Abstract
NADH:ubiquinone oxidoreductase core subunit S8 (NDUFS8) is an essential core subunit and component of the iron-sulfur (FeS) fragment of mitochondrial complex I directly involved in the electron transfer process and energy metabolism. Pathogenic variants of the NDUFS8 are relevant to infantile-onset and severe diseases, including Leigh syndrome, cancer, and diabetes mellitus. With over 1000 nuclear genes potentially causing a mitochondrial disorder, the current diagnostic approach requires targeted molecular analysis, guided by a combination of clinical and biochemical features. Currently, there are only several studies on pathogenic variants of the NDUFS8 in Leigh syndrome, and a lack of literature on its precise mechanism in cancer and diabetes mellitus exists. Therefore, NDUFS8-related diseases should be extensively explored and precisely diagnosed at the molecular level with the application of next-generation sequencing technologies. A more distinct comprehension will be needed to shed light on NDUFS8 and its related diseases for further research. In this review, a comprehensive summary of the current knowledge about NDUFS8 structural function, its pathogenic mutations in Leigh syndrome, as well as its underlying roles in cancer and diabetes mellitus is provided, offering potential pathogenesis, progress, and therapeutic target of different diseases. We also put forward some problems and solutions for the following investigations.
1. Introduction
Mitochondria in eukaryotes play a vital role in bioenergetics, metabolism, as well as signal transduction, and are related to various diseases [1]. Mitochondria contain at least 1500 different proteins in humans, functioning as core parts in energy metabolism, the fusion and fission of mitochondria, and the regulation of cell function [1,2]. From 20 to 25% of these proteins were used to express, regulate, and maintain the mitochondrial genome that codes for only 1% of mitochondrial proteins, and only up to 15% were directly involved in energy metabolism [3]. About 99% of mitochondrial proteins are encoded by nuclear genes, synthesized in the cytosol, and directed onto the membrane and then into the subcompartments of mitochondria by different signal pathways [4]. The invaginations of the mitochondrial inner membrane contain the respiratory complexes I to IV and the F1F0-ATP synthase, which constitute the oxidative phosphorylation system [1].
Mitochondrial complex I is also named mitochondrial NADH: ubiquinone oxidoreductase is the largest multi-subunit enzyme of the mitochondrial respiratory chain with 38 nuclear-encoded subunits and 7 subunits encoded by the mitochondrial genome [5]. Mitochondrial complex I, the initial and key limiting enzyme, can catalyze the transfer of electrons from Nicotinamide adenine dinucleotide (NADH) to ubiquinone through the respiratory chain, whose changes in the structure and function lead to wide varieties of clinical syndromes ranging from lethal encephalopathies to neurodegenerative disorders [6,7]. Complex I also consists of the I–III–IV supercomplexes, which may coordinate the activity of respiratory complexes, affect the assembly and stability of the complexes, and/or reduce the formation of reactive oxygen species (ROS) [8].
NADH:ubiquinone oxidoreductase core subunit S8 (NDUFS8), also called TYKY, is a nuclear-encoded subunit of human mitochondrial complex I with a molecular size of about 23 kDa. NDUFS8 is one of the core subunits of complex I, directly involved in the electron transfer process catalyzed by complex I owing to its special structure of containing tetranuclear FeS clusters (N6a and N6b) [9,10]. Based on this, pathogenic variants of the NDUFS8 are relevant to infantile-onset and severe diseases, including Leigh syndrome, cancer, and diabetes mellitus [6,9]. The first nuclear-encoded subunit mutations of complex I in a patient with Leigh syndrome are missense mutations in the NDUFS8, followed by several patients with different pathogenic variants [11]. In addition, with the development of the link between other subunits of complex I and cancer or diabetes mellitus, the results have gradually emerged, while the relation between NDUFS8 and these diseases is poorly studied [12,13,14,15]. Therefore, we summarize the structure, function, and possible roles or mechanisms of NDUFS8 in diseases in this review, establishing the foundation for future research.
2. The Structures and Functions of Complex I and NDUFS8
As mentioned above, complex I (CI), a redox-driven proton pump, can transfer electrons from NADH to ubiquinone, and translocate vectorial proton across the inner mitochondrial membrane [16,17]. The structure of CI has been described by X-ray crystallography and cryo-EM [8,18,19,20,21,22] (Figure 1A). It is an L-shaped molecule containing two domains: a hydrophilic peripheral domain extending into the mitochondrial matrix and a hydrophobic membrane domain [23,24]. The hydrophilic arm comprises the NADH oxidation module (N-module), which provides electron input into the chain of [FeS] clusters, and the ubiquinone reduction module (Q-module), which conducts electrons to the ubiquinone-binding site. There is a chain of seven FeS clusters that can transfer electrons from flavin mononucleotide (FMN) to the ubiquinone reduction site [25]. The hydrophobic arm comprises the proton translocation module (P-module), which catalyzes proton transport (Figure 1C) [26,27].
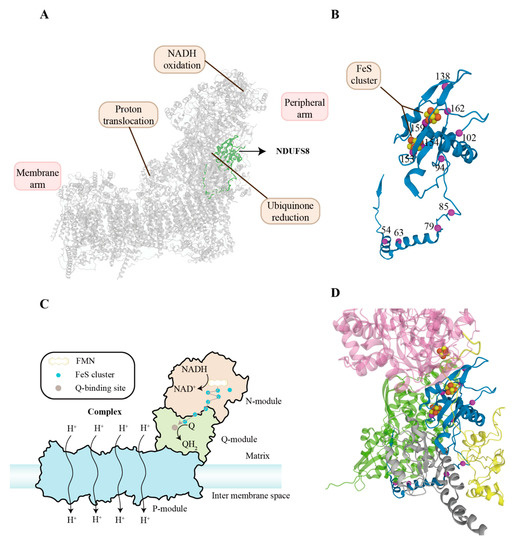
Figure 1.
Location, structure, functional mechanism, and clinical pathogenic variants of NDUFS8. (A) Cryo-EM structure of human respiratory complex I (CI) with other subunits in grey from 5XTD (EMD-6773) [8]. The ribbon highlighted in green represents NDUFS8. (B) The folds of NDUFS8 are shown in ribbon representation. NDUFS8 contains two [4Fe–4S]2+/1+ clusters represented by a combination of red and yellow balls. The pathogenic mutation sites of NDUFS8 are presented by magenta balls. (C) Schematic representation of components and functions of CI. CI consists of three functional modules: NADH oxidation (N module), ubiquinone reduction (Q module), and proton translocation (P module). They form an L-shape: one hydrophobic arm oriented parallel to the membrane and a hydrophilic arm extending into the mitochondrial matrix. There is a chain of seven FeS clusters that can transfer electrons from FMN to the ubiquinone reduction site. The P module harbors proton-translocation pathways connecting the mitochondrial matrix and the intermembrane space. (D) NDUFS8 (blue), NDUFS1 (pink), NDUFS2 (green), NDUFS7 (grey), and NDUFA12 (yellow) of complex I are shown. The annotated pathogenic mutation residues probably have effects on NDUFS8 and its surroundings, leading to complex I dysfunction [28]. Among them, p.Cys153Arg, p.Gly154Ser, and p.Val162Met are associated with the surroundings of two [4Fe–4S]2+/1+ clusters in NDUFS8, possibly disrupting electron transfer. The p.Ala159Asp lies next to Cys126, a component of the surroundings of two [4Fe–4S]2+/1+ clusters in NDUFS8. The p.Arg138His is linked to the disruption of electron transfer at the N5 cluster in NDUFS1. Moreover, the p.Arg94Cys is responsible for the impaired interaction of NDUFS8 and NDUFS2, while the p.Pro85Leu is associated with the impaired interaction of NDUFS8, NDUFA12, and NDUFS7. Abbreviations: NDUFS8: NADH:ubiquinone oxidoreductase core subunit S8; NDUFA12: NADH:ubiquinone oxidoreductase subunit A12; FMN: flavine mononucleotide; CI: complex I; NADH: Nicotinamide adenine dinucleotide; Q: ubiquinone.
NDUFS8 and homologues are highly conserved among eukaryotes and prokaryotes and extensively observed at various levels in all tissues, especially the heart and skeletal muscle [9,29,30,31,32,33]. The NDUFS8 is located on chromosome 11q13 immediately downstream of the aldehyde dehydrogenase 7 isoform gene, splitting into seven exons and six introns [9]. Its cDNA contains an open reading frame of 633 bp, coding for 210 amino acids [11]. The general transcription factors nuclear respiratory factor 1/2 (NRF1/2), Specificity Protein 1 (Sp1), and Yin Yang 1 (YY1) can activate transcription of the NDUFS8 [34]. As the electrical driving unit for a proton pump, NDUFS8 is a 23 kDa special ferredoxin and contains two [4Fe–4S]2+/1+ ferredoxin consensus patterns (N6a and N6b) (Figure 1B), which have long been thought to provide the binding site for the iron-sulfur ending cluster N-2 [35,36]. and there are two four-cysteine patterns (PROSITE pattern PS00198: CIACKLCEAICP and CIYCGFCQEACP) that ligand the two [4Fe–4S]2+/1+ clusters in NDUFS8 [34]. NDUFS8 is located within the ubiquinone-binding module (Q-module) involved in the early steps of respiratory complex I (CI) assembly, which implies its indispensable role in mitochondrial function [37]. A recent study has demonstrated in detail that NDUFS8 plays a crucial role in the global regulation of the cell growth and secondary metabolism of Monascus, revealing that its exact molecular mechanism is via changing the intracellular ROS and ATP levels [38].
3. Roles of NDUFS8 in Diseases
3.1. Pathogenic Variants of the NDUFS8 and Leigh Syndrome
Complex I deficiency caused by complex I gene mutation is highly relevant to mitochondrial disorders, leading to neuromuscular symptoms and various clinical manifestations [39]. Among them, Leigh syndrome (LS, OMIM: 256000) is the most common pediatric presentation of the mitochondrial disorder [40]. Typically first seen before 12 months of age, LS was first described by Denis Leigh in 1951 and characterized by focal, bilaterally symmetrical, and subacute necrotic lesions in the brainstem, the thalamus, and the posterior columns of the spinal cord [41]. Devastatingly, this neurodegenerative disorder is both phenotypically and genetically heterogeneous, and so far, pathogenic mutations in more than 75 genes have been proven, encoded by nuclear and mitochondrial genomes, especially the mutations referring to the mitochondrial respiratory enzyme complex or pyruvate dehydrogenase complex [40,42,43].
To date, mitochondrial complex I deficiency is the most common biochemical cause of LS, nearly constituting one-third of its etiology [40], and pathogenic variants of the NDUFS8 can be responsible for LS, including homozygous (c.236C > T/p.Pro79Leu, c.460G > A/p.Gly154Ser, c.187G > C/p.Glu63Gln, c.160C > T/p.Arg54Trp, c.281C > T het/p.Arg94Cys) and compound heterozygous (c.236C > T/p.Pro79Leu and c.305G > A/p.Arg102His, c.254C > T/p.Pro85Leu and c.413G > A/p.Arg138His, c.229C > T/p.Arg77Trp and c.476C > A/p.Ala159Asp, c.52C > T/p.Arg18Cys, c.484G > T/p.Val162Met, c.457T > C/p.Cys153Arg) variations in NDUFS8 (Figure 1D) [6,44,45,46]. These pathogenic variations influence assembly of complex I not only because of the misfolding of NDUFS8 but also owing to impaired interaction of other subunits and NDUFS8 [28]. Among them, c.236C > T/p.Pro79Leu and c.305G > A/p.Arg102His along with c.364G > A in the NDUFS7 gene have successfully been reconstructed by using the obligate aerobic yeast Yarrowia lipolytica, exhibiting similar complex I defects and possibly sharing their common characteristics in the pathogenesis of LS [47].
By searching the literature, 16 patients with Leigh syndrome carrying NDUFS8 mutations and 1 patient with encephalomyopathy carrying NDUFS8 mutations have been displayed in Table 1 below [48]. To date, all identified mutations were missense mutations, and no patients were reported with nonsense mutations in NDUFS8, which suggested that a complete absence of this protein might lead to intrauterine lethality [6]. The clinical manifestations of LS shown in Table 2 were intricate and manifold and varied from person to person [49]. Of the 13 patients, some showed severe manifestations with respiratory problems, feeding difficulties, epilepsy, and hypertrophic cardiomyopathy, and died within three months of life [11,50]. Others had normal or mildly impaired motor development in the first year of life and developed a slowly progressive neurological disease during childhood, such as slowly progressive muscle weakness, dysarthria, ataxic gait, and severe myopia [6,51]. According to a study in the Drosophila Model of LS, the severity of manifestations varied possibly depending on the maternally inherited mitochondrial background, and mitochondria–nuclear interactions affected lifespan and neurodegeneration [52]. In terms of genetic diagnosis of LS at present, the strategy is rapidly evolving as next-generation sequencing (NGS) technologies, and whole-exome sequencing (WGS), as well as whole-exon sequencing (WES), is becoming available [43,53]. This will make the molecular diagnosis of LS by screening patients’ genes possible [40].

Table 1.
Patients carrying pathogenic variants of the NDUFS8.
Table 2.
Clinical manifestations of Leigh syndrome.
Currently, symptomatic rather than etiological treatments are available for LS, which has been a challenge for researchers [58]. With the improvement of therapeutic technology, a cell-penetrating peptide derived from the HIV-1 trans-activator of transcription protein (TAT) has been successfully applied as a carrier to bring fusion proteins into cells without compromising the biological function of the cargoes. Therefore, Lin et al. successfully developed a TAT-mediated protein transduction system to rescue complex I deficiency caused by NDUFS8 defects and proved that treatment with TAT-NDUFS8 not only significantly ameliorated the assembly of complex I in a NDUFS8-deficient cell line but also partially rescued complex I function both in the in-gel activity assay and the oxygen consumption assay [10]. This study has provided us with a new approach to the treatment of Leigh syndrome.
3.2. NDUFS8 and Cancer
Functional mitochondria are essential for the cancer cell [59], and their dysfunction influences the balance of the intracellular environment through complicated signal pathways during cancer oncogenesis and progression [60]. Among all mitochondrial biochemical steps, complex I is the first entrance step of oxidative phosphorylation [61]. Complex I has also been recognized as one of the main sources of ROS, which is linked to cancer cell survival, proliferation, transformation, and malignancy progression [62,63,64,65]. Contrary to the increasing attention to mtDNA mutation in carcinogenesis [66,67,68], the roles of core nuclear-encoded subunits have not yet been much explored, such as NDUFS1, NDUFS2, NDUFS3, NDUFS7, NDUFS8, NDUFV1, and NDUFV2.
Among the 7 nuclear-encoded subunits above, the overexpression of NDUFS1 could augment the activity of complex I, reverse glycolysis, and enhance the radiosensitivity of colorectal cancer cells in vivo and in vitro [69]. NDUFS2, which impedes anticancer immune surveillance, is the primary target of the antitumor compound SMIP004-7 [70]. Not only core subunits but also accessory subunits are highly associated with the biological function of cancer cells. NDUFAF5 is one of the potential targets of NMS-873, which has been verified to reduce drug resistance in colon cancer cells [71]. The decrease of micropeptide in mitochondria could promote hepatoma metastasis by enhancing complex I activity, which could be attenuated by knocking down NDUFA7 [72]. Likewise, NDUFS8 is involved in carcinogenesis, tumor metabolism, and progression, as shown in Figure 2 [73]. For instance, owing to the upregulation of NDUFS8 mediated by mitochondrial Lon, their overexpression facilitated ROS generation, causing carcinogenesis and progression [62,74,75,76]. Meanwhile, deleterious mtDNA mutations were able to increase ROS, providing a proliferative advantage to cancer [77].
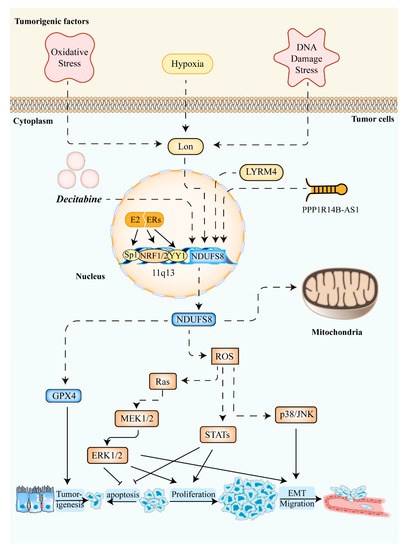
Figure 2.
The roles of NDUFS8 in cancers. Several cancerogenic factors, such as oxidative stress, hypoxia, and DNA damage stress upregulate the level of mitochondrial Lon. Then, the elevated expression of NDUFS8 via the mitochondrial Lon caused an increased level of ROS in mitochondria [62]. Increased ROS concentration in tumor cells improves cell survival, proliferation, transformation, and migration through the activation of Ras-ERK1/2 signaling [78], STATs, and/or p38/JNK [62]. Meanwhile, the expression of NDUFS8 shows a strong positive correlation with GPX4 expression linked to tumorigenesis [79]. Moreover, induced by the E2/ER pathway [80], the general transcription factors NRF1/2, Sp1, and YY1 can activate the transcription of NDUFS8. LYRM4 [81], long non-coding RNA PPP1R14B-AS1 [82], and decitabine [83] can also increase NDUFS8 expression in some cancer cells. Abbreviations: NDUFS8: NADH:ubiquinone oxidoreductase core subunit S8; ROS: Reactive Oxygen Species; GPX4: Glutathione peroxidase 4; EMT: epithelial–mesenchymal transition; MEK1/2: mitogen-activated protein kinase kinases 1/2; ERK1/2: extracellular signal-regulated kinase 1/2; STATs: signal transducers and activators of transcription; JNK: c-Jun N-terminal kinase; E2: estrogen; ER: estrogen receptor; NRF1/2: nuclear respiratory factor 1/2; Sp1: Specificity Protein 1; YY1: Yin Yang 1; LYRM4: LYR (leucine/tyrosine/arginine) motif protein 4.
Studies verified that high NDUFS8 and low NDUFS1 expressions were correlated with poor prognosis in patients and played a leading prognostic role in non-small-cell lung cancer (NSCLC), suggesting that their expressions might predict lung cancer prognosis [73,84]. This result could be possibly explained due to their different locations in mitochondria. NDUFS8 is located within the Q-module involved in the early steps of respiratory complex I (CI) assembly, while NDUFS1 is located within the N-module incorporated in the last step [37]. In this frame, reduced expression of Q-module subunits might have a more severe impact on the CI function than the N-module [73,85]. Consistent with high NDUFS8 expressions in NSCLC linking significantly reduced overall survival, studies suggested using mitochondrial markers as companion diagnostics in NSCLC patients, which was considerable for treatment stratification and personalized medicine [86]. For instance, a study found that NDUFS8 was one of the targets for the treatment of cyclopamine tartrate in NSCLC [87].
The elevated NDUFS8 could also be found in hepatocellular carcinoma (HCC) [81], which was associated with the dedifferentiation of HCC [88]. Likewise, the rise of NDUFS8 in HCC was modulated by LYR (leucine/tyrosine/arginine) motif protein 4 [81], helping HCC cells avoid sorafenib and successfully survive [89]. A combination of lung adenocarcinoma and liver cancer showed that NDUFS8 was overexpressed as well and positively related to long non-coding RNA PPP1R14B-AS1, promoting tumor cell proliferation and migration via the enhancement of mitochondrial respiration [82].
Similarly, according to the survival analysis, the increased expression level of NDUFS8 was associated with poorer overall survival in patients with acute myeloid leukemia (AML). However, how this gene related to the susceptibility and pathogenesis of AML was not extensively examined [79]. Recently, a rare NDUFS8 R2C complex I variant in the germline of two AML patients was identified as heterozygous, exhibiting a decreased basal and maximal oxygen consumption rate [90]. By testing AML blood samples, 47 genes were annotated within 500 Kb of the association signal and the sentinel variant is a significant expression quantitative trait locus for 12 of these, including NDUFS8 (PBH = 1.69 × 10−4) [91].
In breast cells, persistent overstimulation of estrogens and estrogen receptors contributed to the increase of NDUFS8 and ROS, gradually developing estrogen carcinogenesis (Figure 2) [80,92]. Importantly, NDUFS8 has been reported to have an association with tumor relapse in patients with estrogen receptor α-positive breast cancer [93]. Despite the pathogenesis described previously, compared to non-TNBC subtypes, NDUFS8 was downregulated in triple-negative breast cancer (TNBCs) with a reduction in mitochondrial respiratory capacity [94,95].
It has been reported that loss of heterozygosity at chromosome 11q13 existed in up to 70% to 100% of adrenocortical carcinoma (ACC) [96,97]. NDUFS8 was significantly differentially expressed between benign and malignant adrenocortical tumors (p < 0.05) with an overall accuracy of 87–91%, suggesting that NDUFS8 was a good diagnostic marker for distinguishing ACC from adenoma [98]. Based on the underexpression of NDUFS8 in ACC, decitabine, an inhibitor of DNA promoter methylation, recovered the expression of NDUFS8, which revealed a possible role of epigenetic gene silencing in adrenocortical carcinogenesis [83]. However, it was uncertain to confirm that NDUFS8 was an independent adverse predictor of shorter overall survival [99].
Besides, compared with the normal corresponding tissue, NDUFS8 expression was upregulated in some tumors, such as non-functional pituitary adenoma [100], while its expression was significantly decreased in others, such as clear-cell renal cell carcinoma [101] and ovarian clear-cell carcinoma [102]. However, there were still other cancer models not showing the change in NDUFS8 expression, possibly presenting that dysregulation of NDUFS8 may be specific to a few kinds of cancers [83,88,103,104,105,106].
All in all, research on NDUFS8 expression in cancers is mainly cell experiments and animal models, suggesting different expression levels and impacts in different cancer cells or even unworthiness, as shown in Table 3, and the main mechanism between cancer and NDUFS8 refers to mitochondrial dysfunction but lacks specific evidence [99]. Therefore, more extensive studies concerning NDUFS8 in different cancers need to be done with different approaches and aspects. For example, there are more than 50,000 sequenced and accumulated cancer genomes around the world by using WGS, WES, and RNA sequencing (RNA-Seq) [107,108,109]. When it comes to NDUFS8 and cancer, the techniques above, with a whole landscape of driver mutations [110], are of great importance to identify whether the NDUFS8 is a structural variant and pathogen of cancer or only the changing levels of NDUFS8 is a symbol of mitochondrial dysfunction.
Table 3.
NDUFS8 expression in different types of tumors.
3.3. NDUFS8 and Diabetes Mellitus
Diabetes mellitus is one of the metabolic disorders, tightly associated with energy metabolism and mitochondrial function [111,112]. Some trials have been made in patients with diabetes mellitus, in which they discovered the possible role of NDUFS8 change. It has been found that a higher serum concentration of NDUFS8 was linked to higher insulin sensitivity among people with type 1 diabetes mellitus, which might reflect better mitochondrial function [113]. Conversely, the NDUFS8 gene was highly expressed in the skeletal muscle tissue of patients with type 2 diabetes mellitus (T2DM), which might hint that upregulation of NDUFS8 expression would affect the normal glucose metabolism of skeletal muscle tissue, causing insulin resistance and then promoting diabetes. This study also concluded that, through the bioinformatic analysis, NDUFS8 might be a potential therapeutic target, which needed further studies together with molecular experiments [114]. During the formation of T2DM, high glucose, as a strong metabolic stressor, also induced the upregulation of NDUFS8 [115]. Additionally, NDUFS8 exhibited increased interaction with insulin receptor substrate 1, which was linked to inflammation-mediated insulin resistance in T2DM patients [116]. There was a similar result that showed that NDUFS8 expression was upregulation in high-fat diet-induced obesity-dependent diabetes mouse models, especially in the liver tissue [117]. In other studies, some factors, such as Sirtuin 1, leucine, and berberine, could directly or indirectly regulate the expression of the NDUFS8 and then change the secretory pathways and quantity of insulin, suggesting the agent role of NDUFS8 [118,119,120].
Since maternally inherited diabetes and deafness (MIDD) was first described [121], many studies on mitochondrial pathogenic variants have appeared successively. Currently, diabetes mellitus has also been reported in mitochondrial diseases caused by autosomal recessive mutations in the nuclear genes mtDNA polymerase gamma (1399G --> A), and mitochondrial inner membrane protein 17 (p.LysMet88-89MetLeu and p.Leu143 *) [122,123]. The m.14577T > C mutation in MT-ND6, which encodes NADH-ubiquinone oxidoreductase chain 6, a core subunit of complex I, was associated with diabetes mellitus [123]. When it comes to pathogenic variants of the NDUFS8 and diabetes mellitus, there is no study available, which implies that we can use WGS and WES to explore if the mutations exist [124].
3.4. NDUFS8 and Other Diseases
Owing to the importance of NDUFS8 in metabolism, it has relations with different diseases. As a vital component of the mitochondrial electron transport chain, expressions of NDUFS8 were detected or knocked out in many studies to reflect mitochondrial function and indicate the underlying mechanism [125,126,127,128,129]. For example, by measuring expressions of NDUFS8, microRNA-34a accelerated apoptosis of human lens epithelial cells via mitochondrial dysfunction [126]. Throughout the progression of sarcopenia, reduced NDUFS8 caused dysregulation of mitochondrial quality control in skeletal muscle in the senescence-accelerated mouse prone 8 [130]. The decreased NDUFS8 in sepsis was implicated in oxidative phosphorylation, showing disturbances in energy metabolism [131,132]. Embryos lacking NDUFS8 displayed the formation of an egg cylinder but lack of gastrulation features in mice, illustrating the initial value of NDUFS8 during the peri-gastrulation [133].
Besides, many substances can influence the kidney through NDUFS8. Paraoxonase1 deficiency and hyperhomocysteinemia changed the expression of mouse kidney proteins related to renal diseases, and downregulation of NDUFS8 could account for the involvement of hyperhomocysteinemia and reduced Paraoxonase1 in kidney disease through energy metabolism [134,135]. In the kidney of lipopolysaccharide-treated animals, NDUFS8 decreased and then led to decreased fatty acids oxidation following lipopolysaccharide treatment [136]. Because of the mitochondrial structural disorder after acute kidney injury, the level of NDUFS8 was down about 50%, which was ameliorated by some materials, such as mitochondria-targeted antioxidant Mito-2,2,6,6-tetramethylpiperidine-N-oxyl by injectable self-assembling peptide hydrogel, a novel cyclic helix B peptide, 5-methoxyindole-2-carboxylic acid, and curcumin [137,138,139,140]. Interestingly, NDUFS8 played a role in the process that perfusion of isolated rat kidneys with mesenchymal stromal cells/extracellular vesicles prevents ischemic damage, creating a possible application of NDUFS8 in organ transplantation [141].
What’s more, a group studied the mitochondrial role in the development of doxorubicin-induced cardiotoxicity and tested a decrease of NDUFS8 in this process, which implicated a possible mechanism of doxorubicin-induced cardiotoxicity [142], and similarly, downregulation of the NDUFS8 expression was also involved in the mechanism of Propofol-induced cytotoxicity in human-induced pluripotent stem cell-derived cardiomyocytes [143].
Moreover, NDUFS8 can have a relation with neural and psychiatric diseases. Studies showed that chronic stress affected the mitochondrial respiratory chain, and mitochondrial dysfunction might be a potential mechanism of psychiatric pathophysiology [144,145]. Compared with middle-aged individuals, there were obvious upexpressions of NDUFS8 in the frontal cortex in those with Parkinson’s disease, but notable downexpressions in those with Parkinson’s disease with dementia [146]. With the measurement of a significant upregulation of NDUFS8 in the resilient group compared with controls in rat models, it could be speculated that the regulation of this gene protected against stress [147]. Additionally, there was an appealing study about an antioxidant fruit named Euterpe oleracea which improved the expression of NDUFS8 to recover rotenone-caused mitochondrial dysfunction in neuronal-like cells SH-SY5Y [148].
4. Conclusions and Prospects
NDUFS8 is a highly conserved multi-functional subunit of mitochondrial complex I, involved in the electron transfer process and energy production. During mitochondrial respiration, electron transfer through this protein is linked to ROS production, activating the downstream signal pathways and then making much difference to multiple cellular activities. Owing to its vital structure of tetranuclear FeS clusters (N6a and N6b) and function of electron transfer, NDUFS8 plays a crucial role in different diseases and clinical processes, although there is no exact mechanism for cancer and diabetes mellitus.
Since Denis Leigh’s first description of Leigh syndrome in 1951 [41], many pathogenic mutations causing Leigh syndrome have been revealed, mostly linked to pathways of energy generation, but only 14 pathogenic variants of NDUFS8 causing Leigh syndrome have been reported. Pathogenic variants of NDUFS8 contribute to the onset of Leigh syndrome primarily by affecting the assembly of complex I, not only because of the misfolding of NDUFS8, but also owing to impaired interaction of other subunits and NDUFS8 [28]. On account of no effective treatment for Leigh syndrome, therapeutic intervention can benefit several biochemical and genetic forms of Leigh syndrome. Consequently, determining the molecular basis should be taken into great account, indicating applications of WGS and WES and enabling the progress of novel therapeutics.
Mitochondrial function plays an essential role in cancer cells. We have recognized mutations of mtDNA in cancer cells for more than two decades [149]. General attention paid to mitochondrial nuclear-encoded gene mutations has greatly emerged since the defects of fumarate hydratase, succinate dehydrogenase, isocitrate dehydrogenase 1 (IDH1), and IDH2 in cancer cells were established [150]. Currently, studies have demonstrated that tumor growth requires an electron transport chain (ETC) whose inhibition has an anti-tumor effect together with targeted treatment [151,152]. Under this circumstance, it is of vital significance to explore the proteins and genes of ETC. Therefore, we summarize the relations between cancer and NDUFS8 of complex I. On the one hand, the level of NDUFS8 shows a prognostic factor for NSCLC and AML in a combination of other subunits in complex I, possibly pointing out a therapeutic target for cancer. On the other hand, the expressions of NDUFS8 vary from cancer to cancer, showing its specific roles in different cancer cells. However, most of these studies have focused on the expressions of NDUFS8 rather than the genetic level, which lacks molecular mechanisms of oncogenesis, tumor formation, and progression. RNA interference techniques can be utilized to manipulate the expression of disease-related genes, including small interfering RNA (siRNA) and short hairpin RNA (shRNA) [153,154], which inspires people to develop novel methods of disease treatment [155]. Providing an efficient and accurate technology, CRISPR/Cas9 and modified versions have become a powerful tool for etiological analysis, drug development, and cell therapy through altering the genomes [156]. Therefore, the use of siRNA, shRNA, and CRISPR/Cas9 technology could be applied in the future research of NDUFS8-related disease modeling, diagnosis, and treatment.
As a complex metabolic disorder, diabetes mellitus has an impact on many tissues where mitochondrial dysfunction can also be observed. The interactions of diabetes and mitochondria have been under discussion, especially mitochondrial roles in the pathology of diabetes complications [157]. Based on such a situation, we concentrate on NDUFS8 and its potential influence on diabetes. According to recent studies, different types of diabetes mellitus show diverse relevance to NDUFS8, from reflecting mitochondrial function to promoting diabetes formation. Nevertheless, there is no published literature on pathogenic variants of NDUFS8 in diabetes but pathogenic mutations exist in other mitochondrial subunits in diabetes. To further study their links, proteomic and genetic data should be carried out.
Besides Leigh syndrome, cancer, and diabetes mellitus, as a core subunit of complex I, NDUFS8 has a relation with neural and psychiatric diseases. However, when it comes to mitochondrial diseases, which are some of the most common inherited neurometabolic disorders, the involvement of NDUFS8 should not be overlooked, which might help understand the elucidation of mitochondrial disease mechanisms, gene therapies, and new drugs [158].
Regardless of the upregulation or downregulation of NDUFS8, the change may contribute to the metabolic disorder of mitochondria, causing related diseases. Although there are many diseases related to NDUFS8, the unknown significance of some pathogenic variants in NDUFS8 remained, such as the variants of exertional rhabdomyolysis in NDUFS8 [159]. Studies wholly on NDUFS8 are rare and there are mainly research works referring to NDUFS8 that lack more specific data and principally treat NDUFS8 as an agent for their mechanism. Further studies are required to determine the accurate mechanism between NDUFS8 and its related diseases and the potential value for prevention, diagnosis, prognosis, and therapeutics.
Author Contributions
Writing, review, and editing were carried out by all the authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (project No. 81602167), the Hunan Provincial Natural Science Foundation of China (project No. 2017JJ3494 and 2021JJ31100), the Science and Technology Program Foundation of Changsha City (project No. kq2004085), and the Undergraduate Training Programs for Innovation and Entrepreneurship (UTPIE, S2021105330991 (S.W.) and S2021105330018 (Y.K.)).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interests
The authors declare no conflict of interest.
References
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Pagniez-Mammeri, H.; Loublier, S.; Legrand, A.; Benit, P.; Rustin, P.; Slama, A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol. Genet. Metab. 2012, 105, 163–172. [Google Scholar] [CrossRef]
- Marina, A.D.; Schara, U.; Pyle, A.; Moller-Hartmann, C.; Holinski-Feder, E.; Abicht, A.; Czermin, B.; Lochmuller, H.; Griffin, H.; Santibanez-Koref, M.; et al. NDUFS8-related Complex I Deficiency Extends Phenotype from “PEO Plus” to Leigh Syndrome. JIMD Rep. 2013, 10, 17–22. [Google Scholar]
- Koopman, W.J.; Visch, H.J.; Verkaart, S.; van den Heuvel, L.W.; Smeitink, J.A.; Willems, P.H. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am. J. Physiol. Cell Physiol. 2005, 289, C881–C890. [Google Scholar] [CrossRef]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e1212. [Google Scholar] [CrossRef]
- de Sury, R.; Martinez, P.; Procaccio, V.; Lunardi, J.; Issartel, J.P. Genomic structure of the human NDUFS8 gene coding for the iron-sulfur TYKY subunit of the mitochondrial NADH:ubiquinone oxidoreductase. Gene 1998, 215, 1–10. [Google Scholar] [CrossRef]
- Lin, B.Y.; Zheng, G.T.; Teng, K.W.; Chang, J.Y.; Lee, C.C.; Liao, P.C.; Kao, M.C. TAT-Conjugated NDUFS8 Can Be Transduced into Mitochondria in a Membrane-Potential-Independent Manner and Rescue Complex I Deficiency. Int. J. Mol. Sci. 2021, 22, 6524. [Google Scholar] [CrossRef]
- Loeffen, J.; Smeitink, J.; Triepels, R.; Smeets, R.; Schuelke, M.; Sengers, R.; Trijbels, F.; Hamel, B.; Mullaart, R.; van den Heuvel, L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998, 63, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; He, L.; Zhao, Y.; Zhang, L.; He, Y.; Li, J.; Li, C.; Zhang, B.; Huang, Q.; Xing, J.; et al. Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia 2020, 63, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Castellanos, G.; Carrier, A. A novel anticancer pharmacological agent targeting mitochondrial complex I. Trends Pharmacol. Sci. 2022, 43, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kurelac, I.; Iommarini, L.; Vatrinet, R.; Amato, L.B.; De Luise, M.; Leone, G.; Girolimetti, G.; Umesh Ganesh, N.; Bridgeman, V.L.; Ombrato, L.; et al. Inducing cancer indolence by targeting mitochondrial Complex I is potentiated by blocking macrophage-mediated adaptive responses. Nat. Commun. 2019, 10, 903. [Google Scholar] [CrossRef] [PubMed]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Nunez de Arenas, C.; Gomez de Cedron, M.; Formentini, L.; Cuezva, J.M. Coordinate beta-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef] [PubMed]
- Maklashina, E.; Kotlyar, A.B.; Cecchini, G. Active/de-active transition of respiratory complex I in bacteria, fungi, and animals. Biochim. Biophys. Acta 2003, 1606, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kahlhofer, F.; Gansen, M.; Zickermann, V. Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly. Life 2021, 11, 455. [Google Scholar] [CrossRef]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef]
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory complex I—structure, mechanism and evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef]
- Agip, A.A.; Blaza, J.N.; Fedor, J.G.; Hirst, J. Mammalian Respiratory Complex I Through the Lens of Cryo-EM. Annu. Rev. Biophys. 2019, 48, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Grba, D.N.; Hirst, J. Mitochondrial complex I structure reveals ordered water molecules for catalysis and proton translocation. Nat. Struct. Mol. Biol. 2020, 27, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.J.; Sazanov, L.A. Three-dimensional structure of respiratory complex I from Escherichia coli in ice in the presence of nucleotides. Biochim. Biophys. Acta 2008, 1777, 711–718. [Google Scholar] [CrossRef]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 872–883. [Google Scholar] [CrossRef]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Brandt, U. Energy converting NADH:quinone oxidoreductase (complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Sazanov, L.A. Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations. Trends Cell Biol. 2018, 28, 835–867. [Google Scholar] [CrossRef]
- Walker, J.E. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 1992, 25, 253–324. [Google Scholar] [CrossRef]
- Duarte, M.; Finel, M.; Videira, A. Primary structure of a ferredoxin-like iron-sulfur subunit of complex I from Neurospora crassa. Biochim. Biophys. Acta 1996, 1275, 151–153. [Google Scholar] [CrossRef][Green Version]
- Dupuis, A.; Chevallet, M.; Darrouzet, E.; Duborjal, H.; Lunardi, J.; Issartel, J.P. The complex I from Rhodobacter capsulatus. Biochim. Biophys. Acta 1998, 1364, 147–165. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weidner, U.; Geier, S.; Ptock, A.; Friedrich, T.; Leif, H.; Weiss, H. The gene locus of the proton-translocating NADH: Ubiquinone oxidoreductase in Escherichia coli. Organization of the 14 genes and relationship between the derived proteins and subunits of mitochondrial complex I. J. Mol. Biol. 1993, 233, 109–122. [Google Scholar] [CrossRef]
- Chevallet, M.; Dupuis, A.; Lunardi, J.; van Belzen, R.; Albracht, S.P.; Issartel, J.P. The NuoI subunit of the Rhodobacter capsulatus respiratory Complex I (equivalent to the bovine TYKY subunit) is required for proper assembly of the membraneous and peripheral domains of the enzyme. Eur. J. Biochem. 1997, 250, 451–458. [Google Scholar] [CrossRef]
- Lescuyer, P.; Martinez, P.; Lunardi, J. YY1 and Sp1 activate transcription of the human NDUFS8 gene encoding the mitochondrial complex I TYKY subunit. Biochim. Biophys. Acta 2002, 1574, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Procaccio, V.; Depetris, D.; Soularue, P.; Mattei, M.G.; Lunardi, J.; Issartel, J.P. cDNA sequence and chromosomal localization of the NDUFS8 human gene coding for the 23 kDa subunit of the mitochondrial complex I. Biochim. Biophys. Acta 1997, 1351, 37–41. [Google Scholar] [CrossRef]
- Albracht, S.P.; Hedderich, R. Learning from hydrogenases: Location of a proton pump and of a second FMN in bovine NADH--ubiquinone oxidoreductase (Complex I). FEBS Lett. 2000, 485, 1–6. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhang, S.; Lin, J.; Wang, Y.; Ye, F.; Zhou, B.; Lin, Q.; Liu, J. Role of the Gene ndufs8 Located in Respiratory Complex I from Monascus purpureus in the Cell Growth and Secondary Metabolites Biosynthesis. J. Fungi 2022, 8, 655. [Google Scholar] [CrossRef]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 2008, 39, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Nagappa, M.; Bindu, P.S.; Sinha, S.; Bharath, R.D.; Sandhya, M.; Saini, J.; Mathuranath, P.S.; Taly, A.B. Palatal Tremor Revisited: Disorder with Nosological Diversity and Etiological Heterogeneity. Can. J. Neurol. Sci. 2018, 45, 243–247. [Google Scholar] [CrossRef]
- Stenton, S.L.; Tesarova, M.; Sheremet, N.L.; Catarino, C.B.; Carelli, V.; Ciara, E.; Curry, K.; Engvall, M.; Fleming, L.R.; Freisinger, P.; et al. DNAJC30 defect: A frequent cause of recessive Leber hereditary optic neuropathy and Leigh syndrome. Brain 2022, 145, 1624–1631. [Google Scholar] [CrossRef]
- Zawadzka, M.; Krygier, M.; Pawlowicz, M.; Wilke, M.; Rutkowska, K.; Gueguen, N.; Desquiret-Dumas, V.; Klee, E.W.; Schimmenti, L.A.; Slawek, J.; et al. Expanding the phenotype of DNAJC30-associated Leigh syndrome. Clin. Genet. 2022, 102, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Ahlers, P.M.; Garofano, A.; Kerscher, S.J.; Brandt, U. Application of the obligate aerobic yeast Yarrowia lipolytica as a eucaryotic model to analyse Leigh syndrome mutations in the complex I core subunits PSST and TYKY. Biochim. Biophys. Acta 2000, 1459, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hinttala, R.; Uusimaa, J.; Remes, A.M.; Rantala, H.; Hassinen, I.E.; Majamaa, K. Sequence analysis of nuclear genes encoding functionally important complex I subunits in children with encephalomyopathy. J. Mol. Med. 2005, 83, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar]
- Tuppen, H.A.; Hogan, V.E.; He, L.; Blakely, E.L.; Worgan, L.; Al-Dosary, M.; Saretzki, G.; Alston, C.L.; Morris, A.A.; Clarke, M.; et al. The p.M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain 2010, 133, 2952–2963. [Google Scholar] [CrossRef]
- Procaccio, V.; Wallace, D.C. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology 2004, 62, 1899–1901. [Google Scholar] [CrossRef]
- Loewen, C.A.; Ganetzky, B. Mito-Nuclear Interactions Affecting Lifespan and Neurodegeneration in a Drosophila Model of Leigh Syndrome. Genetics 2018, 208, 1535–1552. [Google Scholar] [CrossRef]
- Grody, W.W.; Thompson, B.H.; Hudgins, L. Whole-exome/genome sequencing and genomics. Pediatrics 2013, 132, S211–S215. [Google Scholar] [CrossRef]
- Distelmaier, F.; Koopman, W.J.; van den Heuvel, L.P.; Rodenburg, R.J.; Mayatepek, E.; Willems, P.H.; Smeitink, J.A. Mitochondrial complex I deficiency: From organelle dysfunction to clinical disease. Brain 2009, 132, 833–842. [Google Scholar] [CrossRef]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Haberberger, B.; Frisch, E.M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef]
- Sofou, K.; De Coo, I.F.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lonnqvist, T.; et al. A multicenter study on Leigh syndrome: Disease course and predictors of survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Bakare, A.B.; Lesnefsky, E.J.; Iyer, S. Leigh Syndrome: A Tale of Two Genomes. Front. Physiol. 2021, 12, 693734. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Iommarini, L.; Calvaruso, M.A.; Kurelac, I.; Gasparre, G.; Porcelli, A.M. Complex I impairment in mitochondrial diseases and cancer: Parallel roads leading to different outcomes. Int. J. Biochem. Cell Biol. 2013, 45, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Wang, T.Y.; Kao, M.C.; Lee, A.Y. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Cancer Genome Atlas Research, N.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Jiang, H.; Sun, X.; Xu, H.; Wei, X.; Wei, Y.; Xiao, G.; Song, Z.; Zhou, F. Mitochondrial dysfunction induces radioresistance in colorectal cancer by activating [Ca(2+)]m-PDP1-PDH-histone acetylation retrograde signaling. Cell Death Dis. 2021, 12, 837. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Hu, C.; Kluza, J.; Ke, W.; Tian, G.; Giurgiu, M.; Bleilevens, A.; Campos, A.R.; Charbono, A.; Stickeler, E.; et al. Metabolic targeting of cancer by a ubiquinone uncompetitive inhibitor of mitochondrial complex I. Cell Chem. Biol. 2022, 29, 436–450.e415. [Google Scholar] [CrossRef]
- Li, S.; Wang, F.; Zhang, G.; Chou, T.F. NMS-873 Leads to Dysfunctional Glycometabolism in A p97-Independent Manner in HCT116 Colon Cancer Cells. Pharmaceutics 2022, 14, 764. [Google Scholar] [CrossRef]
- Xiao, M.H.; Lin, Y.F.; Xie, P.P.; Chen, H.X.; Deng, J.W.; Zhang, W.; Zhao, N.; Xie, C.; Meng, Y.; Liu, X.; et al. Downregulation of a mitochondrial micropeptide, MPM, promotes hepatoma metastasis by enhancing mitochondrial complex I activity. Mol. Ther. 2022, 30, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Su, C.Y.; Chang, Y.C.; Yang, C.J.; Huang, M.S.; Hsiao, M. The opposite prognostic effect of NDUFS1 and NDUFS8 in lung cancer reflects the oncojanus role of mitochondrial complex I. Sci. Rep. 2016, 6, 31357. [Google Scholar] [CrossRef]
- Sung, Y.J.; Kao, T.Y.; Kuo, C.L.; Fan, C.C.; Cheng, A.N.; Fang, W.C.; Chou, H.Y.; Lo, Y.K.; Chen, C.H.; Jiang, S.S.; et al. Mitochondrial Lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. 2018, 9, 697. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Lin, C.S.; Potluri, P.; Procaccio, V.; Wallace, D.C. mtDNA lineage analysis of mouse L-cell lines reveals the accumulation of multiple mtDNA mutants and intermolecular recombination. Genes Dev. 2012, 26, 384–394. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Wei, J.; Xie, Q.; Liu, X.; Wan, C.; Wu, W.; Fang, K.; Yao, Y.; Cheng, P.; Deng, D.; Liu, Z. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann. Transl. Med. 2020, 8, 678. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef]
- Pang, Y.; Tan, G.; Yang, X.; Lin, Y.; Chen, Y.; Zhang, J.; Xie, T.; Zhou, H.; Fang, J.; Zhao, Q.; et al. Iron-sulphur cluster biogenesis factor LYRM4 is a novel prognostic biomarker associated with immune infiltrates in hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 463. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Miao, L.; Liao, W.; Liao, W. LncRNA PPP1R14B-AS1 Promotes Tumor Cell Proliferation and Migration via the Enhancement of Mitochondrial Respiration. Front. Genet. 2020, 11, 557614. [Google Scholar] [CrossRef] [PubMed]
- Suh, I.; Weng, J.; Fernandez-Ranvier, G.; Shen, W.T.; Duh, Q.Y.; Clark, O.H.; Kebebew, E. Antineoplastic effects of decitabine, an inhibitor of DNA promoter methylation, in adrenocortical carcinoma cells. Arch. Surg. 2010, 145, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Lisanti, M.P. Mitochondrial markers predict survival and progression in non-small cell lung cancer (NSCLC) patients: Use as companion diagnostics. Oncotarget 2017, 8, 68095–68107. [Google Scholar] [CrossRef]
- Leone, G.; Abla, H.; Gasparre, G.; Porcelli, A.M.; Iommarini, L. The Oncojanus Paradigm of Respiratory Complex I. Genes 2018, 9, 243. [Google Scholar] [CrossRef]
- Christianson, H.C.; Menard, J.A.; Chandran, V.I.; Bourseau-Guilmain, E.; Shevela, D.; Lidfeldt, J.; Mansson, A.S.; Pastorekova, S.; Messinger, J.; Belting, M. Tumor antigen glycosaminoglycan modification regulates antibody-drug conjugate delivery and cytotoxicity. Oncotarget 2017, 8, 66960–66974. [Google Scholar] [CrossRef]
- Alam, M.M.; Sohoni, S.; Kalainayakan, S.P.; Garrossian, M.; Zhang, L. Cyclopamine tartrate, an inhibitor of Hedgehog signaling, strongly interferes with mitochondrial function and suppresses aerobic respiration in lung cancer cells. BMC Cancer 2016, 16, 150. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Tsutsumi, S.; Taniguchi, H.; Ishii, M.; Kobune, Y.; Kodama, T.; Makuuchi, M.; Aburatani, H. Identification of genes associated with dedifferentiation of hepatocellular carcinoma with expression profiling analysis. Jpn. J. Cancer Res. 2002, 93, 636–643. [Google Scholar] [CrossRef]
- Bai, J.; Liu, Z.; Liu, J.; Zhang, S.; Tian, Y.; Zhang, Y.; Ren, L.; Kong, D. Mitochondrial metabolic study guided by proteomics analysis in hepatocellular carcinoma cells surviving long-term incubation with the highest dose of sorafenib. Aging 2019, 11, 12452–12475. [Google Scholar] [CrossRef]
- Bassal, M.A.; Samaraweera, S.E.; Lim, K.; Benard, B.A.; Bailey, S.; Kaur, S.; Leo, P.; Toubia, J.; Thompson-Peach, C.; Nguyen, T.; et al. Germline mutations in mitochondrial complex I reveal genetic and targetable vulnerability in IDH1-mutant acute myeloid leukaemia. Nat. Commun. 2022, 13, 2614. [Google Scholar] [CrossRef]
- Lin, W.Y.; Fordham, S.E.; Hungate, E.; Sunter, N.J.; Elstob, C.; Xu, Y.; Park, C.; Quante, A.; Strauch, K.; Gieger, C.; et al. Genome-wide association study identifies susceptibility loci for acute myeloid leukemia. Nat. Commun. 2021, 12, 6233. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.W.; Fels, D.R.; West, M.; Allen, J.D.; Broadwater, G.; Barry, W.T.; Wilke, L.G.; Masko, E.; Douglas, P.S.; Dash, R.C.; et al. Modulation of circulating angiogenic factors and tumor biology by aerobic training in breast cancer patients receiving neoadjuvant chemotherapy. Cancer Prev. Res. 2013, 6, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Tozlu-Kara, S.; Roux, V.; Andrieu, C.; Vendrell, J.; Vacher, S.; Lazar, V.; Spyratos, F.; Tubiana-Hulin, M.; Cohen, P.; Dessen, P.; et al. Oligonucleotide microarray analysis of estrogen receptor alpha-positive postmenopausal breast carcinomas: Identification of HRPAP20 and TIMELESS as outstanding candidate markers to predict the response to tamoxifen. J. Mol. Endocrinol. 2007, 39, 305–318. [Google Scholar] [CrossRef]
- Guha, M.; Srinivasan, S.; Raman, P.; Jiang, Y.; Kaufman, B.A.; Taylor, D.; Dong, D.; Chakrabarti, R.; Picard, M.; Carstens, R.P.; et al. Aggressive triple negative breast cancers have unique molecular signature on the basis of mitochondrial genetic and functional defects. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1060–1071. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Su, Y.A.; Tsai, H.D.; Mason, J.T.; Chou, M.C.; Man, Y.G. Increased invasiveness and aggressiveness in breast epithelia with cytoplasmic p63 expression. Int. J. Biol. Sci. 2010, 6, 428–442. [Google Scholar] [CrossRef]
- Schulte, K.M.; Mengel, M.; Heinze, M.; Simon, D.; Scheuring, S.; Kohrer, K.; Roher, H.D. Complete sequencing and messenger ribonucleic acid expression analysis of the MEN I gene in adrenal cancer. J. Clin. Endocrinol. Metab. 2000, 85, 441–448. [Google Scholar] [CrossRef]
- Kjellman, M.; Roshani, L.; I, B.T.; Kallioniemi, O.P.; Hoog, A.; Gray, S.; Farnebo, L.O.; Holst, M.; Backdahl, M.; Larsson, C. Genotyping of adrenocortical tumors: Very frequent deletions of the MEN1 locus in 11q13 and of a 1-centimorgan region in 2p16. J. Clin. Endocrinol. Metab. 1999, 84, 730–735. [Google Scholar]
- Fernandez-Ranvier, G.G.; Weng, J.; Yeh, R.F.; Shibru, D.; Khafnashar, E.; Chung, K.W.; Hwang, J.; Duh, Q.Y.; Clark, O.H.; Kebebew, E. Candidate diagnostic markers and tumor suppressor genes for adrenocortical carcinoma by expression profile of genes on chromosome 11q13. World J. Surg. 2008, 32, 873–881. [Google Scholar] [CrossRef]
- Ellinger, J.; Poss, M.; Bruggemann, M.; Gromes, A.; Schmidt, D.; Ellinger, N.; Tolkach, Y.; Dietrich, D.; Kristiansen, G.; Muller, S.C. Systematic Expression Analysis of Mitochondrial Complex I Identifies NDUFS1 as a Biomarker in Clear-Cell Renal-Cell Carcinoma. Clin. Genitourin. Cancer 2017, 15, e551–e562. [Google Scholar] [CrossRef]
- Long, Y.; Lu, M.; Cheng, T.; Zhan, X.; Zhan, X. Multiomics-Based Signaling Pathway Network Alterations in Human Non-functional Pituitary Adenomas. Front. Endocrinol. 2019, 10, 835. [Google Scholar] [CrossRef]
- Stein, J.; Tenbrock, J.; Kristiansen, G.; Muller, S.C.; Ellinger, J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. Int. J. Urol. 2019, 26, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, J.; Randall, L.; De Robles, G.; Agrawal, A.; Mercola, D.; Liu, M.; Randhawa, P.; Edwards, R.; McClelland, M.; Rahmatpanah, F. Transcriptome Analysis of Ovarian and Uterine Clear Cell Malignancies. Front. Oncol. 2020, 10, 598579. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, S.; Goemaere, J.; Knoops, B. Silencing of peroxiredoxin 3 and peroxiredoxin 5 reveals the role of mitochondrial peroxiredoxins in the protection of human neuroblastoma SH-SY5Y cells toward MPP+. Neurosci. Lett. 2008, 433, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Kropotov, A.; Gogvadze, V.; Shupliakov, O.; Tomilin, N.; Serikov, V.B.; Tomilin, N.V.; Zhivotovsky, B. Peroxiredoxin V is essential for protection against apoptosis in human lung carcinoma cells. Exp. Cell Res. 2006, 312, 2806–2815. [Google Scholar] [CrossRef]
- Sensi, M.; Nicolini, G.; Zanon, M.; Colombo, C.; Molla, A.; Bersani, I.; Lupetti, R.; Parmiani, G.; Anichini, A. Immunogenicity without immunoselection: A mutant but functional antioxidant enzyme retained in a human metastatic melanoma and targeted by CD8(+) T cells with a memory phenotype. Cancer Res. 2005, 65, 632–640. [Google Scholar] [CrossRef]
- Shiota, M.; Izumi, H.; Miyamoto, N.; Onitsuka, T.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Takahashi, M.; Ono, M.; Kuwano, M.; et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high-mobility group protein B1. Cancer Sci. 2008, 99, 1950–1959. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wardell, C.P.; Furuta, M.; Taniguchi, H.; Fujimoto, A. Cancer whole-genome sequencing: Present and future. Oncogene 2015, 34, 5943–5950. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef]
- Kupriyanova, Y.; Zaharia, O.P.; Bobrov, P.; Karusheva, Y.; Burkart, V.; Szendroedi, J.; Hwang, J.H.; Roden, M.; the GDS group. Early changes in hepatic energy metabolism and lipid content in recent-onset type 1 and 2 diabetes mellitus. J. Hepatol. 2021, 74, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Flotyńska, J.; Klause, D.; Kulecki, M.; Cieluch, A.; Uruska, A. Higher NDUFS8 Serum Levels Correlate with Better Insulin Sensitivity in Type 1 Diabetes. Curr. Issues Mol. Biol. 2022, 44, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Zheng, W.L.; Ma, W.L. Bioinformatic analysis of genes related to type 2 diabetes mellitus. Basic Clin. Med. 2015, 35, 749–753. [Google Scholar]
- Jimenez-Sanchez, C.; Brun, T.; Maechler, P. Mitochondrial Carriers Regulating Insulin Secretion Profiled in Human Islets upon Metabolic Stress. Biomolecules 2020, 10, 1543. [Google Scholar] [CrossRef]
- Caruso, M.; Ma, D.; Msallaty, Z.; Lewis, M.; Seyoum, B.; Al-janabi, W.; Diamond, M.; Abou-Samra, A.B.; Hojlund, K.; Tagett, R.; et al. Increased interaction with insulin receptor substrate 1, a novel abnormality in insulin resistance and type 2 diabetes. Diabetes 2014, 63, 1933–1947. [Google Scholar] [CrossRef]
- Guo, Y.; Darshi, M.; Ma, Y.; Perkins, G.A.; Shen, Z.; Haushalter, K.J.; Saito, R.; Chen, A.; Lee, Y.S.; Patel, H.H.; et al. Quantitative proteomic and functional analysis of liver mitochondria from high fat diet (HFD) diabetic mice. Mol. Cell. Proteomics 2013, 12, 3744–3758. [Google Scholar] [CrossRef]
- Wollam, J.; Mahata, S.; Riopel, M.; Hernandez-Carretero, A.; Biswas, A.; Bandyopadhyay, G.K.; Chi, N.W.; Eiden, L.E.; Mahapatra, N.R.; Corti, A.; et al. Chromogranin A regulates vesicle storage and mitochondrial dynamics to influence insulin secretion. Cell Tissue Res. 2017, 368, 487–501. [Google Scholar] [CrossRef]
- Li, H.; Xu, M.; Lee, J.; He, C.; Xie, Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1234–E1244. [Google Scholar] [CrossRef]
- Gomes, A.P.; Duarte, F.V.; Nunes, P.; Hubbard, B.P.; Teodoro, J.S.; Varela, A.T.; Jones, J.G.; Sinclair, D.A.; Palmeira, C.M.; Rolo, A.P. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim. Biophys. Acta 2012, 1822, 185–195. [Google Scholar] [CrossRef]
- van den Ouweland, J.M.; Lemkes, H.H.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.; van de Kamp, J.J.; Maassen, J.A. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Hudson, G.; Ferrari, G.; Futterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmuller, H.; McFarland, R.; Ramesh, V.; et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, V.; Pitceathly, R.D.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The UK MRC Mitochondrial Disease Patient Cohort Study: Clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Zhuang, J.; Zhou, P.; Liu, X.; Luo, Y. MicroRNA-34a promotes mitochondrial dysfunction-induced apoptosis in human lens epithelial cells by targeting Notch2. Oncotarget 2017, 8, 110209–110220. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, P.; Hidalgo-Gutierrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sanchez-Hernandez, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef]
- Lee, J.M.; Hwang, J.W.; Kim, M.J.; Jung, S.Y.; Kim, K.S.; Ahn, E.H.; Min, K.; Choi, Y.S. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants 2021, 10, 696. [Google Scholar] [CrossRef]
- Liu, H.W.; Chang, Y.C.; Chan, Y.C.; Hu, S.H.; Liu, M.Y.; Chang, S.J. Dysregulations of mitochondrial quality control and autophagic flux at an early age lead to progression of sarcopenia in SAMP8 mice. Biogerontology 2020, 21, 367–380. [Google Scholar] [CrossRef]
- Miao, H.; Chen, S.; Ding, R. Evaluation of the Molecular Mechanisms of Sepsis Using Proteomics. Front. Immunol. 2021, 12, 733537. [Google Scholar] [CrossRef] [PubMed]
- de Azambuja Rodrigues, P.M.; Valente, R.H.; Brunoro, G.V.F.; Nakaya, H.T.I.; Araujo-Pereira, M.; Bozza, P.T.; Bozza, F.A.; Trugilho, M.R.O. Proteomics reveals disturbances in the immune response and energy metabolism of monocytes from patients with septic shock. Sci. Rep. 2021, 11, 15149. [Google Scholar] [CrossRef] [PubMed]
- Cheong, A.; Archambault, D.; Degani, R.; Iverson, E.; Tremblay, K.D.; Mager, J. Nuclear-encoded mitochondrial ribosomal proteins are required to initiate gastrulation. Development 2020, 147, dev188714. [Google Scholar] [CrossRef] [PubMed]
- Suszynska-Zajczyk, J.; Sikora, M.; Jakubowski, H. Paraoxonase 1 deficiency and hyperhomocysteinemia alter the expression of mouse kidney proteins involved in renal disease. Mol. Genet. Metab. 2014, 113, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Suszynska-Zajczyk, J.; Utyro, O.; Jakubowski, H. Methionine-induced hyperhomocysteinemia and bleomycin hydrolase deficiency alter the expression of mouse kidney proteins involved in renal disease. Mol. Genet. Metab. 2014, 112, 339–346. [Google Scholar] [CrossRef]
- Feingold, K.R.; Wang, Y.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J. Lipid Res. 2008, 49, 2179–2187. [Google Scholar] [CrossRef]
- Zhao, M.; Zhou, Y.; Liu, S.; Li, L.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. Control release of mitochondria-targeted antioxidant by injectable self-assembling peptide hydrogel ameliorated persistent mitochondrial dysfunction and inflammation after acute kidney injury. Drug Deliv. 2018, 25, 546–554. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Zhou, Y.; Zhao, M.; Wang, Y.; Wang, C.; Lou, P.; Huang, R.; Ma, L.; Lu, Y.; et al. Indispensable role of mitochondria in maintaining the therapeutic potential of curcumin in acute kidney injury. J. Cell. Mol. Med. 2021, 25, 9863–9877. [Google Scholar] [CrossRef]
- Yang, C.; Liu, J.; Li, L.; Hu, M.; Long, Y.; Liu, X.; Zhu, T.; Huang, X.; Zhao, S.; Liu, S.; et al. Proteome Analysis of Renoprotection Mediated by a Novel Cyclic Helix B Peptide in Acute Kidney Injury. Sci. Rep. 2015, 5, 18045. [Google Scholar] [CrossRef]
- Wu, J.; Jin, Z.; Yang, X.; Yan, L.J. Post-ischemic administration of 5-methoxyindole-2-carboxylic acid at the onset of reperfusion affords neuroprotection against stroke injury by preserving mitochondrial function and attenuating oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 444–450. [Google Scholar] [CrossRef]
- Gregorini, M.; Corradetti, V.; Pattonieri, E.F.; Rocca, C.; Milanesi, S.; Peloso, A.; Canevari, S.; De Cecco, L.; Dugo, M.; Avanzini, M.A.; et al. Perfusion of isolated rat kidney with Mesenchymal Stromal Cells/Extracellular Vesicles prevents ischaemic injury. J. Cell. Mol. Med. 2017, 21, 3381–3393. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.E.; Callender, R.; Ma, J.; Grove, M.L.; Hildebrandt, M.A. Abstract 892: Doxorubicin-induced cardiotoxicity in iPSC-cardiomyocytes: Altered mitochondrial gene expression and function. Cancer Res. 2018, 78, 892. [Google Scholar] [CrossRef]
- Kido, K.; Ito, H.; Yamamoto, Y.; Makita, K.; Uchida, T. Cytotoxicity of propofol in human induced pluripotent stem cell-derived cardiomyocytes. J. Anesth. 2018, 32, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 2009, 34, 1021–1029. [Google Scholar] [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Dobeln, U.; Hagenfeldt, L.; Hallstrom, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef]
- Garcia-Esparcia, P.; Koneti, A.; Rodriguez-Oroz, M.C.; Gago, B.; Del Rio, J.A.; Ferrer, I. Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson’s disease and Parkinson’s disease with dementia. Brain Pathol. 2018, 28, 43–57. [Google Scholar] [CrossRef]
- Henningsen, K.; Palmfeldt, J.; Christiansen, S.; Baiges, I.; Bak, S.; Jensen, O.N.; Gregersen, N.; Wiborg, O. Candidate hippocampal biomarkers of susceptibility and resilience to stress in a rat model of depression. Mol. Cell. Proteomics 2012, 11, M111-016428. [Google Scholar] [CrossRef]
- Machado, A.K.; Andreazza, A.C.; da Silva, T.M.; Boligon, A.A.; do Nascimento, V.; Scola, G.; Duong, A.; Cadona, F.C.; Ribeiro, E.E.; da Cruz, I.B. Neuroprotective Effects of Acai (Euterpe oleracea Mart.) against Rotenone In Vitro Exposure. Oxid. Med. Cell. Longev. 2016, 2016, 8940850. [Google Scholar] [CrossRef]
- Horton, T.M.; Petros, J.A.; Heddi, A.; Shoffner, J.; Kaufman, A.E.; Graham, S.D., Jr.; Gramlich, T.; Wallace, D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosomes Cancer 1996, 15, 95–101. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, P.H.; Gupta, P.; Li, X.; Zhao, S.L.; Zhou, X.; Li, Z.; Wei, S.; Xu, L.; Han, R.; et al. MG53 suppresses tumor progression and stress granule formation by modulating G3BP2 activity in non-small cell lung cancer. Mol. Cancer 2021, 20, 118. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef]
- Germain, N.D.; Chung, W.K.; Sarmiere, P.D. RNA interference (RNAi)-based therapeutics for treatment of rare neurologic diseases. Mol. Aspects Med. 2022, 101148. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef]
- Chen, X.; Wei, S.; Yang, F. Mitochondria in the pathogenesis of diabetes: A proteomic view. Protein Cell 2012, 3, 648–660. [Google Scholar] [CrossRef]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef]
- Sambuughin, N.; Mungunsukh, O.; Ren, M.; Capacchione, J.F.; Horkayne-Szakaly, I.; Chuang, K.; Muldoon, S.M.; Smith, J.K.; O’Connor, F.G.; Deuster, P.A. Pathogenic and rare deleterious variants in multiple genes suggest oligogenic inheritance in recurrent exertional rhabdomyolysis. Mol. Genet. Metab. Rep. 2018, 16, 76–81. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).