

Tuning the Photophysics of Two-Arm Bis[(dimethylamino)styryl]benzene Derivatives by Heterocyclic Substitution

 , , , and
, , , and
Abstract
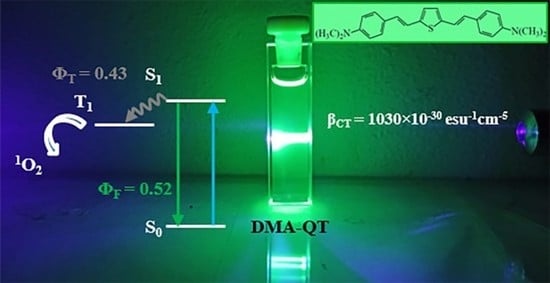
1. Introduction
2. Results
2.1. Solvent Effect on Spectral and Fluorescence Properties
2.2. Quantum Mechanical Calculations
2.3. NLO Properties
2.4. Excited-State Dynamics
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Synthesis Characterization
4.3. Materials
4.4. Photophysical Measurements
4.5. Quantum Mechanical Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Carlotti, B.; Cesaretti, A.; Fortuna, C.G.; Spalletti, A.; Elisei, F. Experimental Evidence of Dual Emission in a Negatively Solvatochromic Push–Pull Pyridinium Derivative. Phys. Chem. Chem. Phys. 2015, 17, 1877–1882. [Google Scholar] [CrossRef]
- Benassi, E.; Carlotti, B.; Segado, M.; Cesaretti, A.; Spalletti, A.; Elisei, F.; Barone, V. Presence of Two Emissive Minima in the Lowest Excited State of a Push–Pull Cationic Dye Unequivocally Proved by Femtosecond Up-Conversion Spectroscopy and Vibronic Quantum-Mechanical Computations. J. Phys. Chem. B 2015, 119, 6035–6040. [Google Scholar] [CrossRef]
- Cesaretti, A.; Carlotti, B.; Consiglio, G.; Del Giacco, T.; Spalletti, A.; Elisei, F. Inclusion of Two Push–Pull N-Methylpyridinium Salts in Anionic Surfactant Solutions: A Comprehensive Photophysical Investigation. J. Phys. Chem. B 2015, 119, 6658–6667. [Google Scholar] [CrossRef]
- Cesaretti, A.; Carlotti, B.; Germani, R.; Spalletti, A.; Elisei, F. Inclusion of Push–Pull N -Methylpyridinium Salts within Surfactant Hydrogels: Is Their Excited State Intramolecular Charge Transfer Mediated by Twisting? Phys. Chem. Chem. Phys. 2015, 17, 17214–17220. [Google Scholar] [CrossRef]
- Carlotti, B.; Benassi, E.; Cesaretti, A.; Fortuna, C.G.; Spalletti, A.; Barone, V.; Elisei, F. An Ultrafast Spectroscopic and Quantum Mechanical Investigation of Multiple Emissions in Push–Pull Pyridinium Derivatives Bearing Different Electron Donors. Phys. Chem. Chem. Phys. 2015, 17, 20981–20989. [Google Scholar] [CrossRef]
- Carlotti, B.; Cesaretti, A.; Gentili, P.L.; Marrocchi, A.; Elisei, F.; Spalletti, A. A Two Excited State Model to Explain the Peculiar Photobehaviour of a Flexible Quadrupolar D–π–D Anthracene Derivative. Phys. Chem. Chem. Phys. 2016, 18, 23389–23399. [Google Scholar] [CrossRef]
- Abraham, E.; Oberlé, J.; Jonusauskas, G.; Lapouyade, R.; Rullière, C. Photophysics of 4-Dimethylamino 4′-Cyanostilbene and Model Compounds: Dual Excited States Revealed by Sub-Picosecond Transient Absorption and Kerr Ellipsometry. Chem. Phys. 1997, 214, 409–423. [Google Scholar] [CrossRef]
- Singh, A.K.; Darshi, M.; Kanvah, S. α,ω-Diphenylpolyenes Cabable of Exhibiting Twisted Intramolecular Charge Transfer Fluorescence: A Fluorescence and Fluorescence Probe Study of Nitro- and Nitrocyano-Substituted 1,4-Diphenylbutadienes. J. Phys. Chem. A 2000, 104, 464–471. [Google Scholar] [CrossRef]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Pines, D.; Pines, E.; Rettig, W. Dual Fluorescence and Excited-State Structural Relaxations in Donor−Acceptor Stilbenes. J. Phys. Chem. A 2003, 107, 236–242. [Google Scholar] [CrossRef]
- Cesaretti, A.; Bonaccorso, C.; Elisei, F.; Fortuna, C.G.; Mencaroni, L.; Spalletti, A. Photoinduced Intramolecular Charge Transfer and Hyperpolarizability Coefficient in Push-Pull Pyridinium Salts with Increasing Strength of the Acceptor Group. ChemPlusChem 2018, 83, 1021–1031. [Google Scholar] [CrossRef]
- Carlotti, B.; Cesaretti, A.; Cannelli, O.; Giovannini, T.; Cappelli, C.; Bonaccorso, C.; Fortuna, C.G.; Elisei, F.; Spalletti, A. Evaluation of Hyperpolarizability from the Solvatochromic Method: Thiophene Containing Push–Pull Cationic Dyes as a Case Study. J. Phys. Chem. C 2018, 122, 2285–2296. [Google Scholar] [CrossRef]
- Oudar, J.L.; Chemla, D.S. Hyperpolarizabilities of the Nitroanilines and Their Relations to the Excited State Dipole Moment. J. Chem. Phys. 1977, 66, 2664–2668. [Google Scholar] [CrossRef]
- Prasad, P.N.; Williams, D.J. 1943-Introduction to Nonlinear Optical Effects in Molecules and Polymers; Wiley: Hoboken, NJ, USA, 1991. [Google Scholar]
- Kanis, D.R.; Ratner, M.A.; Marks, T.J. Design and Construction of Molecular Assemblies with Large Second-Order Optical Nonlinearities. Quantum Chemical Aspects. Chem. Rev. 1994, 94, 195–242. [Google Scholar] [CrossRef]
- Forrest, S.R.; Thompson, M.E. Introduction: Organic Electronics and Optoelectronics. Chem. Rev. 2007, 107, 923–925. [Google Scholar] [CrossRef]
- Kim, H.M.; Cho, B.R. Small-Molecule Two-Photon Probes for Bioimaging Applications. Chem. Rev. 2015, 115, 5014–5055. [Google Scholar] [CrossRef]
- Terenziani, F.; Painelli, A.; Katan, C.; Charlot, M.; Blanchard-Desce, M. Charge Instability in Quadrupolar Chromophores: Symmetry Breaking and Solvatochromism. J. Am. Chem. Soc. 2006, 128, 15742–15755. [Google Scholar] [CrossRef]
- Easwaramoorthi, S.; Shin, J.-Y.; Cho, S.; Kim, P.; Inokuma, Y.; Tsurumaki, E.; Osuka, A.; Kim, D. Versatile Photophysical Properties of Meso-Aryl-Substituted Subporphyrins: Dipolar and Octupolar Charge-Transfer Interactions. Chem.-Eur. J. 2009, 15, 12005–12017. [Google Scholar] [CrossRef]
- Huang, T.; Wang, Y.; Kang, Z.; Yao, J.; Lu, R.; Zhang, H. Investigation on Photophysical Properties of D–π–A–π–D-Type Fluorenone-Based Linear Conjugated Oligomers by Using Femtosecond Transient Absorption Spectroscopy. Photochem. Photobiol. 2014, 90, 29–34. [Google Scholar] [CrossRef]
- Lavanya Devi, C.; Yesudas, K.; Makarov, N.S.; Jayathirtha Rao, V.; Bhanuprakash, K.; Perry, J.W. Combined Experimental and Theoretical Study of One- and Two-Photon Absorption Properties of D–π–A–π–D Type Bis (Carbazolylfluorenylethynyl) Arene Derivatives: Influence of Aromatic Acceptor Bridge. Dyes Pigment. 2015, 113, 682–691. [Google Scholar] [CrossRef]
- Breukers, R.D.; Janssens, S.; Raymond, S.G.; Bhuiyan, M.D.H.; Kay, A.J. Synthesis and Characterization of Strongly Two Photon Absorbing and Photoswitchable Azo Molecules. Dyes Pigment. 2015, 112, 17–23. [Google Scholar] [CrossRef]
- Ricci, F.; Carlotti, B.; Keller, B.; Bonaccorso, C.; Fortuna, C.G.; Goodson, T.I.; Elisei, F.; Spalletti, A. Enhancement of Two-Photon Absorption Parallels Intramolecular Charge-Transfer Efficiency in Quadrupolar versus Dipolar Cationic Chromophores. J. Phys. Chem. C 2017, 121, 3987–4001. [Google Scholar] [CrossRef]
- Bonaccorso, C.; Cesaretti, A.; Elisei, F.; Mencaroni, L.; Spalletti, A.; Fortuna, C.G. New Styryl Phenanthroline Derivatives as Model D−π−A−π−D Materials for Non-Linear Optics. ChemPhysChem 2018, 19, 1917–1929. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, B.; Cesaretti, A.; Cacioppa, G.; Elisei, F.; Odak, I.; Škorić, I.; Spalletti, A. Fluorosolvatochromism and Hyperpolarizability of One-Arm and Two-Arms Nitro-Compounds Bearing Heterocyclic Rings. J. Photochem. Photobiol. Chem. 2019, 368, 190–199. [Google Scholar] [CrossRef]
- Mencaroni, L.; Carlotti, B.; Cesaretti, A.; Elisei, F.; Grgičević, A.; Škorić, I.; Spalletti, A. Competition between Fluorescence and Triplet Production Ruled by Nitro Groups in One-Arm and Two-Arm Styrylbenzene Heteroanalogues. Photochem. Photobiol. Sci. 2020, 19, 1665–1676. [Google Scholar] [CrossRef]
- Carlotti, B.; Spalletti, A.; Šindler-Kulyk, M.; Elisei, F. Ultrafast Photoinduced Intramolecular Charge Transfer in Push–Pull Distyryl Furan and Benzofuran: Solvent and Molecular Structure Effect. Phys. Chem. Chem. Phys. 2011, 13, 4519–4528. [Google Scholar] [CrossRef] [PubMed]
- Kikaš, I.; Carlotti, B.; Škorić, I.; Šindler-Kulyk, M.; Mazzucato, U.; Spalletti, A. Synthesis, Spectral Properties and Photobehaviour of Push–Pull Distyrylbenzene Nitro-Derivatives. J. Photochem. Photobiol. Chem. 2012, 244, 38–46. [Google Scholar] [CrossRef]
- Carlotti, B.; Kikaš, I.; Škorić, I.; Spalletti, A.; Elisei, F. Photophysics of Push–Pull Distyrylfurans, Thiophenes and Pyridines by Fast and Ultrafast Techniques. ChemPhysChem 2013, 14, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Bradamante, S.; Facchetti, A.; Pagani, G.A. Heterocycles as Donor and Acceptor Units in Push–Pull Conjugated Molecules. Part 1. J. Phys. Org. Chem. 1997, 10, 514–524. [Google Scholar] [CrossRef]
- Bureš, F.; Čermáková, H.; Kulhánek, J.; Ludwig, M.; Kuznik, W.; Kityk, I.V.; Mikysek, T.; Růžička, A. Structure–Property Relationships and Nonlinear Optical Effects in Donor-Substituted Dicyanopyrazine-Derived Push–Pull Chromophores with Enlarged and Varied π-Linkers. Eur. J. Org. Chem. 2012, 2012, 529–538. [Google Scholar] [CrossRef]
- Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Progress in the Enhancement of Electro-Optic Coefficients and Orientation Stability for Organic Second-Order Nonlinear Optical Materials. Dyes Pigment. 2020, 181, 108509. [Google Scholar] [CrossRef]
- Pham, T.T.T.; Chitose, Y.; Tam, T.T.T.; Tseng, W.-L.; Lin, T.-C.; Abe, M. Impact of Five-Membered Heterocyclic Rings on Photophysical Properties Including Two-Photon Absorption Character. Chem. Lett. 2021, 50, 1810–1813. [Google Scholar] [CrossRef]
- Achelle, S.; Verbitskiy, E.V.; Fecková, M.; Bureš, F.; Barsella, A.; Robin-le Guen, F. V-Shaped Methylpyrimidinium Chromophores for Nonlinear Optics. ChemPlusChem 2021, 86, 758–762. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; Achelle, S.; Bureš, F.; le Poul, P.; Barsella, A.; Kvashnin, Y.A.; Rusinov, G.L.; Guen, F.R.; Chupakhin, O.N.; Charushin, V.N. Synthesis, Photophysical and Nonlinear Optical Properties of [1,2,5]Oxadiazolo[3,4-b]Pyrazine-Based Linear Push-Pull Systems. J. Photochem. Photobiol. Chem. 2021, 404, 112900. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; le Poul, P.; Bureš, F.; Achelle, S.; Barsella, A.; Kvashnin, Y.A.; Rusinov, G.L.; Charushin, V.N. Push–Pull Derivatives Based on 2,4′-Biphenylene Linker with Quinoxaline, [1,2,5]Oxadiazolo[3,4-B]Pyrazine and [1,2,5]Thiadiazolo[3,4-B]Pyrazine Electron Withdrawing Parts. Molecules 2022, 27, 4250. [Google Scholar] [CrossRef]
- Görner, H.; Elisei, F.; Mazzucato, U.; Galiazzo, G. Trans → Cis Photoisomerization of 1-(1-Naphthyl)-2-(4-Nitrophenyl)Ethylene. J. Photochem. Photobiol. Chem. 1988, 43, 139–154. [Google Scholar] [CrossRef]
- Crespo-Hernández, C.E.; Burdzinski, G.; Arce, R. Environmental Photochemistry of Nitro-PAHs: Direct Observation of Ultrafast Intersystem Crossing in 1-Nitropyrene. J. Phys. Chem. A 2008, 112, 6313–6319. [Google Scholar] [CrossRef]
- Zugazagoitia, J.S.; Almora-Díaz, C.X.; Peon, J. Ultrafast Intersystem Crossing in 1-Nitronaphthalene. An Experimental and Computational Study. J. Phys. Chem. A 2008, 112, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, B.; Elisei, F.; Spalletti, A. A Peculiar Dependence of Intersystem Crossing of P-Nitro-2,5-Distyrylfuran on the Dielectric Properties of the Solvent. Phys. Chem. Chem. Phys. 2011, 13, 20787–20793. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, B.; Elisei, F.; Mazzucato, U.; Spalletti, A. Unusual High Fluorescence of Two Nitro-Distyrylbenzene-like Compounds Induced by CT Processes Affecting the Fluorescence/Intersystem-Crossing Competition. Phys. Chem. Chem. Phys. 2015, 17, 14740–14749. [Google Scholar] [CrossRef]
- Yang, J.-S.; Lin, C.-J. Fate of Photoexcited Trans-Aminostilbenes. J. Photochem. Photobiol. Chem. 2015, 312, 107–120. [Google Scholar] [CrossRef]
- Tian, X.; Hussain, S.; Wang, H.; Zhang, Q.; Zhao, M.; Chen, J.; Zhang, H.; Zhou, H.; Chen, Y.; Tian, Y. A Series of Water-Soluble Pyridinium Derivatives with Two-Photon Absorption in the near Infrared Region for Mitochondria Targeting under Stimulated Emission Depletion (STED) Nanoscopy. Dyes Pigment. 2017, 147, 90–98. [Google Scholar] [CrossRef]
- Zou, J.; Yin, Z.; Wang, P.; Chen, D.; Shao, J.; Zhang, Q.; Sun, L.; Huang, W.; Dong, X. Photosensitizer Synergistic Effects: D–A–D Structured Organic Molecule with Enhanced Fluorescence and Singlet Oxygen Quantum Yield for Photodynamic Therapy. Chem. Sci. 2018, 9, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xie, M.; Zhao, H.; Tang, Y.; Yao, S.; He, T.; Ye, C.; Wang, Q.; Lu, X.; Huang, W.; et al. Nitric Oxide Activatable Photosensitizer Accompanying Extremely Elevated Two-Photon Absorption for Efficient Fluorescence Imaging and Photodynamic Therapy. Chem. Sci. 2018, 9, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Solvent and Environmental Effects. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2006; pp. 205–235. ISBN 978-0-387-46312-4. [Google Scholar]
- Giglio, L.; Mazzucato, U.; Musumarra, G.; Spalletti, A. Photophysics and Photochemistry of 2,6-Distyrylpyridine and Some Heteroanalogues. Phys. Chem. Chem. Phys. 2000, 2, 4005–4012. [Google Scholar] [CrossRef]
- Spalletti, A.; Cruciani, G.; Mazzucato, U. Conformational Equilibria in EE-2,6-Di-[2-(Furan-2-Yl)Vinyl]Pyridine Controlled by Intramolecular Hydrogen-Type Bonds. J. Mol. Struct. 2002, 612, 339–347. [Google Scholar] [CrossRef]
- Baraldi, I.; Benassi, E.; Spalletti, A. Cis Peak as Probe to Investigate the Molecular Structure: Application to the Rotational Isomerism of 2,5-Diphenylethenyl(Hetero)Arenes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2008, 71, 543–549. [Google Scholar] [CrossRef]
- Alain, V.; Blanchard-Desce, M.; Ledoux-Rak, I.; Zyss, J. Amphiphilic Polyenic Push–Pull Chromophores for Nonlinear Optical Applications. Chem. Commun. 2000, 5, 353–354. [Google Scholar] [CrossRef]
- Cesaretti, A.; Foggi, P.; Fortuna, C.G.; Elisei, F.; Spalletti, A.; Carlotti, B. Uncovering Structure–Property Relationships in Push–Pull Chromophores: A Promising Route to Large Hyperpolarizability and Two-Photon Absorption. J. Phys. Chem. C 2020, 124, 15739–15748. [Google Scholar] [CrossRef]
- Wan, Y.; Guo, Q.; Wang, X.; Xia, A. Photophysical Properties of Rhodamine Isomers: A Two-Photon Excited Fluorescent Sensor for Trivalent Chromium Cation (Cr3+). Anal. Chim. Acta 2010, 665, 215–220. [Google Scholar] [CrossRef]
- Kita, H.; Yamakado, R.; Fukuuchi, R.; Konishi, T.; Kamada, K.; Haketa, Y.; Maeda, H. Switching of Two-Photon Optical Properties by Anion Binding of Pyrrole-Based Boron Diketonates through Conformation Change. Chem.-Eur. J. 2020, 26, 3404–3410. [Google Scholar] [CrossRef] [PubMed]
- Horng, M.L.; Gardecki, J.A.; Papazyan, A.; Maroncelli, M. Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited. J. Phys. Chem. 1995, 99, 17311–17337. [Google Scholar] [CrossRef]
- Reynolds, L.; Gardecki, J.A.; Frankland, S.J.V.; Horng, M.L.; Maroncelli, M. Dipole Solvation in Nondipolar Solvents: Experimental Studies of Reorganization Energies and Solvation Dynamics. J. Phys. Chem. 1996, 100, 10337–10354. [Google Scholar] [CrossRef]
- Kong, J.; Zhang, W.; Guo, Y.; Niu, X.; Yamao, T.; Yamashita, K.; Xia, A. Comprehensive Photophysical Properties of Thiophene/Phenylene Co-Oligomers Investigated by Theoretical and Experimental Studies. J. Phys. Chem. C 2020, 124, 18946–18955. [Google Scholar] [CrossRef]
- Carlotti, B.; Flamini, R.; Kikaš, I.; Mazzucato, U.; Spalletti, A. Intramolecular Charge Transfer, Solvatochromism and Hyperpolarizability of Compounds Bearing Ethenylene or Ethynylene Bridges. Chem. Phys. 2012, 407, 9–19. [Google Scholar] [CrossRef]
- Mencaroni, L.; Cesaretti, A.; Elisei, F.; Škorić, I.; Mlakić, M.; Spalletti, A. Acid–Base Strength and Acido(Fluoro)Chromism of Three Push–Pull Derivatives of 2,6-Distyrylpyridine. Photochem. Photobiol. Sci. 2022, 21, 935–947. [Google Scholar] [CrossRef]
- Bartocci, G.; Masetti, F.; Mazzucato, U.; Spalletti, A.; Baraldi, I.; Momicchioli, F. Photophysical and Theoretical Studies of Photoisomerism and Rotamerism of Trans-Styrylphenanthrenes. J. Phys. Chem. 1987, 91, 4733–4743. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Press: Boca Raton, FL, USA, 2006; ISBN 978-1-4200-1519-5. [Google Scholar]
- Schmidt, R.; Tanielian, C.; Dunsbach, R.; Wolff, C. Phenalenone, a Universal Reference Compound for the Determination of Quantum Yields of Singlet Oxygen O2(1Δg) Sensitization. J. Photochem. Photobiol. Chem. 1994, 79, 11–17. [Google Scholar] [CrossRef]
- Makitra, R.G.; Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, Weinheim: Wiley-VCH, 2003, 630p. Russ. J. Gen. Chem. 2005, 75, 664. [Google Scholar] [CrossRef]
- Bünau, G.V.J.B. Birks: Photophysics of Aromatic Molecules. Wiley-Interscience, London 1970. 704 Seiten. Preis: 210s. Berichte Bunsenges. Für Phys. Chem. 1970, 74, 1294–1295. [Google Scholar] [CrossRef]
- Reish, M.E.; Kay, A.J.; Teshome, A.; Asselberghs, I.; Clays, K.; Gordon, K.C. Testing Computational Models of Hyperpolarizability in a Merocyanine Dye Using Spectroscopic and DFT Methods. J. Phys. Chem. A 2012, 116, 5453–5463. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, I.; Hug, G.L. Triplet–Triplet Absorption Spectra of Organic Molecules in Condensed Phases. J. Phys. Chem. Ref. Data 1986, 15, 1–250. [Google Scholar] [CrossRef]
- Bartocci, G.; Spalletti, A.; Becker, R.S.; Elisei, F.; Floridi, S.; Mazzucato, U. Excited-State Behavior of Some All-Trans-α,ω-Dithienylpolyenes. J. Am. Chem. Soc. 1999, 121, 1065–1075. [Google Scholar] [CrossRef]
- Allonas, X.; Ley, C.; Bibaut, C.; Jacques, P.; Fouassier, J.P. Investigation of the Triplet Quantum Yield of Thioxanthone by Time-Resolved Thermal Lens Spectroscopy: Solvent and Population Lens Effects. Chem. Phys. Lett. 2000, 322, 483–490. [Google Scholar] [CrossRef]
- Samanta, A.; Ramachandram, B.; Saroja, G. An Investigation of the Triplet State Properties of 1,8-Naphthalimide: A Laser Flash Photolysis Study. J. Photochem. Photobiol. Chem. 1996, 101, 29–32. [Google Scholar] [CrossRef]
- Xu, C.; Webb, W.W. Measurement of Two-Photon Excitation Cross Sections of Molecular Fluorophores with Data from 690 to 1050 Nm. JOSA B 1996, 13, 481–491. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian16, RevisionB. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tortorella, S.; Talamo, M.M.; Cardone, A.; Pastore, M.; Angelis, F.D. Benchmarking DFT and Semi-Empirical Methods for a Reliable and Cost-Efficient Computational Screening of Benzofulvene Derivatives as Donor Materials for Small-Molecule Organic Solar Cells. J. Phys. Condens. Matter 2016, 28, 074005. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DMA-QP | DMA-QF | DMA-QT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvent | f (ε, n) a | λabs/nm | λem/nm | λabs/nm | λem/nm | λabs/nm | λem/nm | |||
| Tol | 0.02423 | 368 | 430 | 3920 | 428 | 484 | 2700 | 438, 462 sh | 488, 523 | 1200 |
| Tol/An (50/50 v/v) | 0.14278 | 367 | 437 | 4365 | 433 | 504 | 3250 | 440, 462 sh | 496, 528 | 1480 |
| An | 0.22353 | 371 | 448 | 4630 | 435 | 523 | 3570 | 442, 462 sh | 504, 533 | 1800 |
| EtOAc | 0.36011 | 363 | 443 | 4975 | 431 | 527 | 4230 | 430, 455 sh | 496 sh, 521 | 1820 |
| An/DCE (50/50 v/v) | 0.39998 | 369 | 452 | 4980 | 436 | 540 | 4420 | 440, 462 sh | 504, 533 | 1800 |
| DCE | 0.4968 | 368 | 458 | 5340 | 437 | 545 | 4540 | 437, 462 sh | 513 sh, 532 | 2850 |
| DCE/DMF (50/50 v/v) | 0.58324 | 368 | 468 | 5810 | 437 | 568 | 5280 | 438, 462 sh | 536 | 2990 |
| EtOH | 0.6668 | 367 | 480 | 6415 | 452 | 612 | 5780 | 429, 455 sh | 522 | 2820 |
| AcCN | 0.66697 | 364 | 451 | 5300 | 431 | 581 | 5990 | 432,455 sh | 537 | 3360 |
| DMF | 0.71215 | 368 | 474 | 6080 | 437 | 579 | 5610 | 440, 462 sh | 548 | 3400 |
| Calculated Parameter | Experimental Parameter | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | μGS/D | μES/D | a/Å | f | Slope | ΔμCT/D | βCT/10−30 esu−1 cm5 | β0/10−30 esu−1 cm5 | |
| DMA-QP | 3.41 | 6.68 | 11.1 | 1.3168 | 3200 | 27,170 | 20.9 | 248 | 114 |
| DMA-QF | 3.13 | 3.87 | 11.8 | 1.5571 | 4700 | 23,360 | 27.6 | 924 | 273 |
| DMA-QT | 1.56 | 2.40 | 12.9 | 1.9155 | 3200 | 22,830 | 21.1 | 1030 | 276 |
| DMA-QP | DMA-QF | DMA-QT | |||||
|---|---|---|---|---|---|---|---|
| Solvent | τ/ps | Assignment | |||||
| TA | FUC | TA | FUC | TA | FUC | ||
| Tol | 0.90 | 0.39 | Solv.i. | ||||
| 1.7 | 3.5 | 2.7 | 2.8 | 6.4 | 5.5 | Solv.d. | |
| 280 | 130 | SR | |||||
| 88 | 89 | 110 | 93 | 900 | 840 | S1 | |
| Inf | Inf | Inf | T1 | ||||
| DMF | 1.5 | 0.97 | 0.77 | 0.68 | 0.57 | 0.47 | Solv.i., S1,LE |
| 3.3 | 3.0 | 3.1 | 2.2 | Solv.d. | |||
| 150 | 140 | 280 | 270 | 170 | 110 | SR | |
| 1100 | 970 | 860 | 830 | 1000 | 840 | S1, ICT | |
| Inf | Inf | Inf | T1 | ||||
| Compound | Solvent | λT/nm | τT,AIR/μs | τT,N2/μs | kOX/109 s−1 | εT */M−1 cm−1 | ΦT | kT/108 s−1 | ΦΔ |
|---|---|---|---|---|---|---|---|---|---|
| DMA-QP | Tol | 460 | 0.23 | 4.63 | 4.2 | 5900 | 0.11 | 12 | |
| DMF | 450 | 0.09 | 3.14 | 0.28 | <0.05 | ||||
| DMA-QF | Tol | 650 | 0.09 | 0.41 | 8.8 | 4500 | 0.41 | 4.9 | |
| DMF | 600 | 0.13 | <0.01 | ||||||
| DMA-QT | Tol | 610 | 0.135 | 16.4 | 7.5 | 26,600 | 0.80 | 9.6 | 0.69 |
| DMF | 610 | 0.22 | 14.8 | 11.5 | 0.43 | 5.1 | 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mencaroni, L.; Cesaretti, A.; Carlotti, B.; Alebardi, M.; Elisei, F.; Ratković, A.; Škorić, I.; Spalletti, A. Tuning the Photophysics of Two-Arm Bis[(dimethylamino)styryl]benzene Derivatives by Heterocyclic Substitution. Molecules 2022, 27, 8725. https://doi.org/10.3390/molecules27248725
Mencaroni L, Cesaretti A, Carlotti B, Alebardi M, Elisei F, Ratković A, Škorić I, Spalletti A. Tuning the Photophysics of Two-Arm Bis[(dimethylamino)styryl]benzene Derivatives by Heterocyclic Substitution. Molecules. 2022; 27(24):8725. https://doi.org/10.3390/molecules27248725
Chicago/Turabian StyleMencaroni, Letizia, Alessio Cesaretti, Benedetta Carlotti, Martina Alebardi, Fausto Elisei, Ana Ratković, Irena Škorić, and Anna Spalletti. 2022. "Tuning the Photophysics of Two-Arm Bis[(dimethylamino)styryl]benzene Derivatives by Heterocyclic Substitution" Molecules 27, no. 24: 8725. https://doi.org/10.3390/molecules27248725
APA StyleMencaroni, L., Cesaretti, A., Carlotti, B., Alebardi, M., Elisei, F., Ratković, A., Škorić, I., & Spalletti, A. (2022). Tuning the Photophysics of Two-Arm Bis[(dimethylamino)styryl]benzene Derivatives by Heterocyclic Substitution. Molecules, 27(24), 8725. https://doi.org/10.3390/molecules27248725