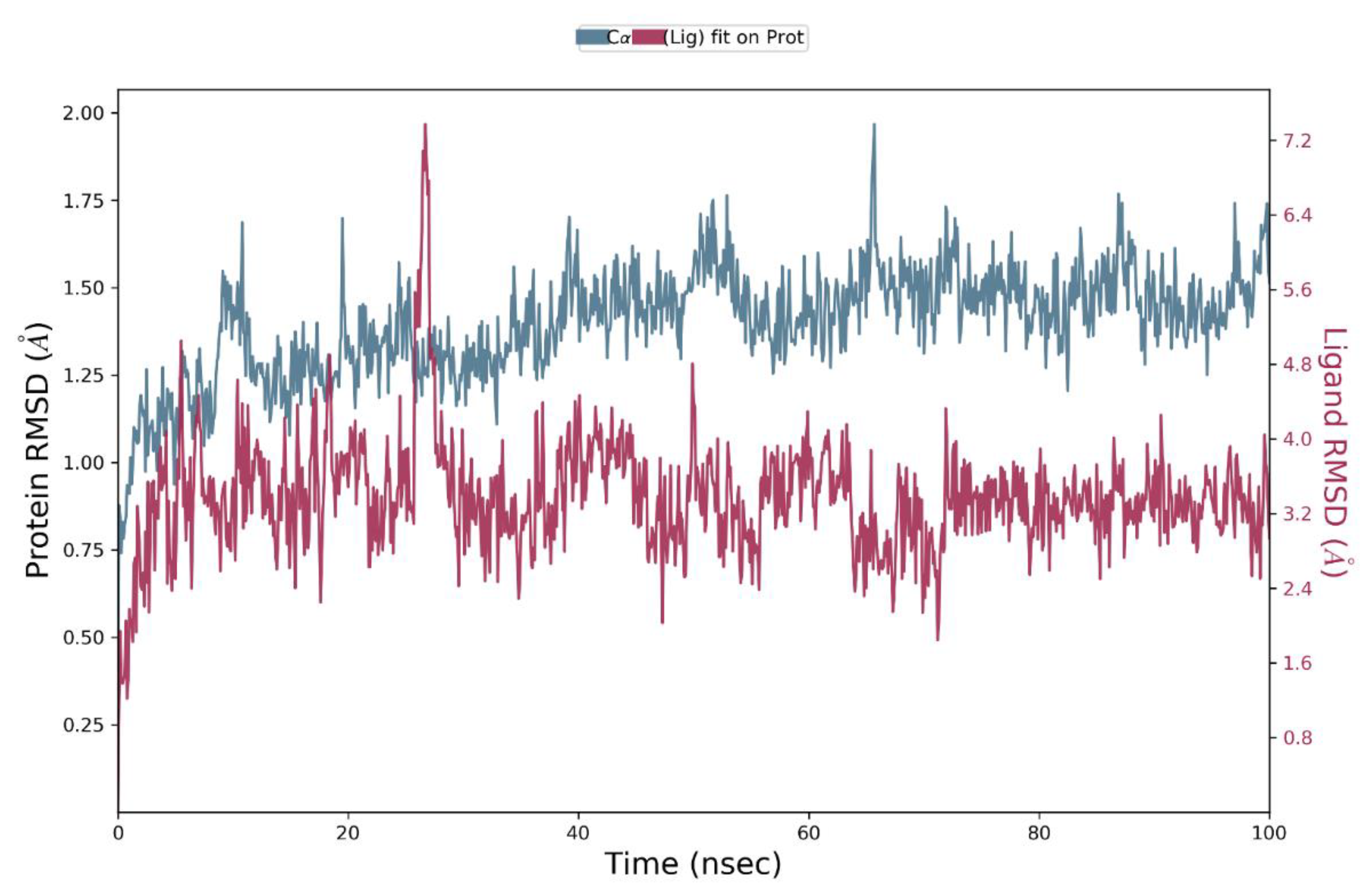
5.3.3. Molecular Dynamics of the CA XII:Ligand 19 Complex
5.3.4. hMAO-A and hMAO-B Protein Structure Preparation and Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E.; Yordi, E.G. Coumarins—An Important Class of Phytochemicals. In Phytochemicals—Isolation, Characterisation and Role in Human Health; Rao, V., Rao, L., Eds.; InTech: Rijeka, Croatia, 2015; Chapter 5; pp. 113–140. [Google Scholar]
- Hoult, J.R.S.; Payá, M. Pharmacological and biochemical actions of simple coumarins: Natural products with therapeutic potential. Gen. Pharmacol. 1996, 27, 713–722. [Google Scholar] [CrossRef]
- Sadeghpour, M.; Olyaei, A.; Adl, A. 4-Aminocoumarin derivatives: Synthesis and applications. New J. Chem. 2021, 45, 5744–5763. [Google Scholar] [CrossRef]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.; Matos, M.J.; Uriarte, E.; Santana, L. Trending Topics on Coumarin and Its Derivatives in 2020. Molecules 2021, 26, 501. [Google Scholar] [PubMed]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Bizzarri, B.; Granese, A.; Carradori, S.; Yáñez, M.; Orallo, F.; Ortuso, F.; et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009, 52, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Carradori, S.; Bolasco, A.; Chimenti, P.; Yáñez, M.; Ortuso, F.; Alcaro, S. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 4846–4852. [Google Scholar]
- Carotti, A.; Altomare, C.; Catto, M.; Gnerre, C.; Summo, L.; De Marco, A.; Rose, S.; Jenner, P.; Testa, B. Lipophilicity plays a major role in modulating the inhibition of monoamine oxidase B by 7-substituted coumarins. Chem. Biodivers. 2006, 3, 134–149. [Google Scholar] [CrossRef]
- De Luca, L.; Mancuso, F.; Ferro, S.; Buemi, M.R.; Angeli, A.; Del Prete, S.; Capasso, C.; Supuran, C.T.; Gitto, R. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur. J. Med. Chem. 2018, 143, 276–282. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Supuran, C.T.; Dedhar, S. Cancer Therapeutic Targeting of Hypoxia Induced Carbonic Anhydrase IX: From Bench to Bedside. Cancers 2022, 14, 3297. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2021, 30, 1197–1208. [Google Scholar] [CrossRef]
- Marconi, G.D.; Gallorini, M.; Carradori, S.; Guglielmi, P.; Cataldi, A.; Zara, S. The Up-Regulation of Oxidative Stress as a Potential Mechanism of Novel MAO-B Inhibitors for Glioblastoma Treatment. Molecules 2019, 24, 2005. [Google Scholar] [CrossRef] [PubMed]
- Shui, X.; Ren, X.; Xu, R.; Xie, Q.; Hu, Y.; Qin, J.; Meng, H.; Zhang, C.; Zhao, J.; Shi, C. Monoamine oxidase A drives neuroendocrine differentiation in prostate cancer. Biochem. Biophys. Res. Commun. 2022, 606, 135–141. [Google Scholar] [PubMed]
- Wu, J.B.; Shao, C.; Li, X.; Li, Q.; Hu, P.; Shi, C.; Li, Y.; Chen, Y.T.; Yin, F.; Liao, C.P.; et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J. Clin. Investig. 2014, 124, 2891–2908. [Google Scholar] [CrossRef] [PubMed]
- Bardaweel, S.; Aljanabi, R.; Sabbah, D.; Sweidan, K. Design, Synthesis, and Biological Evaluation of Novel MAO-A Inhibitors Targeting Lung Cancer. Molecules 2022, 27, 2887. [Google Scholar]
- Küçükbay, F.Z.; Küçükbay, H.; Tanc, M.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory properties of amino acid—Coumarin/quinolinone conjugates incorporating glycine, alanine and phenylalanine moieties. J. Enzym. Inhib. Med. Chem. 2016, 31, 1198–1202. [Google Scholar] [CrossRef]
- Spatola, A.F. Chemistry and Biochemistry of Amino Acids, Peptides and Proteins; Weinstein, B., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1983; Volume 7, pp. 267–357. [Google Scholar]
- Hruby, V.J. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers 1993, 33, 1073–1082. [Google Scholar] [CrossRef]
- Marraud, M.; Aubry, A. Crystal structures of peptides and modified peptides. Biopolymers 1996, 40, 45–83. [Google Scholar] [CrossRef]
- Calcagni, A.; Rossi, D.; Paglialunga Paradisi, M.; Lucente, G.; Luisi, G.; Gavuzzo, E.; Mazza, F.; Pochetti, G.; Paci, M. Peptides containing the sulfonamide junction: Synthesis, structure, and conformation of Z-Tau-Pro-Phe-NHiPr. Biopolymers 1997, 41, 555–567. [Google Scholar] [CrossRef]
- Luisi, G.; Mollica, A.; Carradori, S.; Lenoci, A.; De Luca, A.; Caccuri, A.M. Nitrobenzoxadiazole-based GSTP1-1 inhibitors containing the full peptidyl moiety of (pseudo)glutathione. J. Enzym. Inhib. Med. Chem. 2016, 31, 924–930. [Google Scholar]
- Calcagni, A.; Duprè, S.; Lucente, G.; Luisi, G.; Pinnen, F.; Rossi, D. Synthesis and activity of the glutathione analogue-(L-azaglutamyl)-L-cisteinyl-glycine. Int. J. Peptide Prot. Res. 1995, 46, 434–439. [Google Scholar]
- Semetey, V.; Hemmerlin, C.; Didierjean, C.; Schaffner, A.P.; Giner, A.G.; Aubry, A.; Briand, J.P.; Marraud, M.; Guichard, G. Unexpected stability of the urea cis-trans isomer in urea-containing model pseudopeptides. Org. Lett. 2001, 3, 3843–3846. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.C.; Kowalski, J.A.; Lipton, M.A. Facile incorporation of urea pseudopeptides into protease substrate analogue inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 5219–5222. [Google Scholar] [CrossRef] [PubMed]
- Fayad, A.A.; Pubill-Ulldemolins, C.; Sharma, S.V.; Day, D.; Goss, R.J.M. A One-Pot Synthesis of Symmetrical and Unsymmetrical Dipeptide Ureas. Eur. J. Org. Chem. 2015, 2015, 5603–5609. [Google Scholar] [CrossRef]
- Kumar, A.; Siwach, K.; Supuran, C.T.; Sharma, P.K. A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors. Bioorg. Chem. 2022, 126, 105920. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, A.; Duprè, S.; Lucente, G.; Luisi, G.; Pinnen, F.; Rossi, D.; Spirito, A. Synthesis and activity of the glutathione analogue gamma-(L-gamma-oxaglutamyl)-L-cysteinyl-glycine. Arch. Pharm. Pharm. Med. Chem. 1996, 329, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
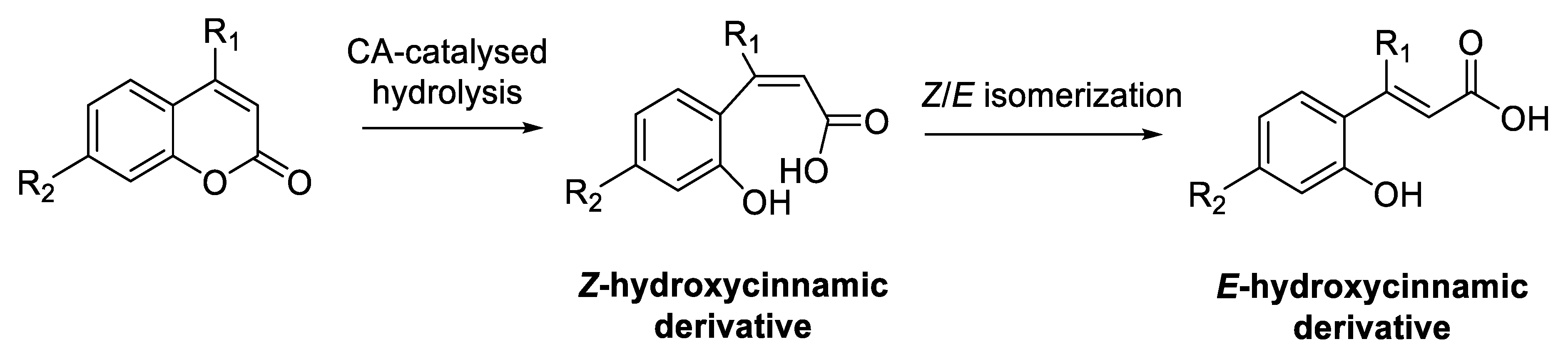
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef]
- Tubert-Brohman, I.; Sherman, W.; Repasky, M.; Beuming, T. Improved docking of polypeptides with Glide. J. Chem. Inf. Model. 2013, 53, 1689–1699. [Google Scholar] [CrossRef]
- Scala, M.C.; Agamennone, M.; Pietrantoni, A.; Di Sarno, V.; Bertamino, A.; Superti, F.; Campiglia, P.; Sala, M. Discovery of a Novel Tetrapeptide Against Influenza A Virus: Rational Design, Synthesis, Bioactivity Evaluation and Computational Studies. Pharmaceuticals 2021, 14, 959. [Google Scholar]
- Agamennone, M.; Superti, F. Broad-Spectrum Activity of Small Molecules Acting against Influenza A Virus: Biological and Computational Studies. Pharmaceuticals 2022, 15, 301. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2012, 20, 2266–2273. [Google Scholar] [PubMed]
- Krishnamurthy, V.M.; Kaufman, G.K.; Urbach, A.R.; Gitlin, I.; Gudiksen, K.L.; Weibel, D.B.; Whitesides, G.M. Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein-Ligand Binding. Chem. Rev. 2008, 108, 946–1051. [Google Scholar] [PubMed]
- Snyder, P.W.; Mecinovic, J.; Moustakas, D.T.; Thomas, S.W., 3rd; Harder, M.; Mack, E.T.; Lockett, M.R.; Héroux, A.; Sherman, W.; Whitesides, G.M. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc. Natl. Acad. Sci. USA 2011, 108, 17889–17894. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef]
- Thacker, P.S.; Mohammed, A.; Supuran, C.T.; Tiwari, P.L.; Goud, N.S.; Srikanth, D.; Angeli, A. Synthesis and biological evaluation of coumarin carboxamides as selective and potent inhibitors of carbonic anhydrases IX and XII. Anticancer Agents Med. Chem. 2022, 22, 2647–2654. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef]
- Mathew, B.; Carradori, S.; Guglielmi, P.; Uddin, M.S.; Kim, H. New aspects of monoamine oxidase B inhibitors: The key role of halogens to open the golden door. Curr. Med. Chem. 2021, 28, 266–283. [Google Scholar]
- Bester, E.; Petzer, A.; Petzer, J.P. Coumarin derivatives as inhibitors of d-amino acid oxidase and monoamine oxidase. Bioorg. Chem. 2022, 123, 105791. [Google Scholar] [CrossRef]
- Koyiparambath, V.P.; Prayaga Rajappan, K.; Rangarajan, T.M.; Al-Sehemi, A.G.; Pannipara, M.; Bhaskar, V.; Nair, A.S.; Sudevan, S.T.; Kumar, S.; Mathew, B. Deciphering the detailed structure-activity relationship of coumarins as Monoamine oxidase enzyme inhibitors—An updated review. Chem. Biol. Drug. Des. 2021, 98, 655–673. [Google Scholar]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Provensi, G.; Costa, A.; Rani, B.; Becagli, M.V.; Vaiano, F.; Passani, M.B.; Tanini, D.; Capperucci, A.; Carradori, S.; Petzer, J.P.; et al. New β-arylchalcogeno amines with procognitive properties targeting Carbonic Anhydrases and Monoamine Oxidases. Eur. J. Med. Chem. 2022, 244, 114828. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [PubMed]
- D’Ascenzio, M.; Secci, D.; Carradori, S.; Zara, S.; Guglielmi, P.; Cirilli, R.; Pierini, M.; Poli, G.; Tuccinardi, T.; Angeli, A.; et al. 1,3-Dipolar Cycloaddition, HPLC Enantioseparation, and Docking Studies of Saccharin/Isoxazole and Saccharin/Isoxazoline Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors. J. Med. Chem. 2020, 63, 2470–2488. [Google Scholar]
- Weissbach, H.; Smith, T.E.; Daly, J.W.; Witkop, B.; Udenfriend, S. A rapid spectrophotometric assay of mono-amine oxidase based on the rate of disappearance of kynuramine. J. Biol. Chem. 1960, 235, 1160–1163. [Google Scholar] [CrossRef]
- Mostert, S.; Petzer, A.; Petzer, J.P. Indanones as high-potency reversible inhibitors of monoamine oxidase. ChemMedChem 2015, 10, 862–873. [Google Scholar] [CrossRef]
- Schrödinger. Schrödinger Release 2021-4: Maestro, Glide, Prime, Desmond, Protein Preparation Wizard, Epik; SiteMap Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Halgren, T. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki hCA I | Ki hCA II | Ki hCA IX | Ki hCA XII | SI hCA IX over hCA I or hCA II b | SI hCA XII over hCA I or hCA II b | SI hCA XII over hCA IX c |
|---|---|---|---|---|---|---|---|
| Z-Ala-AMC (1) | >10,000 | >10,000 | 30.5 | 110.0 d | >327.9 | >90.9 | 0.3 |
| Fmoc-Ala-AMC (2) | >10,000 | >10,000 | 91.0 | 37.2 | >109.9 | >268.8 | 2.4 |
| Ac-Ala-AMC (6) | >10,000 | >10,000 | 23.4 | 30.5 | >427.3 | >327.9 | 0.8 |
| Ac-Ala-AMMC (7) | >10,000 | >10,000 | 29.4 | 46.8 | >340.1 | >213.7 | 0.6 |
| Z-Ile-AMMC (8) | >10,000 | >10,000 | 78.7 | 41.5 | >127.1 | >241.0 | 1.9 |
| Z-Ala-tLeu-AMMC (11) | >10,000 | >10,000 | 171.5 | 336.3 | >58.3 | >29.7 | 0.5 |
| MeO-Gly-CO-Ala-AMC (16) | >10,000 | >10,000 | 183.3 | 38.2 | >54.6 | >261.8 | 4.8 |
| MeO-Ala-CO-Ala-AMC (17) | >10,000 | >10,000 | 163.3 | 9.6 | >61.2 | >1041.7 | 17.0 |
| MeO-Phe-CO-Ala-AMC (18) | >10,000 | >10,000 | 93.6 | 40.0 | >106.8 | >250.0 | 2.3 |
| EtO-βAla-CO-Ala-AMC (19) | >10,000 | >10,000 | 260.5 | 9.5 | >38.4 | >1052.6 | 27.4 |
| MeO-Ala-CO-Ala-AMMC (20) | >10,000 | >10,000 | 27.0 | 54.7 | >370.4 | >182.8 | 0.5 |
| AAZ | 250.0 | 12.1 | 25.8 | 5.7 | 9.7 0.5 | 43.8 2.1 | 4.5 |
| Compound | MAO-A (IC50 μM) | MAO-B (IC50 μM) |
|---|---|---|
| Z-Ala-MAC (1) | 9.31 ± 0.132 | >100 |
| Fmoc-Ala-MAC (2) | 54.8 ± 0.028 | >100 |
| Ac-Ala-MAC (6) | 1.92 ± 0.247 | >100 |
| Ac-Ala-MMAC (7) | 12.0 ± 0.693 | 45.6 ± 1.85 |
| Z-Ile-MMAC (8) | 47.0 ± 13.5 | >100 |
| Z-Ala-tLeu-MMAC (11) | 41.3 ± 17.8 | 79.5 ± 7.24 |
| MeO-Gly-CO-Ala-MAC (16) | 5.910 ± 0.155 | >100 |
| MeO-Ala-CO-Ala-MAC (17) | 69.6 ± 9.28 | 58.6 ± 24.0 |
| MeO-Phe-CO-Ala-MAC (18) | 21.8 ± 0.092 | 78.0 ± 22.3 |
| EtO-βAla-CO-Ala-MAC (19) | >100 | >100 |
| MeO-Ala-CO-Ala-MMAC (20) | >100 | 75.3 ± 0.976 |
| Harmine | 0.0041 ± 0.00007 | - |
| Isatin | 8.43 ± 0.245 | 3.90 ± 0.792 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agamennone, M.; Fantacuzzi, M.; Carradori, S.; Petzer, A.; Petzer, J.P.; Angeli, A.; Supuran, C.T.; Luisi, G. Coumarin-Based Dual Inhibitors of Human Carbonic Anhydrases and Monoamine Oxidases Featuring Amino Acyl and (Pseudo)-Dipeptidyl Appendages: In Vitro and Computational Studies. Molecules 2022, 27, 7884. https://doi.org/10.3390/molecules27227884
Agamennone M, Fantacuzzi M, Carradori S, Petzer A, Petzer JP, Angeli A, Supuran CT, Luisi G. Coumarin-Based Dual Inhibitors of Human Carbonic Anhydrases and Monoamine Oxidases Featuring Amino Acyl and (Pseudo)-Dipeptidyl Appendages: In Vitro and Computational Studies. Molecules. 2022; 27(22):7884. https://doi.org/10.3390/molecules27227884
Chicago/Turabian StyleAgamennone, Mariangela, Marialuigia Fantacuzzi, Simone Carradori, Anél Petzer, Jacobus P. Petzer, Andrea Angeli, Claudiu T. Supuran, and Grazia Luisi. 2022. "Coumarin-Based Dual Inhibitors of Human Carbonic Anhydrases and Monoamine Oxidases Featuring Amino Acyl and (Pseudo)-Dipeptidyl Appendages: In Vitro and Computational Studies" Molecules 27, no. 22: 7884. https://doi.org/10.3390/molecules27227884
APA StyleAgamennone, M., Fantacuzzi, M., Carradori, S., Petzer, A., Petzer, J. P., Angeli, A., Supuran, C. T., & Luisi, G. (2022). Coumarin-Based Dual Inhibitors of Human Carbonic Anhydrases and Monoamine Oxidases Featuring Amino Acyl and (Pseudo)-Dipeptidyl Appendages: In Vitro and Computational Studies. Molecules, 27(22), 7884. https://doi.org/10.3390/molecules27227884