

Development and Validation of a Method for Determination of 43 Antimicrobial Drugs in Western-Style Pork Products by UPLC-MS/MS with the Aid of Experimental Design

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of UPLC-MS/MS Analysis
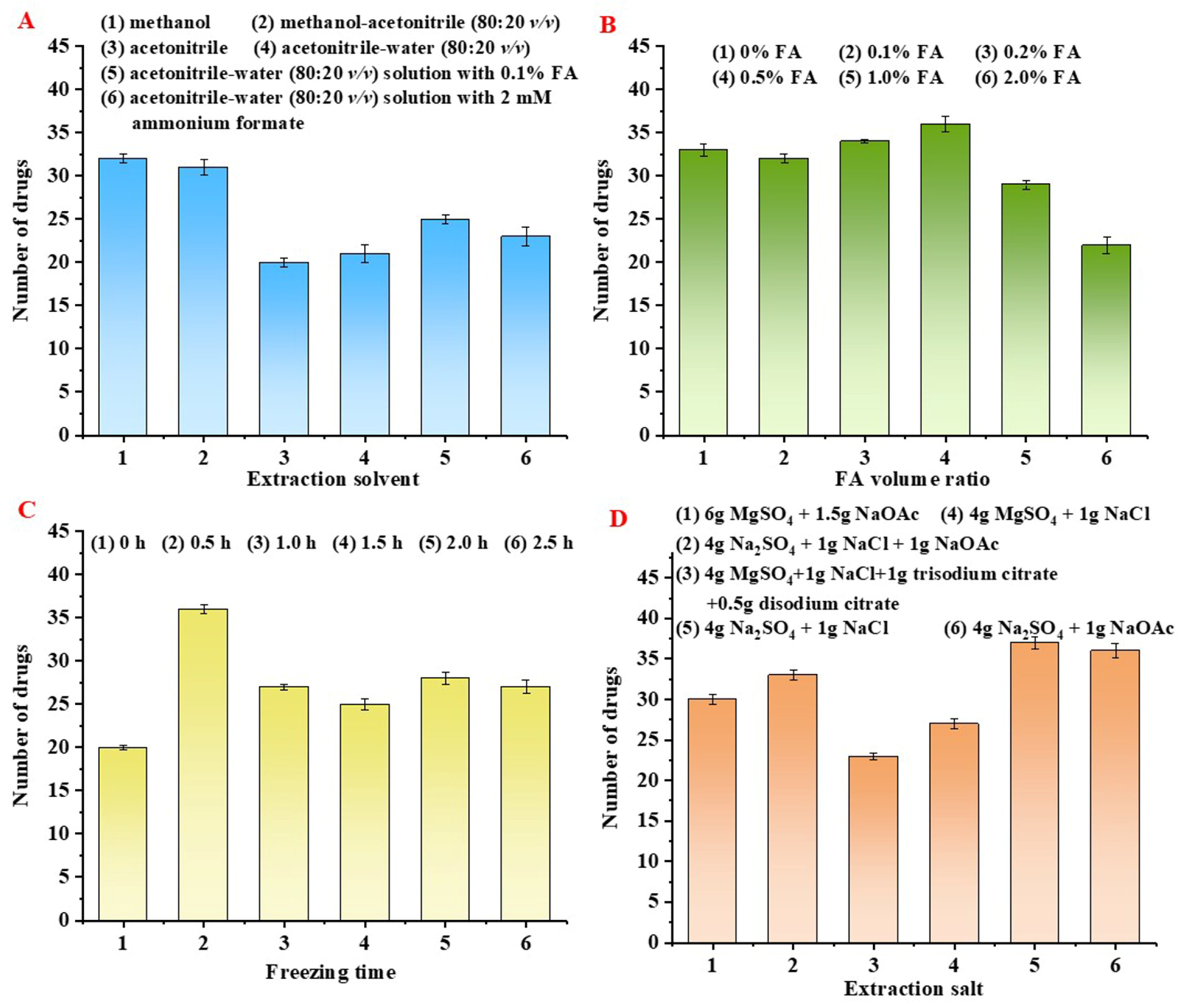
2.2. Optimization of QuEChERS Procedure
2.2.1. Optimization of Extraction Condition
2.2.2. Optimization of Clean-Up Condition
2.3. Method Performance and Validation
2.3.1. Matrix Effect
2.3.2. Selectivity and Linearity
2.3.3. CCα, CCβ, and LOQ
2.3.4. Accuracy and Precision
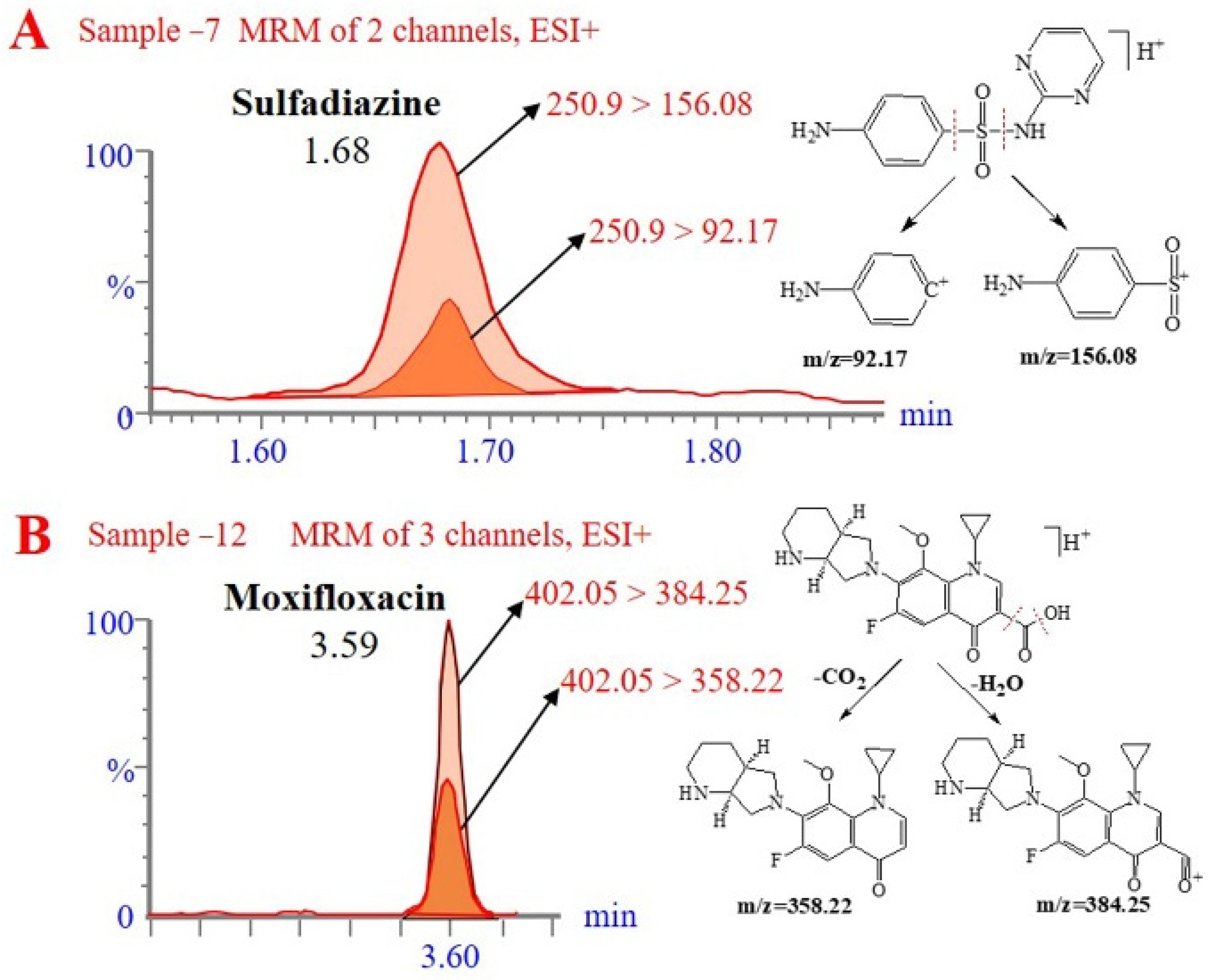
2.4. Analysis of Actual Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation Parameters
3.3. Sample Preparation
3.4. Validation of the Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hong, X.; Li, C.; Bai, J.; Gao, Z.; Wang, L. Chinese Consumers’ Willingness-to-pay for Nutrition Claims on Processed Meat Products, Using Functional Sausages as a Food Medium. China Agric. Econ. Rev. 2021, 13, 495–518. [Google Scholar] [CrossRef]
- Bu, T.; Tang, D.; Liu, Y.; Chen, D. Trends in dietary patterns and diet-related behaviors in China. Am. J. Health Behav. 2021, 45, 371–383. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, K.W.; Seow, W.L.; Mohamed, N.A.; Devaraj, N.K.; Amin-Nordin, S. Effectiveness of integrated technology apps for supporting healthy food purchasing and consumption: A systematic review. Foods 2021, 10, 1861. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.; Mora, L.; Reig, M.; Toldrá, F. Risk assessment of chemical substances of safety concern generated in processed meats. Food Sci. Hum. Well. 2019, 8, 244–251. [Google Scholar] [CrossRef]
- Chen, J.; Ying, G.G.; Deng, W.J. Antibiotic residues in food: Extraction, analysis, and human health concerns. J. Agric. Food Chem. 2019, 67, 7569–7586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, X.; Han, D.; Hou, Y.; Tan, J.; Kim, S.; Li, D.; Yin, Y.; Wang, J. Pork production systems in China: A review of their development, challenges and prospects in green production. Front. Agric. Sci. Eng. 2021, 8, 15–24. [Google Scholar] [CrossRef]
- State Administration for Market Regulation. 2018–2022. Beijing (CN). Available online: https://www.samr.gov.cn/zw/wjfb/ (accessed on 1 November 2022).
- Treiber, F.M.; Beranek-Knauer, H. Antimicrobial residues in food from animal origin—A review of the literature focusing on products collected in stores and markets worldwide. Antibiotics 2021, 10, 534. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Q.; Niu, B. Risk assessment of veterinary drug residues in meat products. Curr. Drug Metab. 2020, 21, 779–789. [Google Scholar] [CrossRef]
- US Food and Drug Administration. CFR-Code of Federal Regulations Title 21 Part 556 Tolerances for Residue of New Animal Drugs in Food; US Food and Drug Administration: Rockville, MD, USA, 2014.
- GB31650-2019; National Food Safety Standard on Maximum Residue Limits for Veterinary Drugs in Foods. Ministry of Agriculture and Rural Affairs of the People’s Republic of China: Beijing, China; National Health Commission of the People’s Republic of China, State Administration for Market Regulation: Beijing, China; National Standard of the People’s Republic of China: Beijing, China, 2019. Available online: https://www.aqsc.agri.cn/tzgg/201910/P02019101257017586-5095.pdf. (accessed on 1 November 2022).
- Commission European, Commission Regulation (EU) No 37/2010 of 22 December 2009 on Pharmacologically Active Substances and their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin (Text with EEA Relevance). Off. J. Eur. Union 2009, 32, 275–346.
- Moga, A.; Vergara-Barberán, M.; Lerma-García, M.J.; Carrasco-Correa, E.J.; Herrero-Martínez, J.M.; Simó-Alfonso, E.F. Determination of antibiotics in meat samples using analytical methodologies: A review. Compr. Rev. Food Sci. Food Saf. 2021, 20, 1681–1716. [Google Scholar] [CrossRef]
- Manimekalai, M.; Rawson, A.; Sengar, A.S.; Kumar, K.S. Development, optimization, and validation of methods for quantification of veterinary drug residues in complex food matrices using liquid-chromatography—A review. Food Anal. Method 2019, 12, 1823–1837. [Google Scholar] [CrossRef]
- Oyedeji, A.O.; Msagati, T.A.; Williams, A.B.; Benson, N.U. Detection and quantification of multiclass antibiotic residues in poultry products using solid-phase extraction and high-performance liquid chromatography with diode array detection. Heliyon 2021, 7, e08469. [Google Scholar] [CrossRef] [PubMed]
- Patyra, E.; Kwiatek, K.; Nebot, C.; Gavilán, R.E. Quantification of Veterinary Antibiotics in Pig and Poultry Feces and Liquid Manure as a Non-Invasive Method to Monitor Antibiotic Usage in Livestock by Liquid Chromatography Mass-Spectrometry. Molecules 2020, 25, 3265. [Google Scholar] [CrossRef] [PubMed]
- Melekhin, A.O.; Tolmacheva, V.V.; Goncharov, N.O.; Apyari, V.V.; Dmitrienko, S.G.; Shubina, E.G.; Grudev, A.I. Multi-class, multi-residue determination of 132 veterinary drugs in milk by magnetic solid-phase extraction based on magnetic hypercrosslinked polystyrene prior to their determination by high-performance liquid chromatography–tandem mass spectrometry. Food Chem. 2022, 387, 132866. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fan, S.; Sun, L.; He, L.; Zhang, Y.; Li, Q. Rapid analysis of fifteen sulfonamide residues in pork and fish samples by automated on-line solid phase extraction coupled to liquid chromatography–tandem mass spectrometry. Food Sci. Hum. Well. 2020, 9, 363–369. [Google Scholar] [CrossRef]
- Jia, W.; Chu, X.; Chang, J.; Wang, P.G.; Chen, Y.; Zhang, F. High-throughput untargeted screening of veterinary drug residues and metabolites in tilapia using high resolution orbitrap mass spectrometry. Anal. Chim. Acta 2017, 957, 29–39. [Google Scholar] [CrossRef]
- Zhao, W.; Jiang, R.; Guo, W.; Li, S.; Wang, S.; Li, Y. Screening and Analysis of Multiclass Veterinary Drug Residues in Animal Source Foods using UPLC-Q-Exactive Orbitrap/MS. B. Environ. Contam. Tox. 2021, 107, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Delatour, T.; Racault, L.; Bessaire, T.; Desmarchelier, A. Screening of veterinary drug residues in food by LC-MS/MS. Background and challenges. Food Addit. Contam. A 2018, 35, 633–646. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Lightfield, A.R. Extract-and-inject analysis of veterinary drug residues in catfish and ready-to-eat meats by ultrahigh-performance liquid chromatography–tandem mass spectrometry. J. AOAC Int. 2020, 103, 584–606. [Google Scholar] [CrossRef]
- Li, F.; Luo, J.; Zhu, B.; Liu, Z. Pretreatment Methods for the Determination of Antibiotics Residues in Food Samples and Detected by Liquid Chromatography Coupled with Mass Spectrometry Detectors: A Review. J. Chromatogr. Sci. 2022, 60, bmac021. [Google Scholar] [CrossRef]
- Zhao, L.; Lucas, D.; Long, D.; Richter, B.; Stevens, J. Multi-class multi-residue analysis of veterinary drugs in meat using enhanced matrix removal lipid cleanup and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1549, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.B.; Molognoni, L.; Deolindo, C.T.P.; Vargas, M.O.; Kleemann, C.R.; Daguer, H. Determination of 62 veterinary drugs in feedingstuffs by novel pressurized liquid extraction methods and LC-MS/MS. J. Chromatogr. B 2020, 1152, 122232. [Google Scholar] [CrossRef]
- Hoff, R.B.; Pizzolato, T.M.; Peralba, M.D.C.R.; Díaz-Cruz, M.S.; Barceló, D. Determination of sulfonamide antibiotics and metabolites in liver, muscle and kidney samples by pressurized liquid extraction or ultrasound-assisted extraction followed by liquid chromatography–quadrupole linear ion trap-tandem mass spectrometry (HPLC–QqLIT-MS/MS). Talanta 2015, 134, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Di, X.; Wang, X.; Liu, Y.; Guo, X.; Di, X. Microwave assisted extraction in combination with solid phase purification and switchable hydrophilicity solvent-based homogeneous liquid-liquid microextraction for the determination of sulfonamides in chicken meat. J. Chromatogr. B 2019, 1118, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Pan, X.; Huang, B.; Xu, J.; Wang, M.; Ren, Y. Determination of 15 β-lactam antibiotics in pork muscle by matrix solid-phase dispersion extraction (MSPD) and ultra-high pressure liquid chromatography tandem mass spectrometry. Food Control 2016, 66, 145–150. [Google Scholar] [CrossRef]
- González-Curbelo, M.Á.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Evolution and applications of the QuEChERS method. Trac-Trend. Anal. Chem. 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [Green Version]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS-Fundamentals, relevant improvements, applications and future trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef]
- Stubbings, G.; Bigwood, T. The development and validation of a multiclass liquid chromatography tandem mass spectrometry (LC–MS/MS) procedure for the determination of veterinary drug residues in animal tissue using a QuEChERS (Quick, Easy, Cheap, Effective, Rugged and Safe) approach. Anal. Chim. Acta 2009, 637, 68–78. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Zheng, J.; Zhang, Y.; Yang, L.; Liao, C.; Su, L.; Zhou, Y.; Gong, D.; Chen, L.; et al. The application of the QuEChERS methodology in the determination of antibiotics in food: A review. Trac-Trends Anal. Chem. 2019, 118, 517–537. [Google Scholar] [CrossRef]
- Ji, B.; Zhao, W.; Xu, X.; Han, Y.; Jie, M.; Xu, G.; Bai, Y. Development of a modified quick, easy, cheap, effective, rugged, and safe method based on melamine sponge for multi-residue analysis of veterinary drugs in milks by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2021, 1651, 462333. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.G.; Santos, F.A.; da Silva, J.C.C.; Augusti, R.; Faria, A.F. Multiresidue determination of fluoroquinolones in poultry muscle and kidney according to the regulation 2002/657/EC. A systematic comparison of two different approaches: Liquid chromatography coupled to high-resolution mass spectrometry or tandem mass spectrometry. J. Chromatogr. A 2015, 1379, 83–91. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Fan, K.; Minh Tien, M.; Savoy, M.C.; Tarres, A.; Fuger, D.; Goyon, A.; Bessaire, T.; Mottier, P. Determination of 105 antibiotic, anti-inflammatory, antiparasitic agents and tranquilizers by LC-MS/MS based on an acidic QuEChERS-like extraction. Food Addit. Contam. A 2018, 35, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tong, K.; Yu, C.; Hou, S.; Xie, Y.; Fan, C.; Chen, H.; Lu, M.; Wang, W. Development of a High-Throughput Screening Analysis for 195 Pesticides in Raw Milk by Modified QuEChERS Sample Preparation and Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry. Separations 2022, 9, 98. [Google Scholar] [CrossRef]
- Petrarca, M.H.; de Campos Braga, P.A.; Reyes, F.G.R.; Bragotto, A.P.A. Exploring miniaturized sample preparation approaches combined with LC-QToF-MS for the analysis of sulfonamide antibiotic residues in meat-and/or egg-based baby foods. Food Chem. 2022, 366, 130587. [Google Scholar] [CrossRef]
- Danezis, G.P.; Anagnostopoulos, C.J.; Liapis, K.; Koupparis, M.A. Multi-residue analysis of pesticides, plant hormones, veterinary drugs and mycotoxins using HILIC chromatography–MS/MS in various food matrices. Anal. Chim. Acta 2016, 942, 121–138. [Google Scholar] [CrossRef]
- Ferreira, S.L.; Junior, M.M.S.; Felix, C.S.; da Silva, D.L.; Santos, A.S.; Neto, J.H.S.; de Souza, C.T.; Junior, R.A.C.; Souza, A.S. Multivariate optimization techniques in food analysis–A review. Food Chem. 2019, 273, 3–8. [Google Scholar] [CrossRef]
- Anon: Commission Decision 2002/657/EC. Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Off. J. Eur. Commun. 2002, 50, 8–36. Available online: https://gfbicfb1d8b84f78a4871sc9w65wnvbf9b6q9bfiac.eds.tju.edu.cn/en/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en (accessed on 1 November 2022).
- Yin, Z.; Chai, T.; Mu, P.; Xu, N.; Song, Y.; Wang, X.; Jia, Q.; Qiu, J. Multi-residue determination of 210 drugs in pork by ultra-high-performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2016, 1463, 49–59. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Sum of Squares | Degrees of Freedom | Mean Square | F-Value | p-Value (Prob > F) | Distinctiveness |
|---|---|---|---|---|---|---|
| Model | 737.76 | 9 | 81.97 | 50.78 | <0.0001 | significant |
| A-C18 | 2 | 1 | 2 | 1.24 | 0.3024 | |
| B-PSA | 18 | 1 | 18 | 11.15 | 0.0124 | * |
| C-Z-Sep+ | 60.5 | 1 | 60.5 | 37.48 | 0.0005 | ** |
| AB | 1 | 1 | 1 | 0.62 | 0.4571 | |
| AC | 4 | 1 | 4 | 2.48 | 0.1595 | |
| BC | 0 | 1 | 0 | 0 | 1 | |
| A2 | 262.78 | 1 | 262.78 | 162.78 | <0.0001 | ** |
| B2 | 297.09 | 1 | 297.09 | 184.04 | <0.0001 | ** |
| C2 | 35.41 | 1 | 35.41 | 21.94 | 0.0023 | * |
| Residual | 11.3 | 7 | 1.61 | |||
| Lack of fit | 8.5 | 3 | 2.83 | 4.05 | 0.1051 | not significant |
| Pure error | 2.8 | 4 | 0.7 | |||
| Cor total | 749.06 | 16 |
| Compound | Bacon | Ham | ||||
|---|---|---|---|---|---|---|
| ccα | ccβ | LOQ | ccα | ccβ | LOQ | |
| (μg/kg) | (μg/kg) | (μg/kg) | (μg/kg) | (μg/kg) | (μg/kg) | |
| Quinolones (21) | ||||||
| cinoxacin | 16.2 | 22.5 | 0.1 | 20.2 | 30.3 | 0.05 |
| ciprofloxacin | 24.7 | 39.4 | 2 | 21.6 | 33.3 | 5 |
| danofloxacin | 12.4 | 14.8 | 0.1 | 24.4 | 38.8 | 0.2 |
| difluoxacin | 22.4 | 34.8 | 1 | 18.9 | 27.8 | 1 |
| enoxacin | 16.4 | 22.9 | 0.5 | 26.3 | 42.6 | 0.1 |
| enrofloxacin | 25.2 | 40.3 | 2 | 20.2 | 30.5 | 0.5 |
| fleroxacin | 24.9 | 39.8 | 2 | 21.7 | 33.4 | 10 |
| flumequine | 15.5 | 21 | 0.1 | 19.4 | 28.7 | 0.05 |
| gatifloxacin | 20.5 | 31 | 0.2 | 27.6 | 45.2 | 1 |
| gemifioxacin | 13.9 | 17.8 | 0.1 | 23.3 | 36.6 | 0.2 |
| lomefloxacin | 20.9 | 31.8 | 1 | 23.3 | 36.6 | 5 |
| marbofloxacin | 13.2 | 16.4 | 0.5 | 27.1 | 44.1 | 0.5 |
| moxifloxacin | 11.8 | 13.7 | 1 | 31.3 | 52.5 | 0.5 |
| nadifloxacin | 14.6 | 19.2 | 0.1 | 21.1 | 32.1 | 0.1 |
| nalidixic acid | 15.1 | 20.2 | 0.2 | 17.3 | 24.6 | 0.1 |
| ofloxacin | 25.9 | 41.8 | 2 | 27.1 | 44.2 | 5 |
| orbifloxacin | 23.8 | 37.7 | 5 | 29.4 | 48.8 | 2 |
| oxolinic acid | 18 | 26 | 0.1 | 19.7 | 29.3 | 0.1 |
| pefloxacin | 14.9 | 19.7 | 0.1 | 24.0 | 38.0 | 0.5 |
| sarafloxacin | 12.7 | 15.4 | 2 | 25.9 | 41.8 | 1 |
| sparfloxacin | 25.7 | 41.3 | 1 | 23.2 | 36.3 | 1 |
| Sulfonamides (22) | ||||||
| sulfabenzamide | 15.4 | 20.8 | 0.05 | 24.7 | 39.5 | 0.1 |
| sulfachloropyridazine | 17.8 | 25.6 | 1 | 25.1 | 40.1 | 2 |
| sulfaclozine | 18.8 | 27.6 | 0.1 | 26.7 | 43.5 | 1 |
| sulfadiazine | 11.6 | 13.3 | 0.05 | 16.5 | 23.0 | 0.1 |
| sulfadimidine | 16.2 | 22.5 | 0.1 | 22.7 | 35.3 | 0.05 |
| sulfadoxine | 12.8 | 15.6 | 0.1 | 24.6 | 39.3 | 0.05 |
| sulfamerazine | 13.9 | 17.8 | 0.2 | 18.4 | 26.8 | 1 |
| sulfameter | 11.9 | 13.9 | 0.2 | 25.7 | 41.5 | 1 |
| sulfamethizole | 17.3 | 24.5 | 0.1 | 18.1 | 26.1 | 0.5 |
| sulfamethoxazole | 14.2 | 18.4 | 0.05 | 20.6 | 31.3 | 0.05 |
| sulfamethoxypyridazine | 16 | 22 | 0.1 | 24.6 | 39.2 | 0.2 |
| sulfamonomethoxine | 21.8 | 33.6 | 0.1 | 21.4 | 32.7 | 0.2 |
| sulfamoxole | 19.4 | 28.9 | 0.1 | 21.6 | 33.2 | 0.2 |
| sulfaphenazole | 12.1 | 14.1 | 0.05 | 20.6 | 31.1 | 0.05 |
| sulfapyrazole | 12.7 | 15.3 | 0.1 | 16.8 | 23.7 | 0.05 |
| sulfapyridine | 15.5 | 20.9 | 0.05 | 19.2 | 28.3 | 0.2 |
| sulfaquinoxaline | 16.3 | 22.5 | 0.05 | 22.1 | 34.2 | 0.05 |
| sulfathiazole | 10.9 | 11.8 | 0.1 | 18.1 | 26.1 | 0.05 |
| sulfisomidine | 13.4 | 16.9 | 0.05 | 17.0 | 24.1 | 0.1 |
| sulfisoxazole | 15.6 | 21.2 | 0.05 | 18.8 | 27.6 | 0.05 |
| sulfadimethoxine | 13.5 | 16.9 | 0.05 | 13.7 | 17.4 | 0.05 |
| trimethoprim | 13.7 | 17.3 | 0.05 | 20.7 | 31.4 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Wu, X.; Xie, Y.; Tong, K.; Wang, M.; Li, J.; Fan, C.; Chen, H. Development and Validation of a Method for Determination of 43 Antimicrobial Drugs in Western-Style Pork Products by UPLC-MS/MS with the Aid of Experimental Design. Molecules 2022, 27, 8283. https://doi.org/10.3390/molecules27238283
Yu X, Wu X, Xie Y, Tong K, Wang M, Li J, Fan C, Chen H. Development and Validation of a Method for Determination of 43 Antimicrobial Drugs in Western-Style Pork Products by UPLC-MS/MS with the Aid of Experimental Design. Molecules. 2022; 27(23):8283. https://doi.org/10.3390/molecules27238283
Chicago/Turabian StyleYu, Xiaoxuan, Xingqiang Wu, Yujie Xie, Kaixuan Tong, Minglin Wang, Jianhui Li, Chunlin Fan, and Hui Chen. 2022. "Development and Validation of a Method for Determination of 43 Antimicrobial Drugs in Western-Style Pork Products by UPLC-MS/MS with the Aid of Experimental Design" Molecules 27, no. 23: 8283. https://doi.org/10.3390/molecules27238283
APA StyleYu, X., Wu, X., Xie, Y., Tong, K., Wang, M., Li, J., Fan, C., & Chen, H. (2022). Development and Validation of a Method for Determination of 43 Antimicrobial Drugs in Western-Style Pork Products by UPLC-MS/MS with the Aid of Experimental Design. Molecules, 27(23), 8283. https://doi.org/10.3390/molecules27238283