
Recent Advances in the Synthesis and Applications of Nitrogen-Containing Macrocycles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
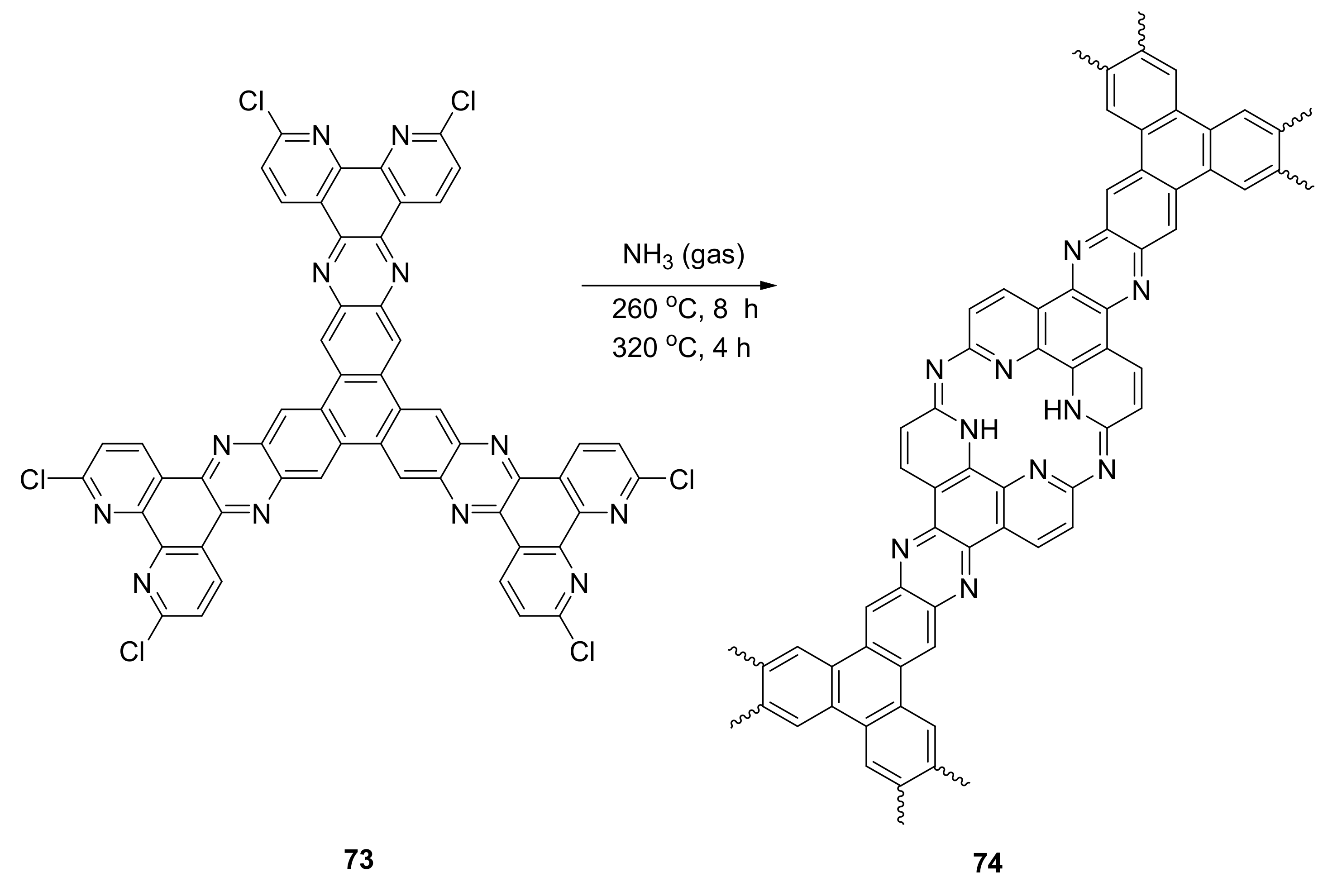{kind=link}
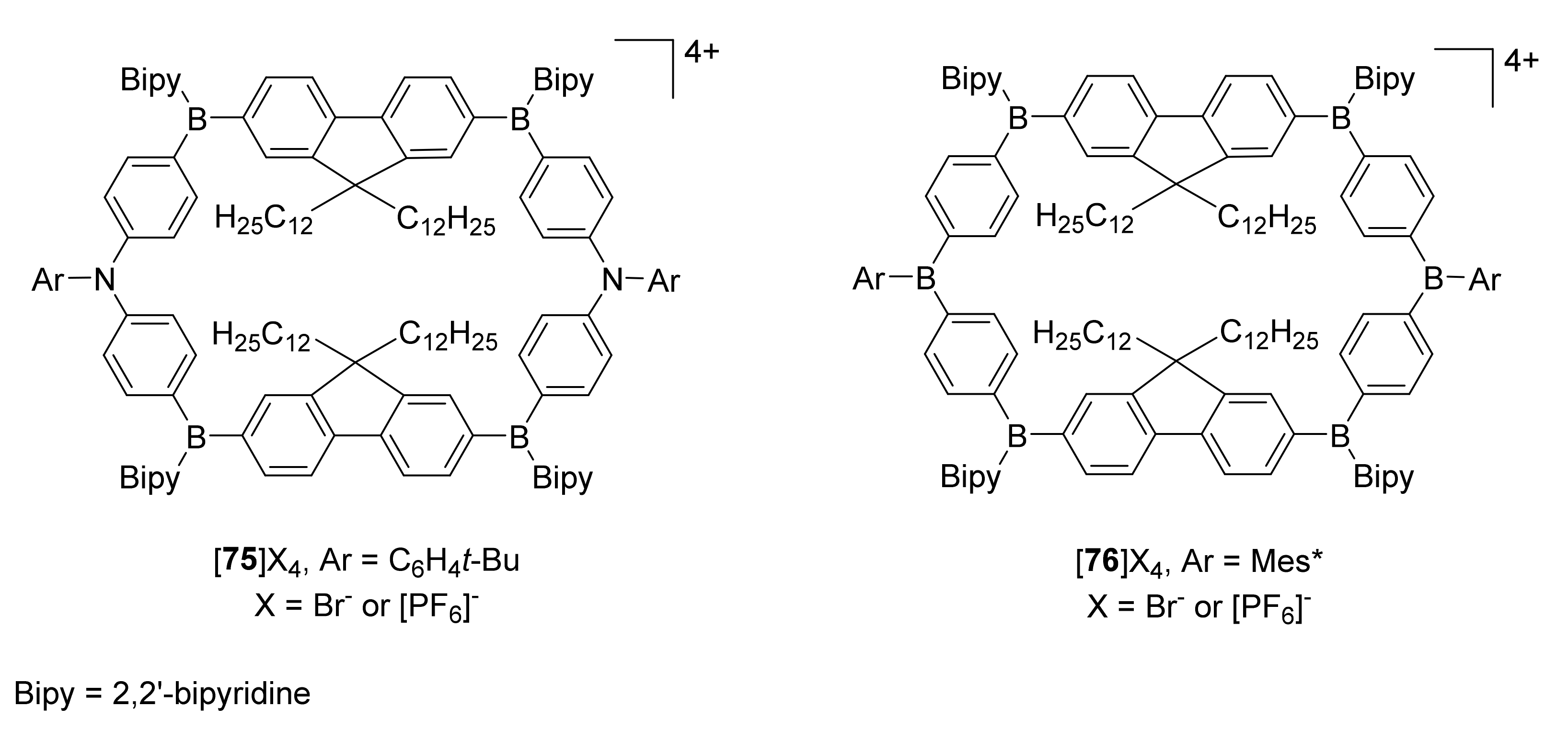{kind=link}
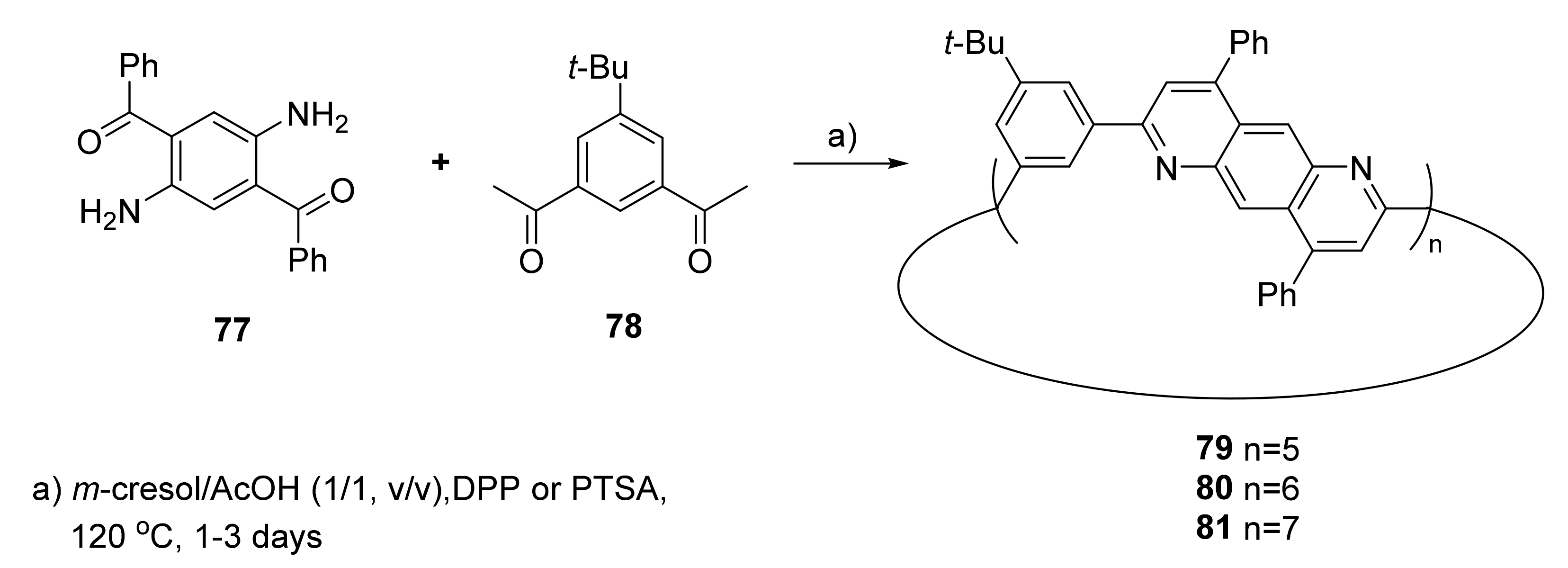{kind=link}
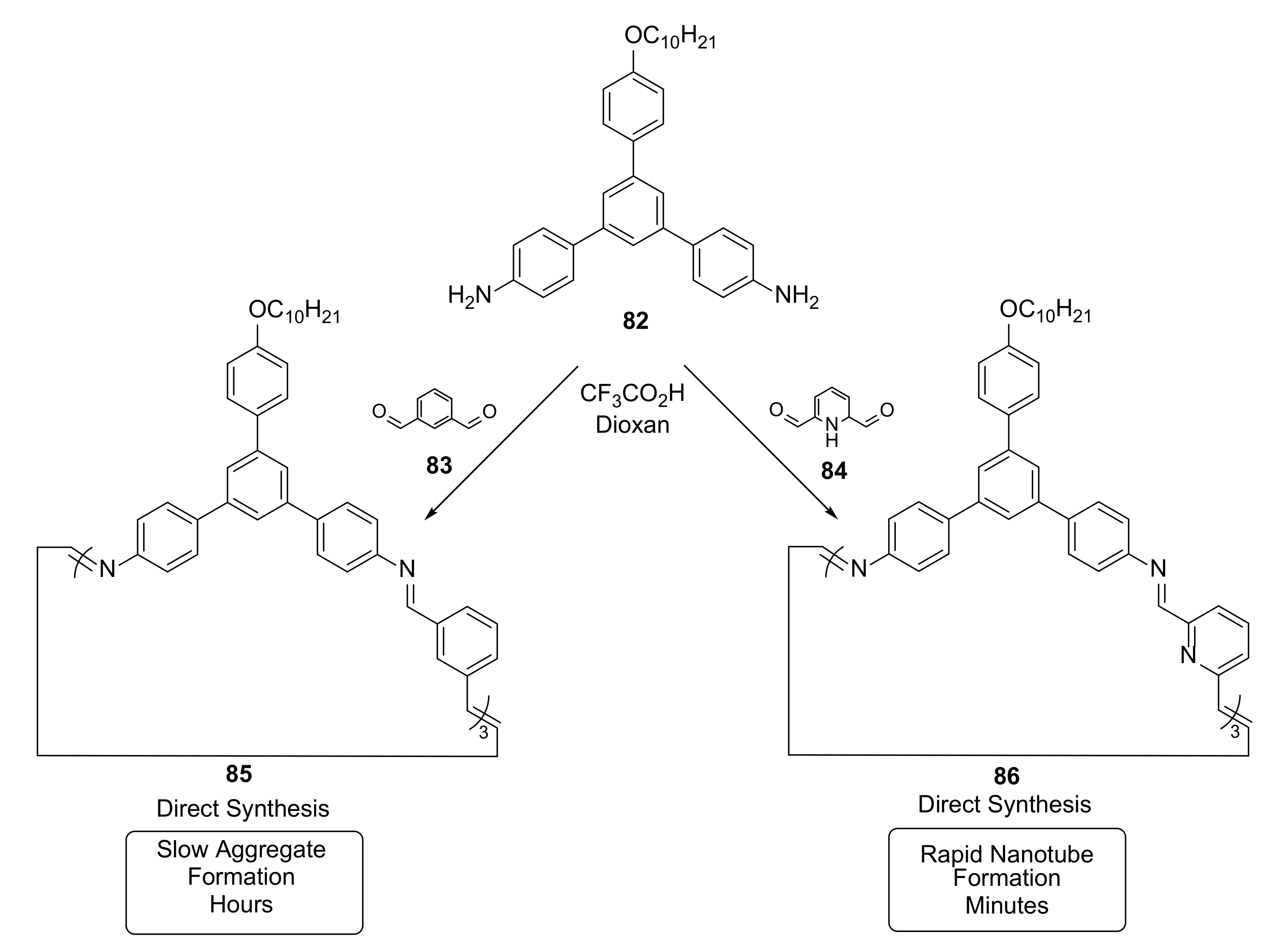{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Grajewski, J. Recent Advances in the Synthesis and Applications of Nitrogen-Containing Macrocycles. Molecules 2022, 27, 1004. https://doi.org/10.3390/molecules27031004
Grajewski J. Recent Advances in the Synthesis and Applications of Nitrogen-Containing Macrocycles. Molecules. 2022; 27(3):1004. https://doi.org/10.3390/molecules27031004
Chicago/Turabian StyleGrajewski, Jakub. 2022. "Recent Advances in the Synthesis and Applications of Nitrogen-Containing Macrocycles" Molecules 27, no. 3: 1004. https://doi.org/10.3390/molecules27031004
APA StyleGrajewski, J. (2022). Recent Advances in the Synthesis and Applications of Nitrogen-Containing Macrocycles. Molecules, 27(3), 1004. https://doi.org/10.3390/molecules27031004