1. Introduction
To date, more than 30,000 marine natural products have been reported [
1]. To use the potentially unique structures and bioactivities of marine natural products in pharmaceutical materials, it is essential to elucidate their mechanisms of action. Probe molecules based on natural products can be powerful tools in establishing efficient methods of identifying target proteins [
2,
3]. Fluorescent probes can be used to visualize subcellular localization, and affinity beads can be used to drag out target proteins. However, probe molecules based on natural compounds need to be custom-made because their chemical properties are greatly influenced by the natural compounds themselves, and as a result, they are not always efficiently prepared. If the number of successful target protein identifications can be increased, although it currently remains limited, we may be able to set up a platform for the efficient analysis of the mechanisms of action of marine natural compounds by integrating the methods developed during the process.
Cyclic peptides are one of the major categories of marine natural products, and many marine cyclic peptides with various bioactivities have been reported to date [
4,
5,
6]. Among them, aurilide—which exhibits strong cytotoxicity—is an example for which the target protein has been successfully identified as prohibitin 1 (PHB1) in mitochondria. The mechanism of action of this compound has been described as the binding of the molecule to PHB1, which leads to mitochondrial fragmentation and the induction of cell apoptosis [
7]. In our continuing study, we are focused on kapakahines, a family of marine cyclic peptides isolated from the sponge
Cribrochalina olemda. Kapakahines share a unique structural motif in which the nitrogen in the indole ring of the
N-terminal tryptophan residue is directly attached to a tetracyclic core containing an α-carboline skeleton. Kapakahine A is the main compound in this family and is cytotoxic to the P388 murine myeloid leukemia cell line in the order of µM and has an
N-terminal reactive amino group that readily binds to active esters [
8]. Compounds such as aurilide—which has cytotoxicity in the order of nM because of its high affinity for the target protein—are generally considered suitable for target identification by means of chemical probes attached to beads. However, if the same approach could be made applicable to the identification of the target proteins of compounds with a weaker cytotoxicity in the order of µM such as kapakahine A, it would pave the way for the mechanistic analysis of many marine natural products that do not have potent activity.
Rocha et al. synthesized a fluorescent probe comprising kapakahine E conjugated with coumarin and found that the fluorescent probe localized to the Golgi apparatus in PC-3M prostate carcinoma cells [
9]. In a previous study, we prepared a fluorescent probe of kapakahine A [Kap A-5FL (
2)] by direct coupling with an FITC derivative with an active ester linker [
10]. Co-localization analysis of Kap A-5FL (
2) and Golgi-binding lectin HPA, or concanavalin A (endoplasmic reticulum marker) in HeLa cells, showed that Kap A-5FL does not localize in the Golgi apparatus or endoplasmic reticulum.
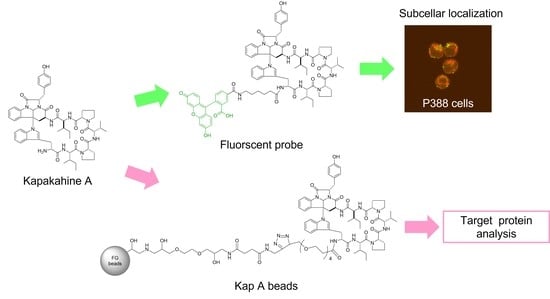
In this study, we describe the preparation of Kap A-beads (6) as well as Kap A-5FL localization with organelles in P388 cells and the target protein candidates of kapakahine A (1).
3. Discussion
In this study, we prepared a linker-conjugated probe, Kap A-probe (3), based on the marine cyclic peptide kapakahine A (1). A pull-down assay using Kap A-beads (6) identified PHB1, PHB2, and ANT2 as target protein candidates. These candidates are all mitochondrial proteins, which is consistent with the finding that the kapakahine A fluorescent probe co-localized with the mitochondria.
PHB1 is known to be the target protein of aurilide, a nanometer-order cytotoxic marine cyclic peptide that induces mitochondrial fragmentation and apoptosis by forming a complex with PHB1 that releases m-AAA involved in the processing of L-OPA1 to S-OPA1 [
7]. PHBs are known to be multifunctional proteins in mitochondria [
11,
12,
13]. It has recently been suggested that PHB2 and ANT2 regulate the uptake of substances such as ATP by forming mitochondrial transmembrane pores, and play an important role in mitophagy, which is involved in maintaining mitochondrial quality [
14,
15].
Sesaminol, an ingredient of sesame that is cytotoxic towards the MCF7 human mammary carcinoma cell line [
16], is known to bind to ANT2 and reduce the protein expression of cyclin D1, which is important for the G
1/S phase transition of the cell cycle. Rabdosianone I, from the medicinal herb
Isodon japonicus, binds to PHB2 and ANT2, leading to a decrease in the expression level of thymidylate synthase, resulting in growth inhibition of the human colon adenocarcinoma cell line HT-29 [
17].
In this study, we successfully prepared Kap A-beads (6) and detected three target protein candidates: PHB1, PHB2, and ANT2. These proteins are known to localize in mitochondria, which is where kapakahine A has been shown to localize. The cytotoxicity mechanisms of sesaminol and rabdosianone I target ANT2 or PHB2; therefore, the cytotoxicity mechanism of action of kapakahine A may be explained by its binding to ANT2 or PHB2. The possibility that PHB1 is a target molecule of kapakahine A can potentially be excluded because the cytotoxicity of kapakahine A was much weaker than that of aurilide and the fragmentation of mitochondria was not observed following the addition of kapakahine A.
Although further study is needed to confirm the mechanism of action of kapakahine A, it was shown that useful mechanistic information can be obtained for moderately active natural products using naturally available quantities.
4. Materials and Methods
4.1. Materials
The confocal microscopy images were acquired using an Olympus FV1000 (Olympus Corporation, Tokyo, Japan). Nuclear magnetic resonance (NMR) spectra were measured with a Bruker Avance (600 MHz, Bruker Corporation, Billerica, MA, USA) spectrometer. HPLC (high-performance liquid chromatography) was conducted using a JASCO PU-1580 pump system equipped with TOSOH UV8011 (TOSOH Corporation, Tokyo, Japan).
All high-resolution mass spectra (HRMS) were recorded on a JEOL JMS-T100CS spectrometer (JEOL Ltd., Tokyo, Japan) at the Materials Characterization Central Laboratory, Waseda University. LC/MS/MS was performed using a Triple TOF 4600 system (AB Sciex, Tokyo, Japan).
4.2. Preparation of the Kap A-Probe (3)
Kapakahine A (
1, 1.0 mg, 0.95 µmol) dissolved in 100 µL of DMF was mixed with 3 µL (9.0 µmol) of compound
4 and allowed to react for 20 h, stirring at room temperature while shielded from light. The collected reaction mixture was concentrated and dried, then subjected to reversed-phase HPLC [COSMOSIL 5C
18-AR II (φ10 × 250 mm), flow rate; 2 mL/min, isocratic elution with 55% acetonitrile, detection; 220 nm JASCO] yielding Kap A-probe (
3, 0.5 mg), which showed a [M-H]
− ion peak at
m/
z 1324.6726 (calcd. C
69H
90N
13O
14 1324.6736). The mass spectrum (
Figure S1),
1H-NMR spectrum (
Figure S2), and HPLC chart (
Figure S3) are shown in
Supplementary Materials.
4.3. Cell Culture
Roswell Park Memorial Institute medium (RPMI, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) supplemented with HEDS solution (2,20-dithiobisethanol) and kanamycin sulfate was used as the culture media for P388 murine leukemia cells. Cells were cultured at 37 °C under an atmosphere of 5% CO2.
4.4. Staining of P388 Cells
A fluorescent control and fluorescently labeled kapakahine A (
2) were prepared as previously described [
4]. P388 cells (10,000 cells/well in 200 µL medium) were plated in each well of a 96-well glass bottom microplate. After incubation for 24 h, the cultural medium in each well was removed and replaced with 100 µL of a solution of fluorescent probe
2 prepared with Lipofectamine 2000 [
4]; then, cells were further incubated at 37 °C under an atmosphere of 5% CO
2 for 2 h. The concentration of compound
2 was 20 μM. After 75 min, 1 mg/mL of MitoTracker Red CMXRos (Thermo Fisher Scientific Inc., Waltham, MA, USA) dissolved in DMSO was diluted with culture medium and 11 µL of the mixture was added to the cells and incubated at 37 °C under an atmosphere of 5% CO
2 for 15 min. The cells were fixed in 100 µL of 4% paraformaldehyde at 37 °C under an atmosphere of 5% CO
2 for 15 min. After fixing and washing with culture medium and PBS, fluorescence cell images were acquired at ×100 magnification using a confocal microscope system.
4.5. Preparation of Protein Extracts Containing Cytoplasmic and Plasma Membrane Fractions of P388 Cells
P388 cells that had reached confluency were collected in 15 mL tubes and centrifuged at 180× g for 5 min at 15 °C. Cold PBS (1 mL) was added to the precipitate obtained by removing the supernatant, then transferred to a 2.0 mL tube and centrifuged at 1500× g for 5 min. This washing procedure was carried out again, and 200 µL of buffer A [10 mM HEPES-NaOH (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF (phenylmethylsulfonyl fluoride), ×1 protease inhibitor cocktail (PI)] was added to the precipitate. After standing on ice for 15 min, 11 µL of 10% NP-40 was added and cells were agitated. The supernatant was then collected following centrifugation at 215× g for 5 min. The supernatant was diluted with buffer B [20 mM HEPES-NaOH (pH 7.9), 100 mM KCl, 1 mM MgCl2, 0.2 mM CaCl2, 0.2 mM EDTA, 10% (v/v) glycerol, 1 mM DTT, 0.5 mM PMSF, ×1 PI] to ensure a 0.1% NP-40 concentration to yield protein extracts including cytoplasmic and plasma membrane fractions.
4.6. Preparation of Kap A-Beads (6)
Magnetic FG beads (5, 1.0 mg) with terminal alkynes (Tamagawa, Seiki Co., Ltd., Nagano, Japan) were mixed with 25 µL of Kap A-probe (3) prepared at 1 mM in tBuOH/DMSO (4/1) and 25 µL of 500 µM TBTA, then sonicated. To this solution, 25 µL each of 5 mM aqueous CuSO4 solution and 5 mM aqueous sodium L-ascorbate solution were added, and the reaction was allowed to proceed at room temperature for 20 h, being shaken by a microtube mixer. The reaction solution was centrifuged at 18,800× g for 5 min, and then the supernatant was collected. The precipitated beads were washed with 200 µL of tBuOH/DMSO/H2O (4/1/2) and centrifuged at 18,800× g for 5 min, followed by washing to remove the supernatant three times. The washing operation was then repeated three times with 200 µL of 50% MeOH. Finally, 40 µL of 50% MeOH was added to yield Kap A-beads (6).
4.7. Binding of Kap A-Beads (6) to Proteins
Protein extracts containing cytoplasmic and membrane fractions from P388 cells were mixed with binding buffer (20 mM HEPES-NaOH, pH 7.9, 100 mM KCl, 1 mM MgCl2, 0.2 mM CaCl2, 0.2 mM EDTA, 10% glycerol, 0.1% NP-40, 1 mM DTT, 0.2 mM PMSF, ×1 PI) which was used to dispense 200 µg. After centrifugation at 215× g for 30 min at 4 °C, the supernatant was mixed with 0.5 mg of Kap A-beads (6). Alkyne beads (5) were used as a control. The mixed solution of supernatant and FG beads was rotated at 4 °C for 4 h to allow the binding reaction to proceed. After the reaction, the mixture was subjected to magnetic separation for 5 min and the supernatant was discarded. Binding buffer (200 µL) was added to the remaining beads, which were then spun down and subjected to magnetic separation for 5 min, and the supernatant was discarded. This washing procedure was repeated twice more. Salt elution buffer (30 µL; 1 M KCl, 1 mM DTT, 0.2 mM PMSF, ×1 PI) was added to the washed beads, and the beads were dispersed and placed on ice for 5 min. After magnetic separation, the supernatant was collected in a 1.5 mL tube and mixed with 8 µL of ×4 loading buffer (0.25 mM Tris-HCl pH 6.8, 0.02% bromophenol blue, 8% SDS, 40% glycerol) and 2 µL of 2-mercaptoethanol, and then heated at 98 °C for 5 min to obtain the salt-eluted bound protein fraction. The remaining beads were dispersed by adding 38 µL of ×1 loading buffer and 2 µL of 2-mercaptoethanol, and then heated at 98 °C for 5 min to obtain the heat-eluted bound protein fraction.
The obtained protein solutions were subjected to SDS-PAGE using Super Sep™ Ace, 5–20% gradient gel (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) with 25 mM Tris, 192 mM glycine, and 0.1% SDS as the buffer for 90 min at a constant current of 18 mA. Precision Plus Protein Dual Color Standards (Bio-Rad, Hercules, CA, USA) were used as molecular weight markers. Protein bands on the gel were detected by silver staining according to the manufacturer’s protocol.
4.8. Target Protein Identification
The protein bands of interest were cut out of the gel after silver staining and collected in 1.5 mL tubes. Decolorizing solution (100 µL) was added, and the samples were allowed to stand for 15 min. The supernatant was discarded, and washing was repeated twice with water. Acetonitrile (100%, 200 µL) was then added and allowed to stand for 5 min, and the gel fragments were dried using a lyophilizer. Two hundred microliters of 10 mM DTT, 25 mM NH4HCO3 mixed solution was added to the dried gel and the proteins were reduced by heating at 56 °C for 45 min. After discarding the solution, 200 µL of 25 mM NH4HCO3 solution was added, the sample was shaken for 5 min, and the solution removal was repeated twice. Acetonitrile (100%, 200 µL) was then added and allowed to stand for 5 min, and the gel fragments were dried using a lyophilizer. Trypsin (10 µL, Trypsin Gold, Mass Spectrometry Grade, Promega, WI, USA) was added to 10 µg/mL of the dried gel sections, and digestion was allowed to proceed in the gel at 37 °C for 18 h. Acetonitrile (50%) containing 0.1% TFA (trifluoro acetic acid) was added, and the solution was collected following sonication. This procedure was repeated twice, and the recovered solution was concentrated for LC/MS measurement. The LC/MS data were analyzed using ProteinPilot Software 4.5 (AB Sciex, Tokyo, Japan) for the identification of proteins.
4.9. Western Blotting
After separation of the protein bands by SDS-PAGE, the gel was fixed in 100% MeOH. The gel was then immersed in transfer buffer (tris-hydroxyaminomethane 25 mM, glycine 192 mM, 20% MeOH) and the proteins in the gel were transferred to a polyvinylidene fluoride (PVDF) membrane using a transfer system at a constant current of 180 mA for 60 min. The transferred membranes were fixed and stained with Ponceau stain and then decolorized. Blocking was then carried out using 10% skim milk powder dissolved in TBS-T for 60 min at room temperature. Then, primary antibodies anti-prohibitin 1 (1:100, sc-377037, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-prohibitin 2 (1:100, sc-133094; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), and anti-ANT2 (1:1000, ANT2/SLC25A5(E2B9D), Cell Signaling Technology, Danvers, MA, USA), diluted in PBS-T solution containing 1% skim milk, were allowed to react overnight at 4 °C. Shaking and washing with PBS-T for 5 min was performed three times; then, the antibodies were reacted with secondary antibodies for 3 h at room temperature. After 3 rounds of shaking and washing with PBS-T for 5 min, protein detection was performed using SuperSignal West Pico Chemiluminescent Substrate or Super Signal West Femto Maximum Sensitivity Substrate (Bio-Rad, Hercules, CA, USA). An LAS-3000 (FUJIFILM, Tokyo, Japan) was used to photograph the membrane.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}