
Novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazepine Derivatives: Synthesis, Anti-Viral In Vitro Study and Target Validation Activity

 , , and
, , and
Abstract
:
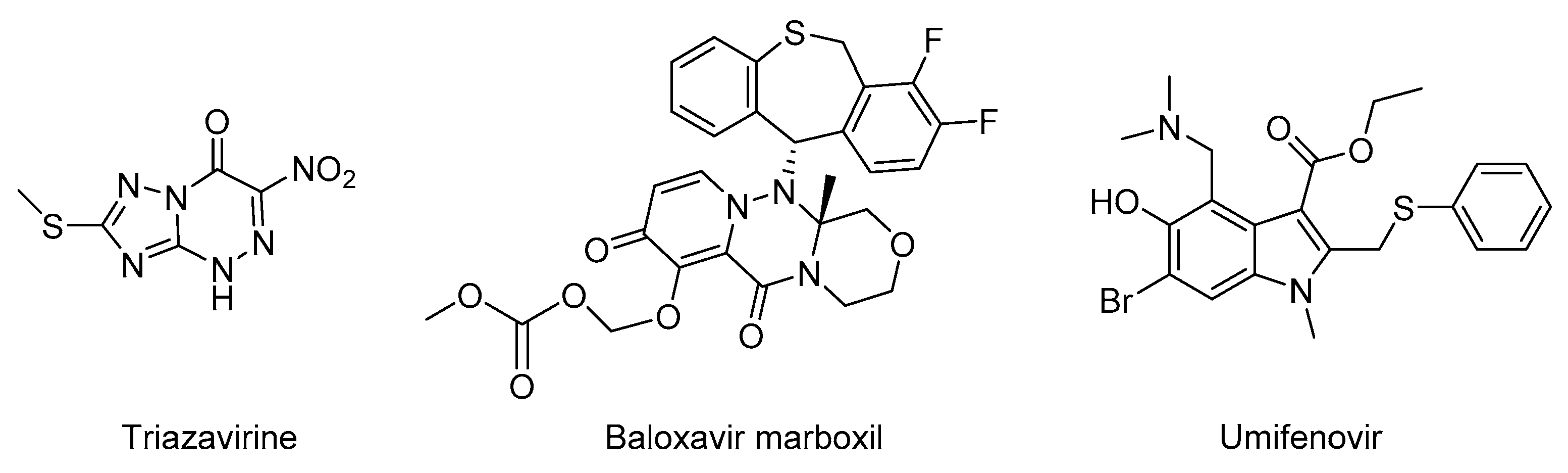
1. Introduction
2. Results and Discussion
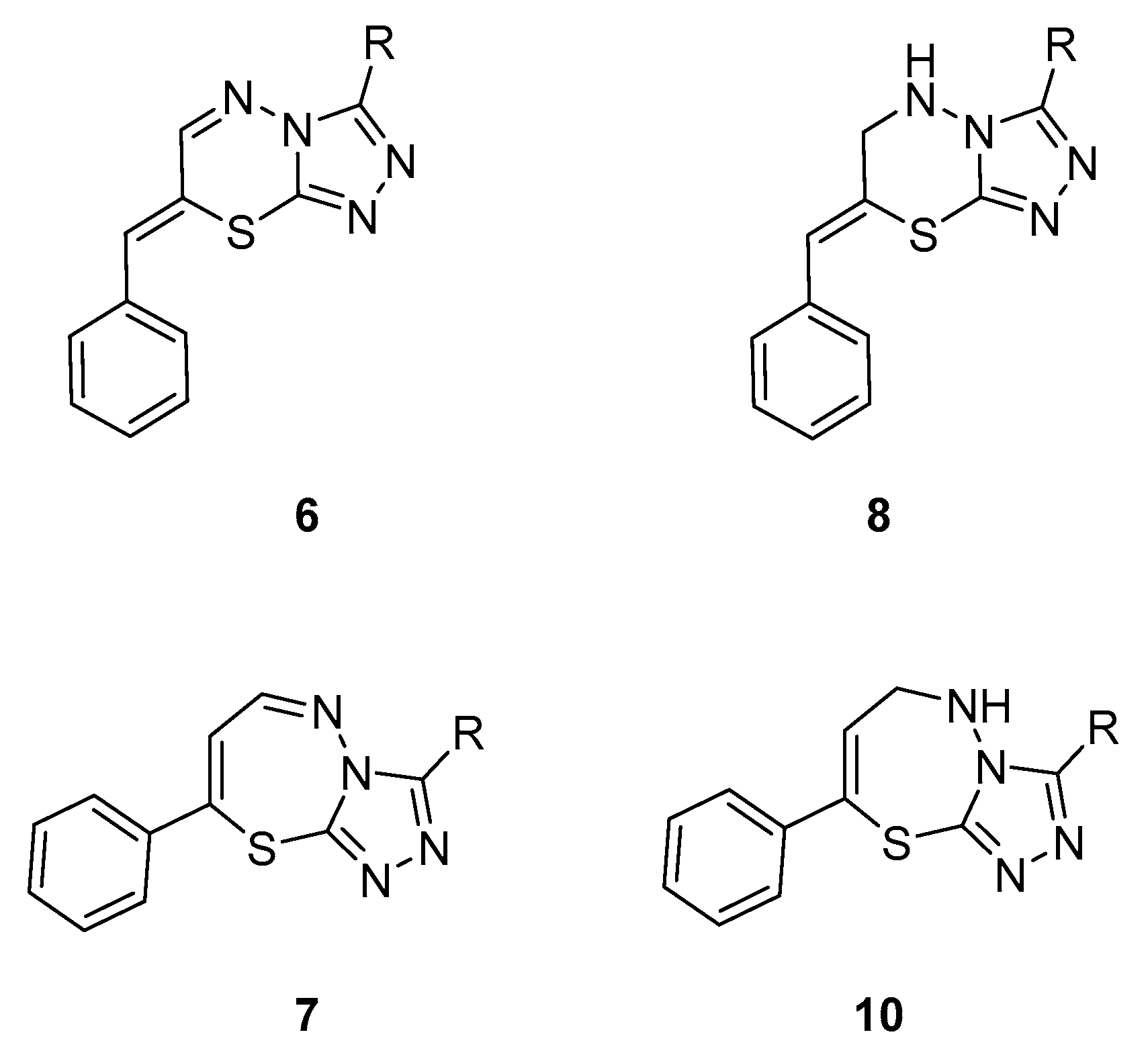
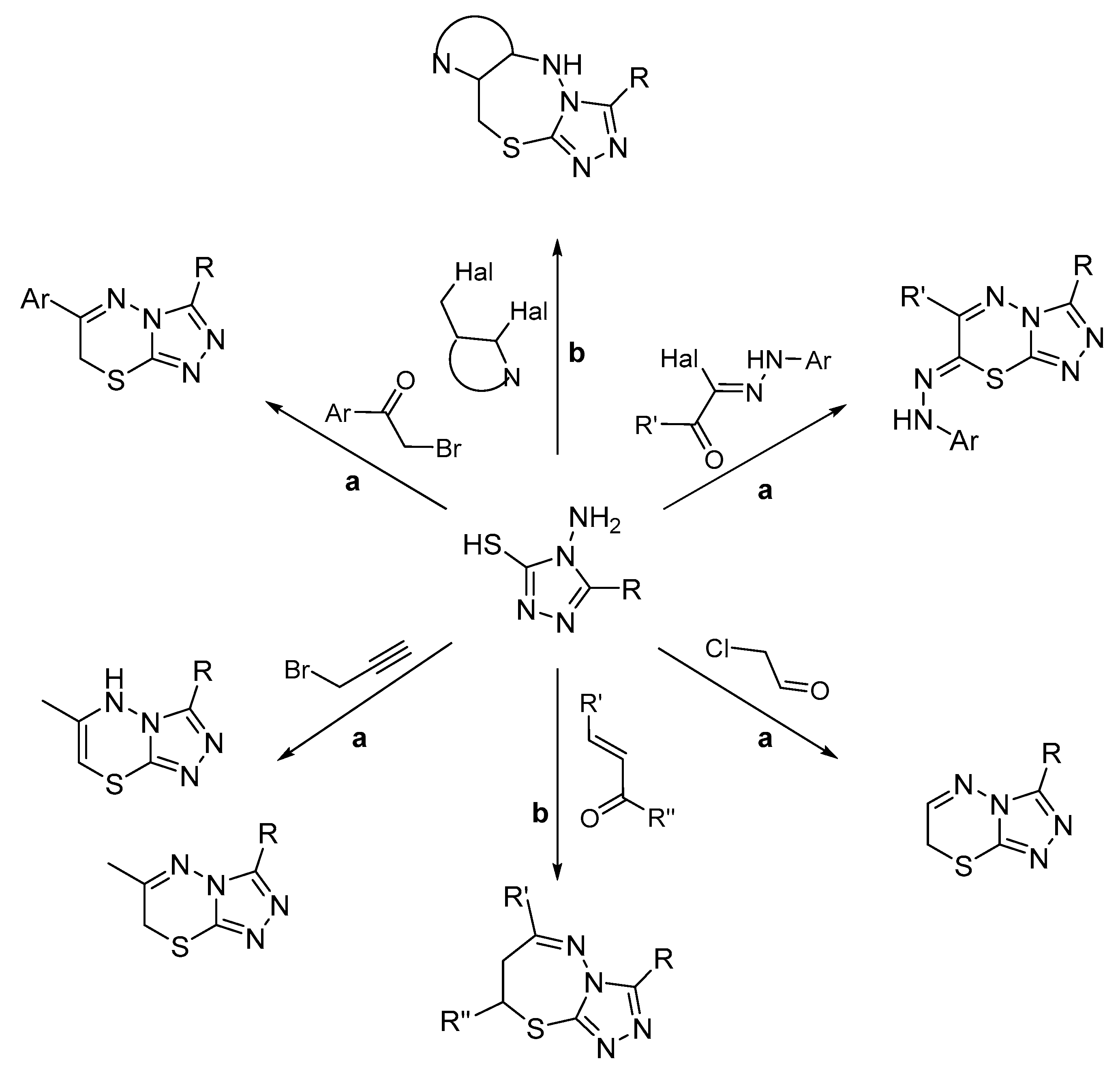
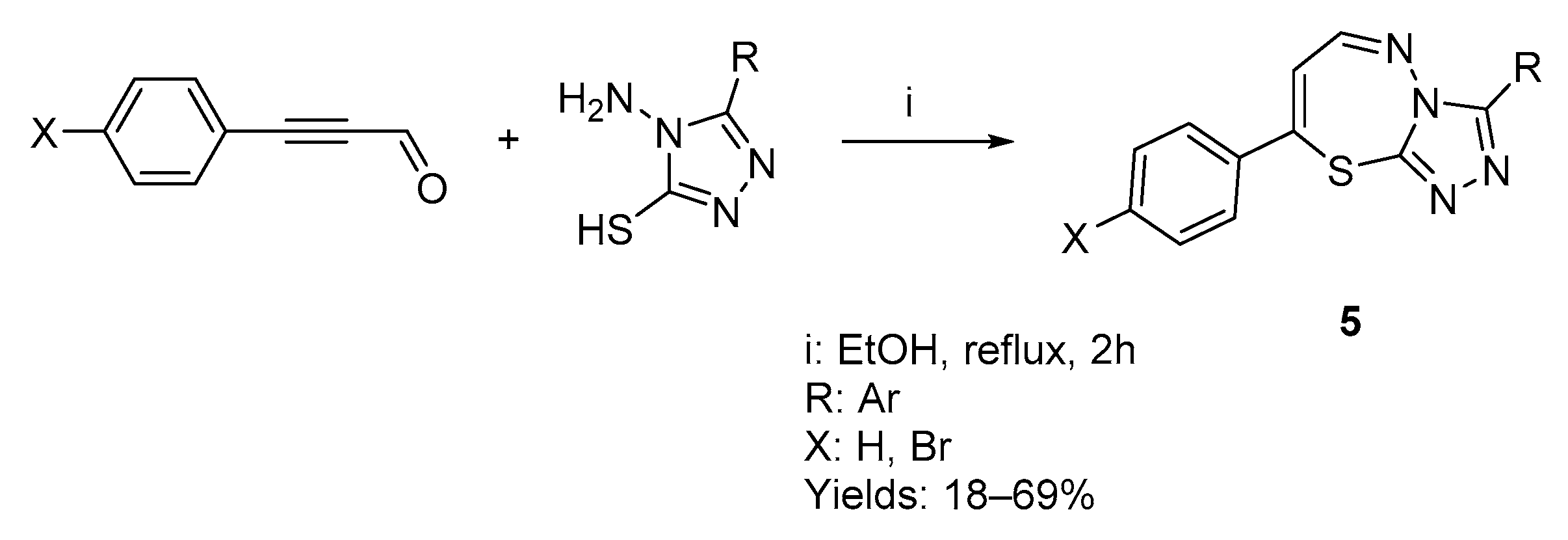
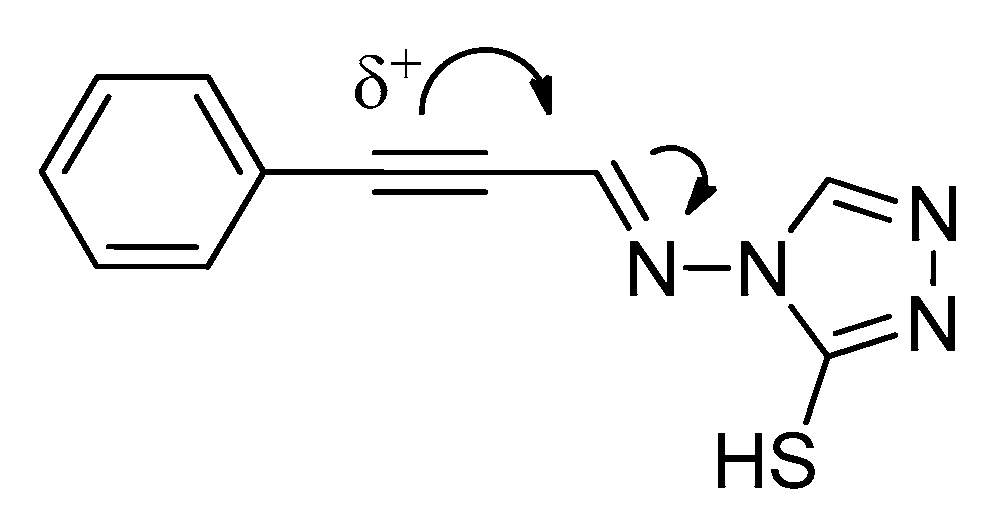
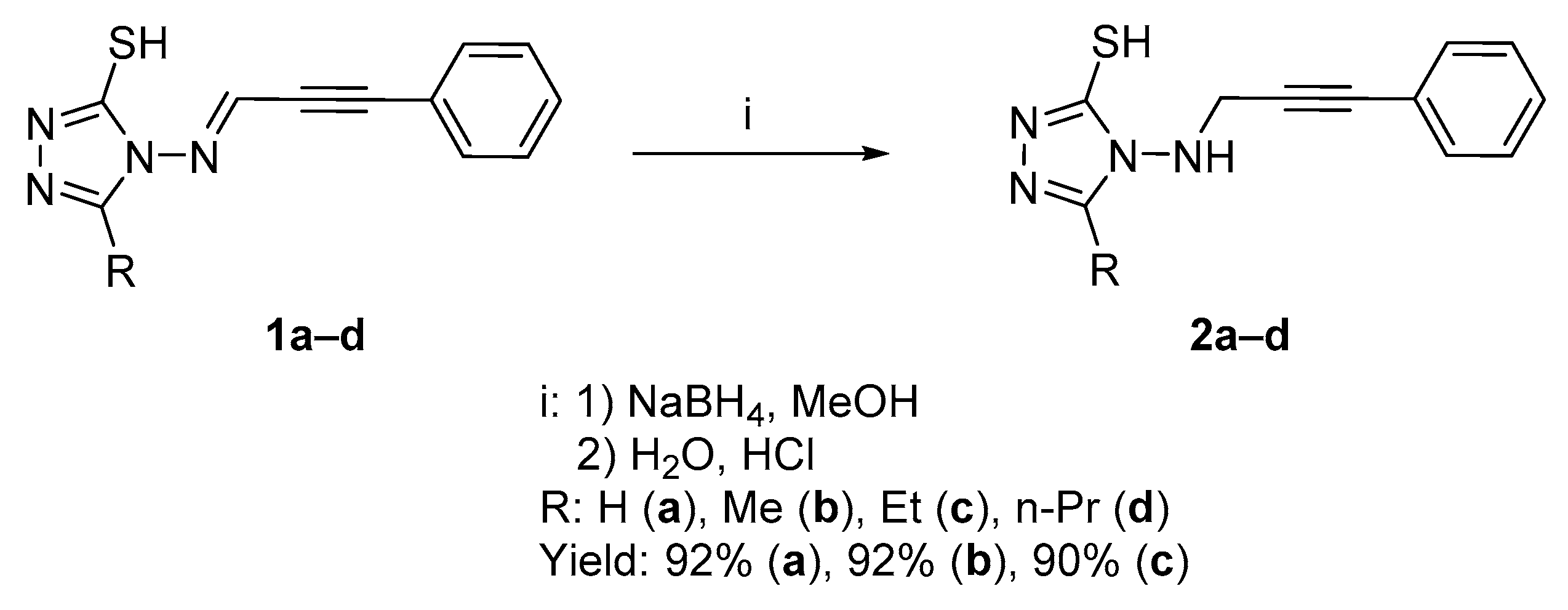
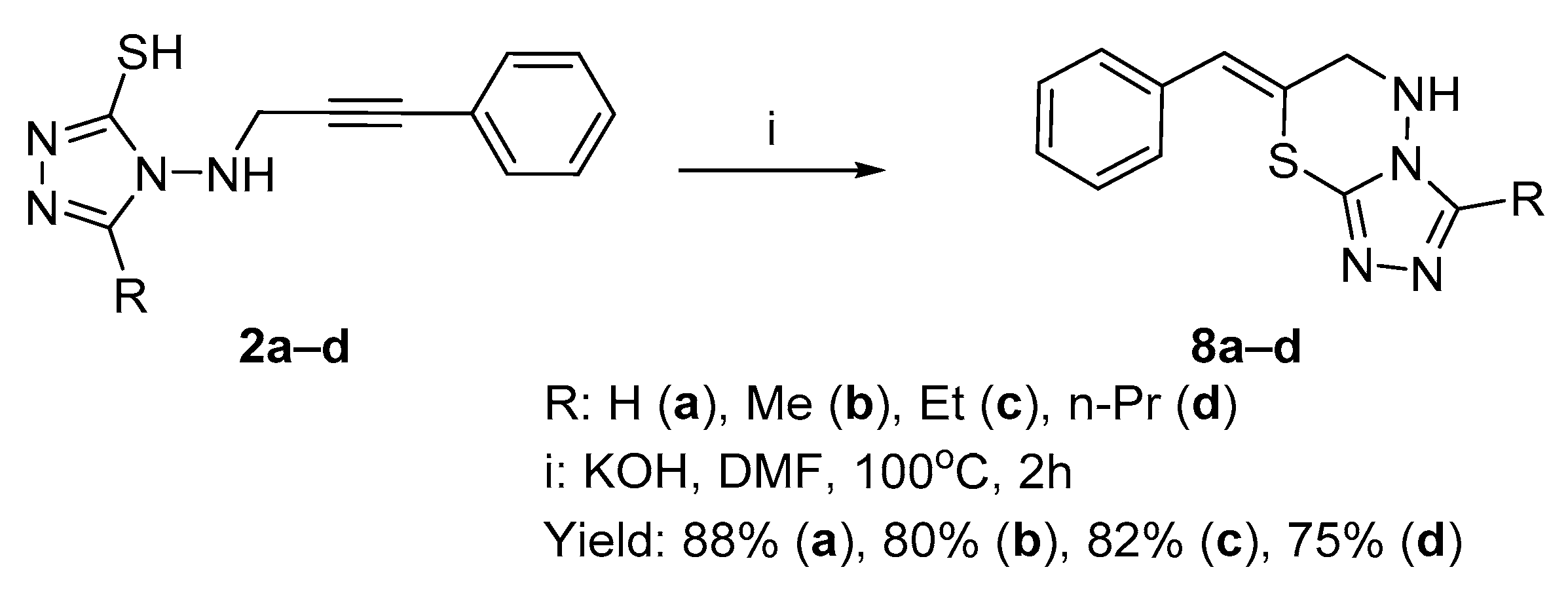
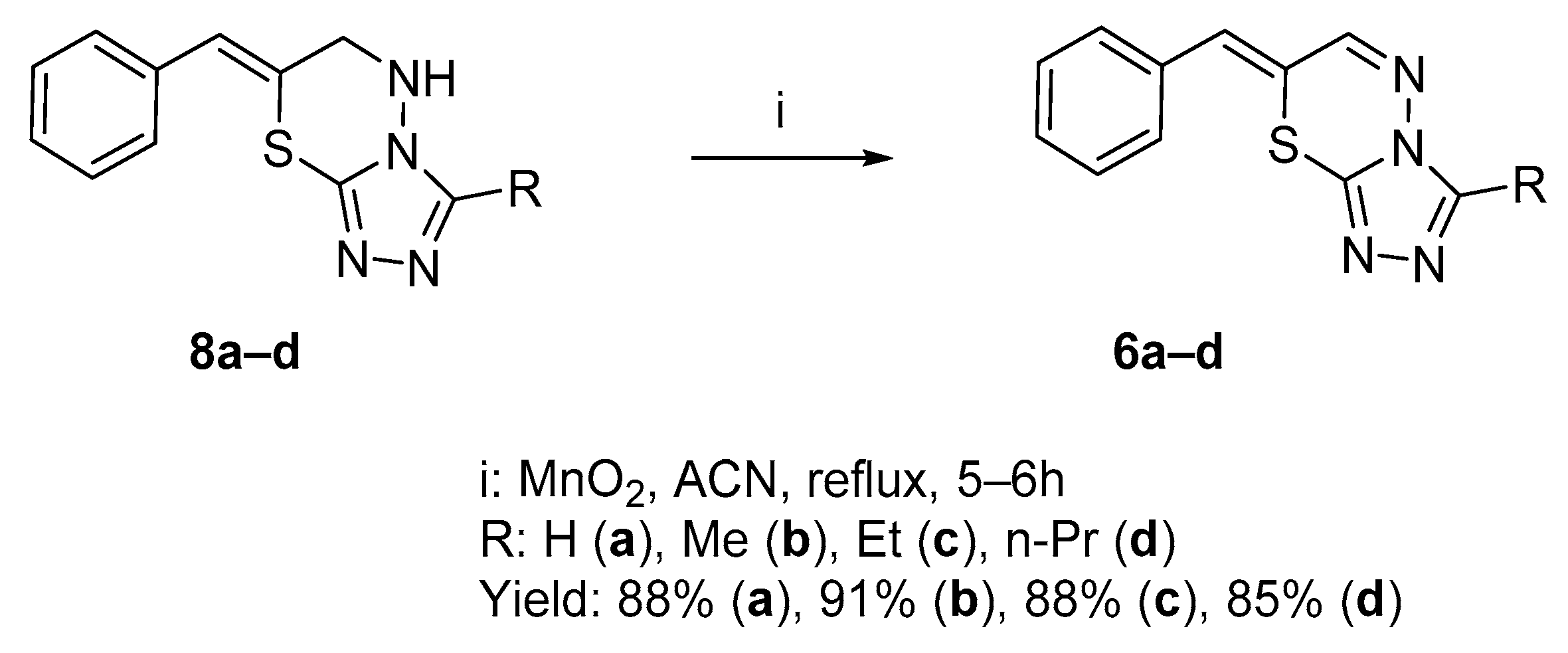
2.1. Chemistry
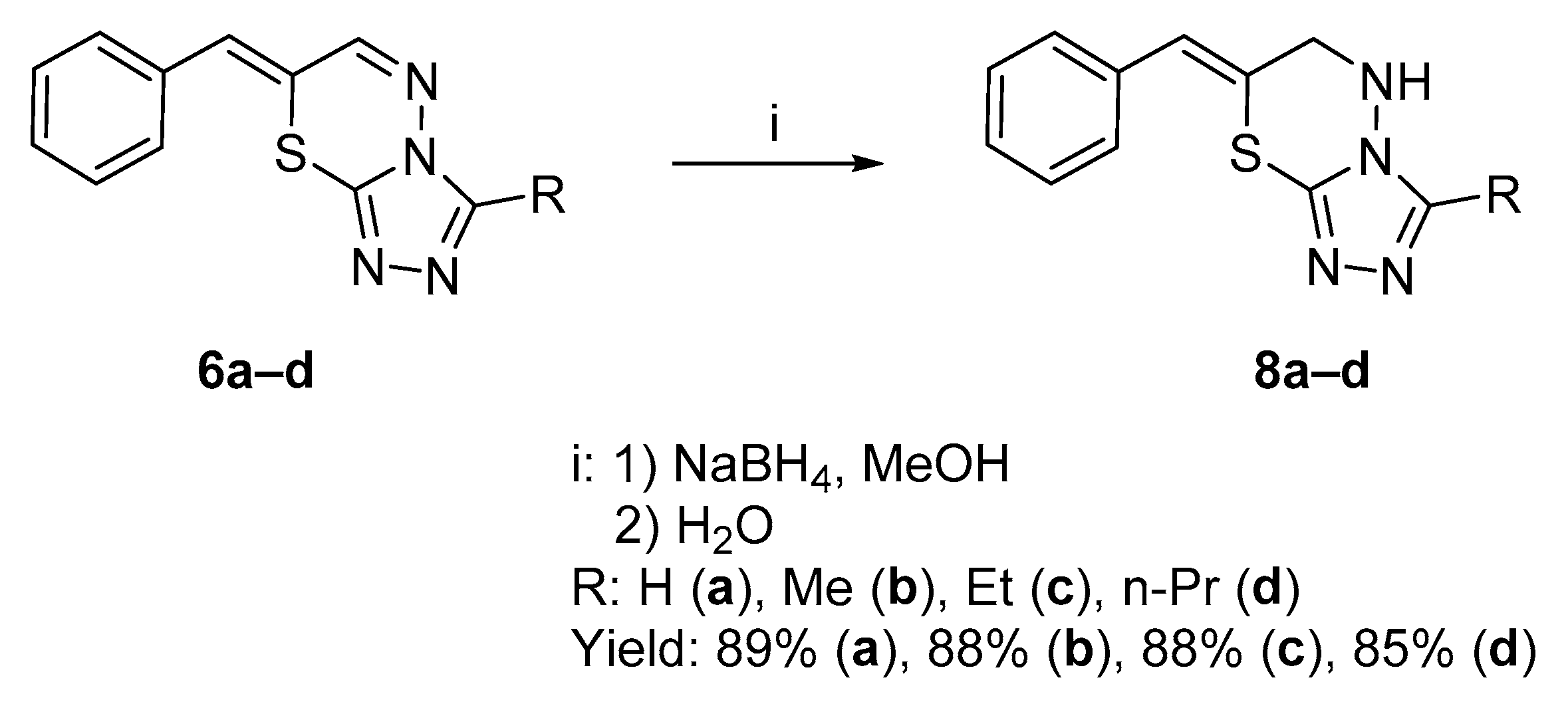
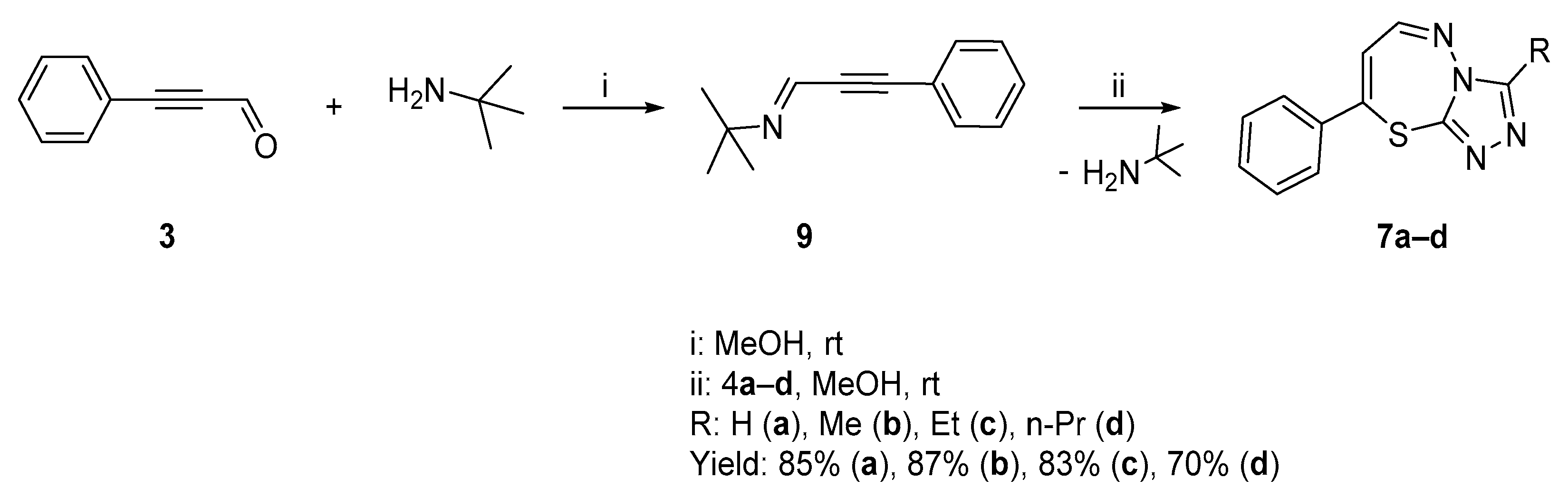
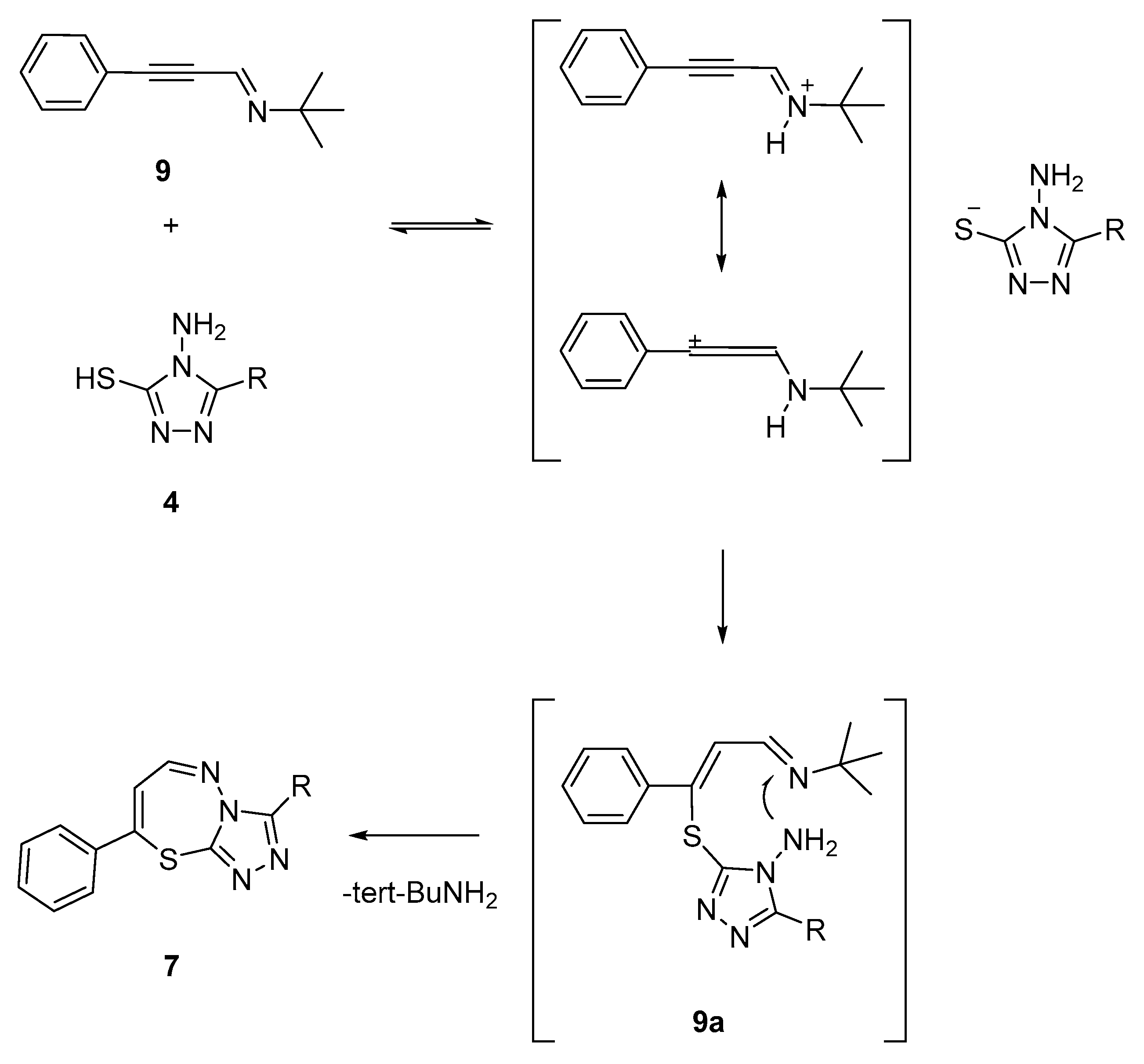
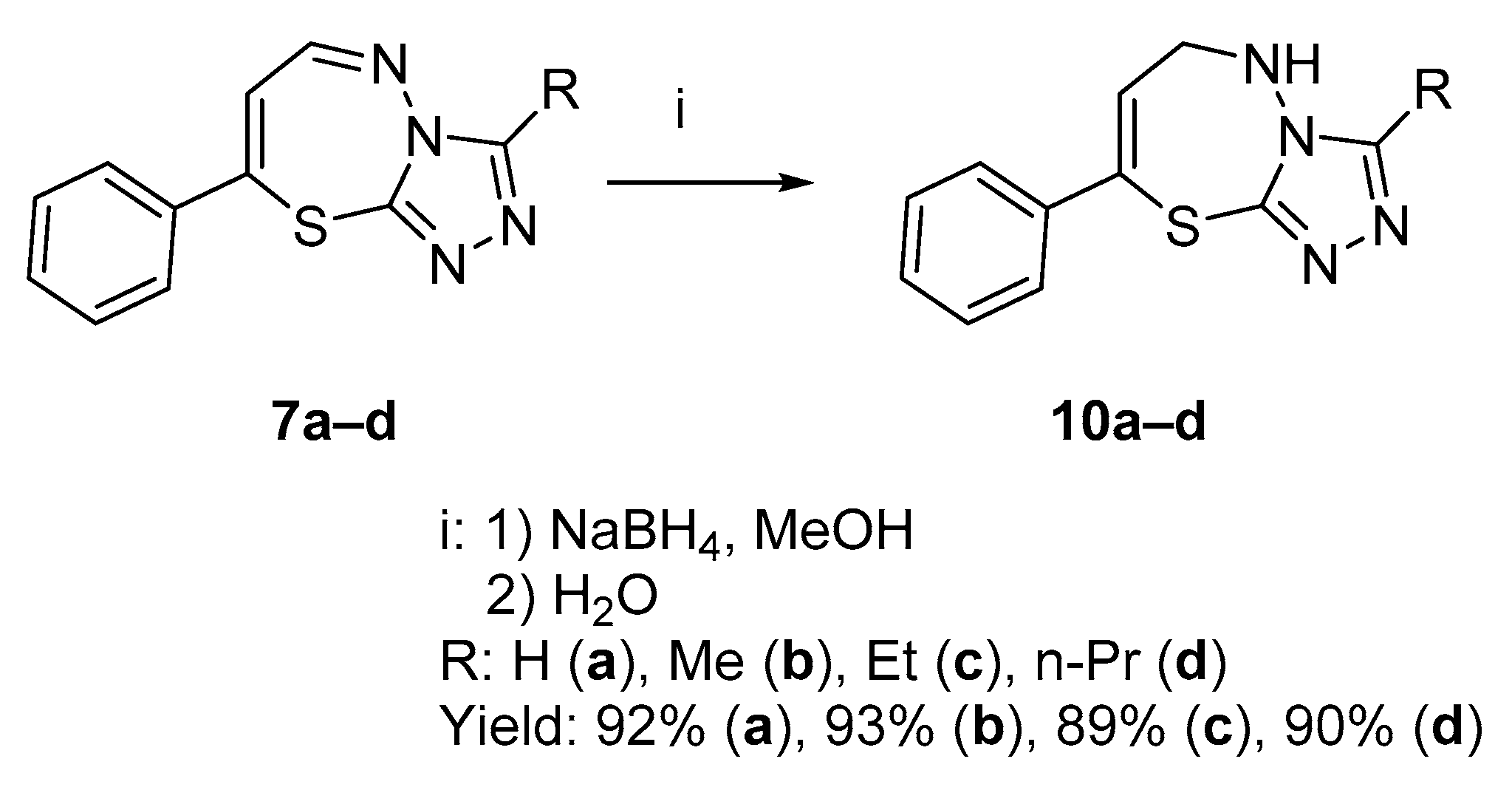
2.1.1. Synthesis of Target Compounds
2.1.2. Structural Analysis
2.2. In Vitro Experiments
2.3. Activity Validation and Target Affinity Prediction
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. X-ray Diffraction
3.4. Molecular Docking
3.5. The Study of the Biological Activity
3.6. The Study of the Anti-Viral Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kiselev, O. Chemo Drugs and Influenza Chemotherapy; Rostok: St. Petersburg, Russia, 2012. [Google Scholar]
- Furuyama, W.; Marzi, A. Annual Review of Virology Ebola Virus: Pathogenesis and Countermeasure Development. Annu. Rev. Virol. 2019, 6, 435–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Zarubaev, V.; Golod, E.; Anfimov, P.; Shtro, A.; Saraev, V.; Gavrilov, A.; Logvinov, A.; Kiselev, O. Synthesis and Anti-Viral Activity of Azolo-Adamantanes against Influenza A Virus. Bioorg. Med. Chem. 2010, 18, 839–848. [Google Scholar] [CrossRef] [PubMed]
- DeClercq, E. Antiviral Agents Active against Influenza A Viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef]
- El-Sebaey, S.A. Recent Advances in 1,2,4-Triazole Scaffolds as Antiviral Agents. ChemistrySelect 2020, 5, 11654–11680. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Farre, C.; Viezzi, S.; Wright, A.; Robin, P.; Lejal, N.; Manzano, M.; Vidic, J.; Chaix, C. Specific and Sensitive Detection of Influenza A Virus Using a Biotin-Coated Nanoparticle Enhanced Immunomagnetic Assay. Anal. Bioanal. Chem. 2022, 414, 265–276. [Google Scholar] [CrossRef]
- De, A.; Sarkar, S.; Majee, A. Recent Advances on Heterocyclic Compounds with Antiviral Properties. Chem. Heterocycl. Compd. 2021, 57, 410–416. [Google Scholar] [CrossRef]
- Ostrovskii, V.; Danagulyan, G.; Nesterova, O.; Pavlyukova, Y.; Tolstyakov, V.; Zarubina, O.; Slepukhin, P.; Esaulkova, Y.; Muryleva, A.; Zarubaev, V.; et al. Synthesis and Antiviral Activity of Nonannulated Tetrazolylpyrimidines. Chem. Heterocycl. Compd. 2021, 57, 448–454. [Google Scholar] [CrossRef]
- Shirley, M. Baloxavir Marboxil: A Review in Acute Uncomplicated Influenza. Drugs 2020, 80, 1109–1118. [Google Scholar] [CrossRef]
- Pécheur, E.-I.; Borisevich, V.; Halfmann, P.; Morrey, J.D.; Smee, D.F.; Prichard, M.; Mire, C.E.; Kawaoka, Y.; Geisbert, T.W.; Polyak, S.J. The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses. J. Virol. 2016, 90, 3086–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karade, N. 1,3,4-Oxadiazines and 1,3,4-Thiadiazines. In Comprehensive Heterocyclic Chemistry IV; Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 9, pp. 402–455. [Google Scholar]
- Kincaid, V.A.; London, N.; Wangkanont, K.; Wesener, D.A.; Marcus, S.A.; Héroux, A.; Nedyalkova, L.; Talaat, A.M.; Forest, K.T.; Shoichet, B.K.; et al. Virtual Screening for UDP-Galactopyranose Mutase Ligands Identifies a New Class of Antimycobacterial Agents. ACS Chem. Biol. 2015, 10, 2209–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Bai, X.; Deng, Q.; Zhang, G.; Zhou, L.; Liu, Y.; Wang, J.; Wang, Y. Preliminary SAR and Biological Evaluation of Antitubercular Triazolothiadiazine Derivatives against Drug-Susceptible and Drug-Resistant Mtb Strains. Bioorg. Med. Chem. 2017, 25, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Altintop, M.D.; Kuş, G.; Özkurt, M.; Özdemir, A.; Kaplancikli, Z.A. Indomethacin Based New Triazolothiadiazine Derivatives: Synthesis, Evaluation of Their Anticancer Effects on T98 Human Glioma Cell Line Related to COX-2 Inhibition and Docking Studies. Eur. J. Med. Chem. 2016, 113, 179–186. [Google Scholar] [CrossRef]
- Mohamady, S.; Gibriel, A.A.; Ahmed, M.S.; Hendy, M.S.; Naguib, B.H. Design and Novel Synthetic Approach Supported with Molecular Docking and Biological Evidence for Naphthoquinone-Hydrazinotriazolothiadiazine Analogs as Potential Anticancer Inhibiting Topoisomerase-IIB. Bioorg. Chem. 2020, 96, 103641. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Kaya Çavuşoğlu, B.; Sağlık, B.N.; Osmaniye, D.; Levent, S.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis, Docking Studies and Biological Activity of New Benzimidazole-Triazolothiadiazine Derivatives as Aromatase Inhibitor. Molecules 2020, 25, 1642. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.; Shokrzadeh, M.; Karima, S.; Rajaei, S.; Hashemi, S.M.; Mirzaei, H.; Fallah, M.; Emami, S. Design, Synthesis and Biological Evaluation of Flexible and Rigid Analogs of 4H-1,2,4-Triazoles Bearing 3,4,5-Trimethoxyphenyl Moiety as New Antiproliferative Agents. Bioorg. Chem. 2019, 93, 103300. [Google Scholar] [CrossRef]
- Penta, S.; Gadidasu, K.K.; Basavoju, S.; Rajeswar Rao, V. An Efficient One-Pot Synthesis of Pyrazolyl-[1,2,4]Triazolo[3,4-b][1,3,4] Thiadiazin-6-Yl)-2H-Pyran-2-One Derivatives via Multicomponent Approach and Their Potential Antimicrobial and Nematicidal Activities. Tetrahedron Lett. 2013, 54, 5663–5666. [Google Scholar] [CrossRef]
- El-Sayed, H.A.; Assy, M.G.; Mohamed, A.S. An Efficient Synthesis and Antimicrobial Activity of N-Bridged Triazolo[3,4-b]Thiadiazine and Triazolo[3,4-b]Thiadiazole Derivatives under Microwave Irradiation. Synth. Commun. 2020, 50, 997–1007. [Google Scholar] [CrossRef]
- Laskin, J.D.; Heindel, N.; Heck, D.; Vetrano, A.M.; Guillon, C.; DeMatteo, P. Fluorescent Fused-Ring Triazoles That Inhibit Cell Proliferation and Uses Thereof. U.S. Patent 7; Application granted, 12 September 2006. [Google Scholar]
- Shaikh, S.K.J.; Kamble, R.R.; Bayannavar, P.K.; Somagond, S.M.; Joshi, S.D. Triazolothiadizepinylquinolines as Potential MetAP-2 and NMT Inhibitors: Microwave-Assisted Synthesis, Pharmacological Evaluation and Molecular Docking Studies. J. Mol. Struct. 2020, 1203, 127445. [Google Scholar] [CrossRef]
- Kalluraya, B.; Rahiman, M.A.; Banji, D. Sydnone Derivatives. Part 5. Synthesis and Pharmacological Properties of Some Novel Triazolothiadiazepines. Indian J. Chem. 2022, 41B, 1712–1717. [Google Scholar] [CrossRef]
- Boraei, A.T.A.; Ghabbour, H.A.; Gomaa, M.S.; el Ashry, E.S.H.; Barakat, A. Synthesis and Anti-Proliferative Assessment of Triazolo-Thiadiazepine and Triazolo-Thiadiazine Scaffolds. Molecules 2019, 24, 4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavurala, S.; Vaarla, K.; Kesharwani, R.; Naesens, L.; Liekens, S.; Vedula, R.R. Bis Coumarinyl Bis Triazolothiadiazinyl Ethane Derivatives: Synthesis, Antiviral Activity Evaluation, and Molecular Docking Studies. Synth. Commun. 2018, 48, 1494–1503. [Google Scholar] [CrossRef]
- Khan, I.; Hameed, S.; Al-Masoudi, N.A.; Abdul-Reda, N.A.; Simpson, J. New Triazolothiadiazole and Triazolothiadiazine Derivatives as Kinesin Eg5 and HIV Inhibitors: Synthesis, QSAR and Modeling Studies. Z. Naturforsch. B Chem. Sci. 2015, 70, 47–58. [Google Scholar] [CrossRef]
- Pandey, V.K.; Tusi, Z.; Tusi, S.; Joshi, M. Synthesis and Biological Evaluation of Some Novel 5-[(3-Aralkyl Amido/Imidoalkyl)Phenyl]-1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazines as Antiviral Agents. ISRN Org. Chem. 2012, 2012, 760517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karabasanagouda, T.; Adhikari, A.V.; Shetty, N.S. Synthesis and Antimicrobial Activities of Some Novel 1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazoles and 1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazines Carrying Thioalkyl and Sulphonyl Phenoxy Moieties. Eur. J. Med. Chem. 2007, 42, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-Y.; Zhang, L.-X.; Chen, X.-X.; Zhang, A.-J.; Zhang, H.-L. Syntheses and Biological Activities of 6-Aryl-3-(3-Hydroxy-Propyl)-7H-1,2,4-Triazolo[3,4-b][1,3,4]Thiadiazines. Molecules 2007, 12, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shawali, A.S.; Zeid, I.F.; Abdelkader, M.H.; Elsherbini, A.A.; Altalbawy, F.M.A. Synthesis, Acidity Constants and Tautomeric Structure of 7-Arylhydrazono[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazines in Ground and Excited States. J. Chin. Chem. Soc. 2001, 48, 65–82. [Google Scholar] [CrossRef]
- Hui, X.-P.; Xu, P.-F.; Wang, Q.; Zhang, Q.; Zhang, Z.-Y. Synthesis and Antibacterial Activity of 3-(5-Methylisoxazol-3-Yl)-1,2,4-Triazolo[3,4-b]-1,3,4-Thiadiazine Derivatives. Chin. J. Chem. 2001, 19, 991–995. [Google Scholar] [CrossRef]
- Motamedi, R.; Heravi, M.M.; Bamoharram, F.F.; Haeri, A. Facile Routes to 1,2,4-Triazolo[3,4-b][1,3,4]Thiadiazines and 1,2,4- Triazino[3,4-b][1,3,4]Thiadiazine by Heteropolyacides. J. Heterocyclic Chem. 2008, 45, 1211–1214. [Google Scholar] [CrossRef]
- Aggarwal, R.; Hooda, M.; Kumar, P.; Sumran, G. Vision on Synthetic and Medicinal Facets of 1,2,4-Triazolo[3,4-b][1,3,4]Thiadiazine Scaffold. Top. Curr. Chem. 2022, 380, 10. [Google Scholar] [CrossRef] [PubMed]
- Heindel, N.; Reid, J. 4-Amino-3-mercapto-4H-1,2,4-triazoles and Propargyl Aldehydes: A New Route to 3-R-8-aryl-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazepines. J. Heterocycl. Chem. 1980, 17, 1087–1088. [Google Scholar] [CrossRef]
- Skryl’nikova, M.A.; Khramchikhin, A.V.; Krivchun, M.N. A New Approach to the Synthesis of 1,2,4-Triazolo[3,4-b][1,3,4]Thiadiazines. Russ. J. Gen. Chem. 2017, 87, 1321–1322. [Google Scholar] [CrossRef]
- Karpov, M.V.; Khramchikhin, A.V.; Stadnichuk, M.D. Reactions of Phenylpropynal N-Tert-Butylimine with 4-Amino-3-Mercapto-1,2,4-Triazole Derivatives. Russ. J. Gen. Chem. 2005, 75, 487–488. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very Fast Prediction and Rationalization of PKa Values for Protein-Ligand Complexes. Proteins Struct. Funct. Genet. 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. In Current Protocols in Bioinformatics; Wiley: Hoboken, NJ, USA, 2007; Volume 18. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 6a | 7c | 8a | 8c | 10a |
|---|---|---|---|---|---|
| Crystal system | orthorhombic | monoclinic | monoclinic | triclinic | monoclinic |
| Space group | P212121 | P21/c | P21/c | P-1 | P21/c |
| a/Å | 3.84480(10) | 22.4518(5) | 22.7514(9) | 9.6530(5) | 9.0504(3) |
| b/Å | 6.6325(2) | 3.92160(10) | 7.8068(3) | 9.7098(5) | 9.9977(3) |
| c/Å | 39.1172(15) | 13.8956(3) | 12.3438(5) | 13.7763(6) | 12.1907(4) |
| α/° | 90 | 90 | 90 | 93.858(4) | 90 |
| β/° | 90 | 94.485(2) | 105.173(4) | 100.186(4) | 111.125(4) |
| γ/° | 90 | 90 | 90 | 91.383(4) | 90 |
| Volume/Å3 | 997.51(6) | 1219.72(5) | 2116.03(15) | 1267.15(11) | 1028.92(6) |
| Z | 4 | 4 | 8 | 4 | 4 |
| ρcalcg/cm3 | 1.520 | 1.396 | 1.446 | 1.354 | 1.487 |
| μ/mm−1 | 2.669 | 2.243 | 0.281 | 0.243 | 2.588 |
| F(000) | 472.0 | 536.0 | 960.0 | 544.0 | 480.0 |
| Crystal size/mm3 | 0.1 × 0.05 × 0.03 | 0.1 × 0.05 × 0.03 | 0.30 × 0.22 × 0.12 | 0.32 × 0.20 × 0.14 | 0.15 × 0.09 × 0.06 |
| Radiation | Cu Kα (λ = 1.54184 Å) | Cu Kα (λ = 1.54184 Å) | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) | Cu Kα (λ = 1.54184 Å) |
| 2Θ range for data collection/° | 4.518 to 139.91 | 3.948 to 138.074 | 5.538 to 54.998 | 5.666 to 54.998 | 10.478 to 152.372 |
| Index ranges | −4 ≤ h ≤ 3, −7 ≤ k ≤ 8, −43 ≤ l ≤ 47 | −27 ≤ h ≤ 24, −4 ≤ k ≤ 4, −16 ≤ l ≤ 15 | −29 ≤ h ≤ 24, −10 ≤ k ≤ 10, −11 ≤ l ≤ 16 | −12 ≤ h ≤ 12, −12 ≤ k ≤ 10, −17 ≤ l ≤ 17 | −11 ≤ h ≤ 10, −12 ≤ k ≤ 12, −15 ≤ l ≤ 15 |
| Reflections collected | 4912 | 9507 | 9608 | 10799 | 8969 |
| Independent reflections | 1892 [Rint = 0.0506, Rsigma = 0.0467] | 2276 [Rint = 0.0334, Rsigma = 0.0288] | 4823 [Rint = 0.0283, Rsigma = 0.0468] | 5805 [Rint = 0.0268, Rsigma = 0.0463] | 2128 [Rint = 0.0367, Rsigma = 0.0270] |
| Data/restraints/ parameters | 1892/126/145 | 2276/194/209 | 4823/0/297 | 5805/0/335 | 2128/0/149 |
| Goodness-of-fit on F2 | 1.112 | 1.081 | 1.177 | 1.028 | 1.061 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0552, wR2 = 0.1376 | R1 = 0.0415, wR2 = 0.1043 | R1 = 0.0710, wR2 = 0.1707 | R1 = 0.0390, wR2 = 0.0862 | R1 = 0.0378, wR2 = 0.0928 |
| Final R indexes [all data] | R1 = 0.0571, wR2 = 0.1384 | R1 = 0.0443, wR2 = 0.1061 | R1 = 0.0809, wR2 = 0.1772 | R1 = 0.0514, wR2 = 0.0943 | R1 = 0.0414, wR2 = 0.0946 |
| Largest diff. peak/hole/e Å−3 | 0.64/−0.50 | 0.43/−0.48 | 1.62/−0.43 | 0.36/−0.29 | 0.39/−0.34 |
| Flack parameter | 0.00(3) | - | - | - | - |
| CCDC | 2212189 | 2212192 | 1813554 | 1813556 | 2212194 |
| Compound | a CC50, µM | b IC50, µM | c SI |
|---|---|---|---|
| 1a | 134 ± 11 | 16.2 ± 2.1 | 8 |
| 1b | 125.5 ± 11.3 | 24.8 ± 2.9 | 5 |
| 1c | 119.4 ± 9.6 | 11.3 ± 2.2 | 11 |
| 2a | >1300 | 4.3 ± 1.0 | >300 |
| 2b | >1200 | 106.4 ± 12.5 | >12 |
| 2c | 487.3 ± 22.5 | 37.2 ± 4.1 | 13 |
| 2d | 138 ± 12.0 | 100.9 ± 12.4 | 1 |
| 6a | 578.2 ± 32.3 | 34.2 ± 4.2 | 17 |
| 6b | 433.3 ± 37.6 | 99.9 ± 11.8 | 4 |
| 6c | 979 ± 55.1 | 117.8 ± 12.7 | 8 |
| 6d | >1100 | 33.6 ± 4.4 | >33 |
| 7a | 42.9 ± 3.2 | >13 | <3 |
| 7b | NT | NT | NT |
| 7c | 6.1 ± 0.3 | >3 | <2 |
| 7d | NT | NT | NT |
| 8a | >1300 | 39.5 ± 5.1 | >33 |
| 8b | >1200 | >1200 | 1 |
| 8c | >1100 | 50.3 ± 6.2 | >23 |
| 8d | >1100 | 58 ± 6.6 | >19 |
| 10a | >1300 | 703 ± 82.9 | >2 |
| 10b | >1100 | 41.4 ± 5.1 | 27 |
| 10c | 971.7 ± 66.4 | >300 | <3 |
| 10d | NT | NT | NT |
| Rimantadine | 312.3 ± 22.8 | 64.1 ± 7.2 | 5 |
| Oseltamivircarboxylate | >100 | 0.17 ± 0.02 | >588 |
| Compound | Glidescore, (kcal/mol) | MMGBSA ΔG, (kcal/mol) | IC50 | ||
|---|---|---|---|---|---|
| Neuraminidase | M2 | M2—Lipo | M2—Strain Energy | ||
| 1a | −4.39 | −6.42 | −12.06 | 0.85 | 16.2 ± 2.1 |
| 1b | −3.72 | −6.66 | −11.49 | 2.57 | 24.8 ± 2.9 |
| 1c | −4.07 | −6.96 | −14.21 | 1.11 | 11.3 ± 2.2 |
| 2a | −4.18 | −6.12 | −16.42 | 0.68 | 4.3 ± 1.0 |
| 2b | −4.57 | −6.38 | −13.41 | 1.56 | 106.4 ± 12.5 |
| 2c | −4.41 | −6.50 | −15.80 | 0.45 | 37.2 ± 4.1 |
| 2d | −3.48 | −5.96 | −14.62 | 1.82 | 100.9 ± 12.4 |
| 6a | −4.52 | −6.09 | −12.25 | 0.41 | 34.2 ± 4.2 |
| 6b | −4.28 | −6.15 | −10.91 | 0.58 | 99.9 ± 11.8 |
| 6c | −4.19 | −6.15 | −11.56 | 1.37 | 117.8 ± 12.7 |
| 6d | −3.85 | −6.00 | −12.20 | 0.70 | 33.6 ± 4.4 |
| 7a | −4.15 | −6.48 | −10.22 | 1.27 | >13 |
| 7c | −3.75 | −6.30 | −13.35 | 0.50 | >3 |
| 8a | −4.53 | −6.70 | −13.75 | 0.38 | 39.5 ± 5.1 |
| 8b | −4.50 | −6.19 | −11.62 | 0.34 | >1200 |
| 8c | −4.42 | −6.43 | −11.99 | 0.42 | 50.3 ± 6.2 |
| 8d | −4.20 | −6.38 | −12.70 | 0.61 | 58 ± 6.6 |
| 10a | −4.78 | −6.54 | −11.51 | 0.51 | 703 ± 82.9 |
| 10b | −3.41 | −6.47 | −14.77 | 0.75 | 41.4 ± 5.1 |
| 10c | −3.40 | −6.37 | −13.02 | 1.70 | >300 |
| Rimantadine | – | −6.60 | −13.25 | 0.78 | 64.1 ± 7.2 |
| Oseltamivir | −5.50 | – | – | – | 0.17 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khramchikhin, A.V.; Skryl’nikova, M.A.; Esaulkova, I.L.; Sinegubova, E.O.; Zarubaev, V.V.; Gureev, M.A.; Puzyk, A.M.; Ostrovskii, V.A. Novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazepine Derivatives: Synthesis, Anti-Viral In Vitro Study and Target Validation Activity. Molecules 2022, 27, 7940. https://doi.org/10.3390/molecules27227940
Khramchikhin AV, Skryl’nikova MA, Esaulkova IL, Sinegubova EO, Zarubaev VV, Gureev MA, Puzyk AM, Ostrovskii VA. Novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazepine Derivatives: Synthesis, Anti-Viral In Vitro Study and Target Validation Activity. Molecules. 2022; 27(22):7940. https://doi.org/10.3390/molecules27227940
Chicago/Turabian StyleKhramchikhin, Andrey V., Mariya A. Skryl’nikova, Iana L. Esaulkova, Ekaterina O. Sinegubova, Vladimir V. Zarubaev, Maxim A. Gureev, Aleksandra M. Puzyk, and Vladimir A. Ostrovskii. 2022. "Novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazepine Derivatives: Synthesis, Anti-Viral In Vitro Study and Target Validation Activity" Molecules 27, no. 22: 7940. https://doi.org/10.3390/molecules27227940