
Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors


Abstract
:1. Introduction
2. Results and Discussion
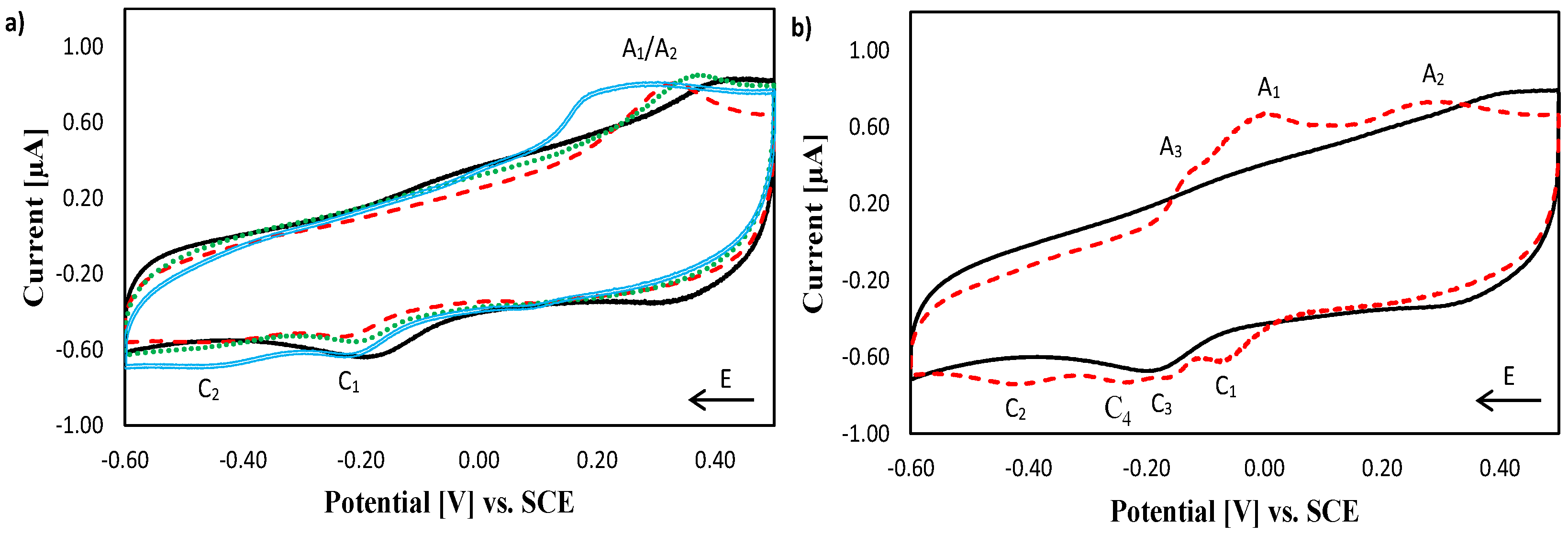
2.1. Electrochemical Characterisation of Copper Complexes at Gold Electrodes
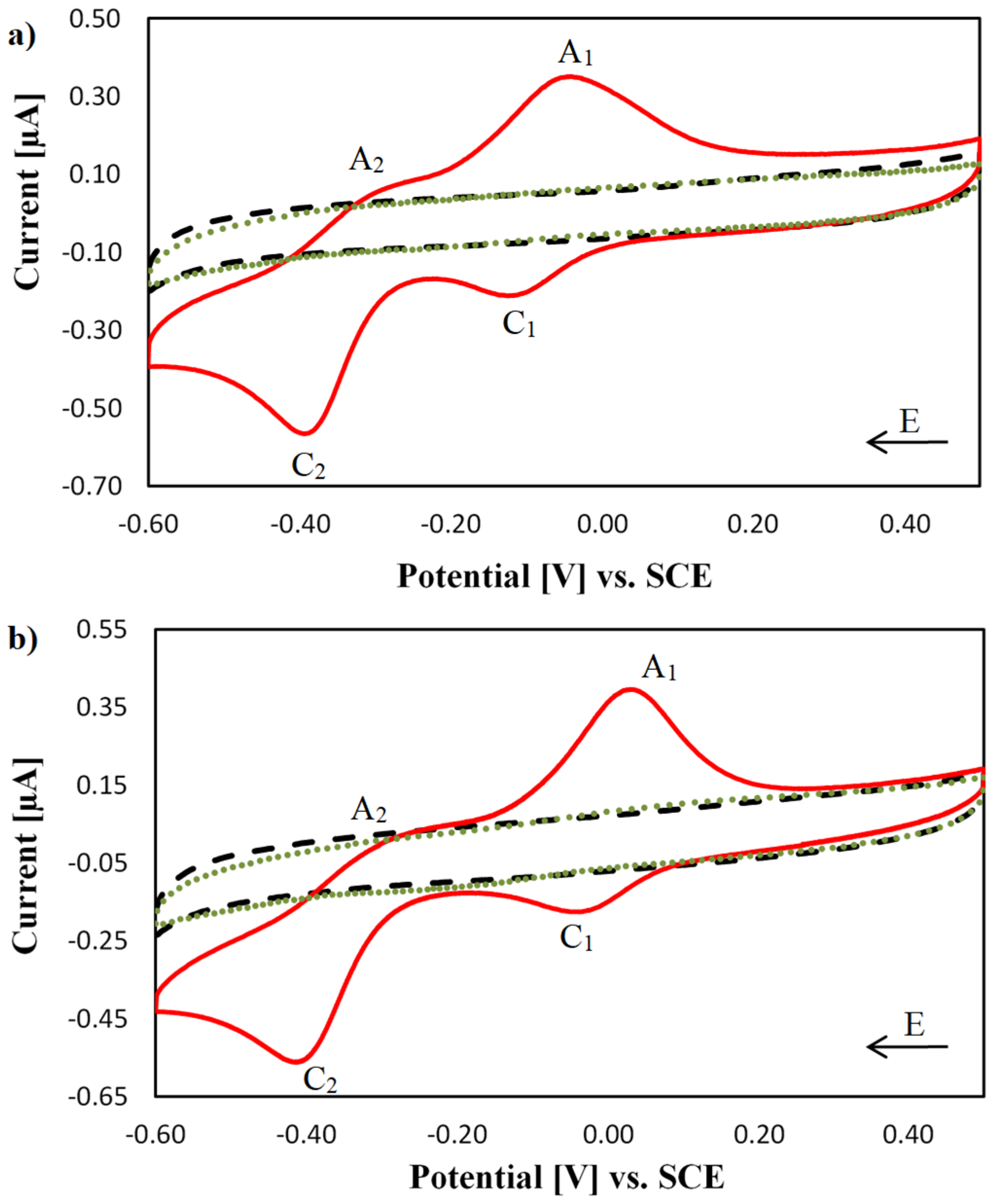
2.2. Electrochemical Characterisation of [Cu(phen)2]2+, [Cu(TPMA)]2+ and the TPMA Ligand at the DNA Biosensor
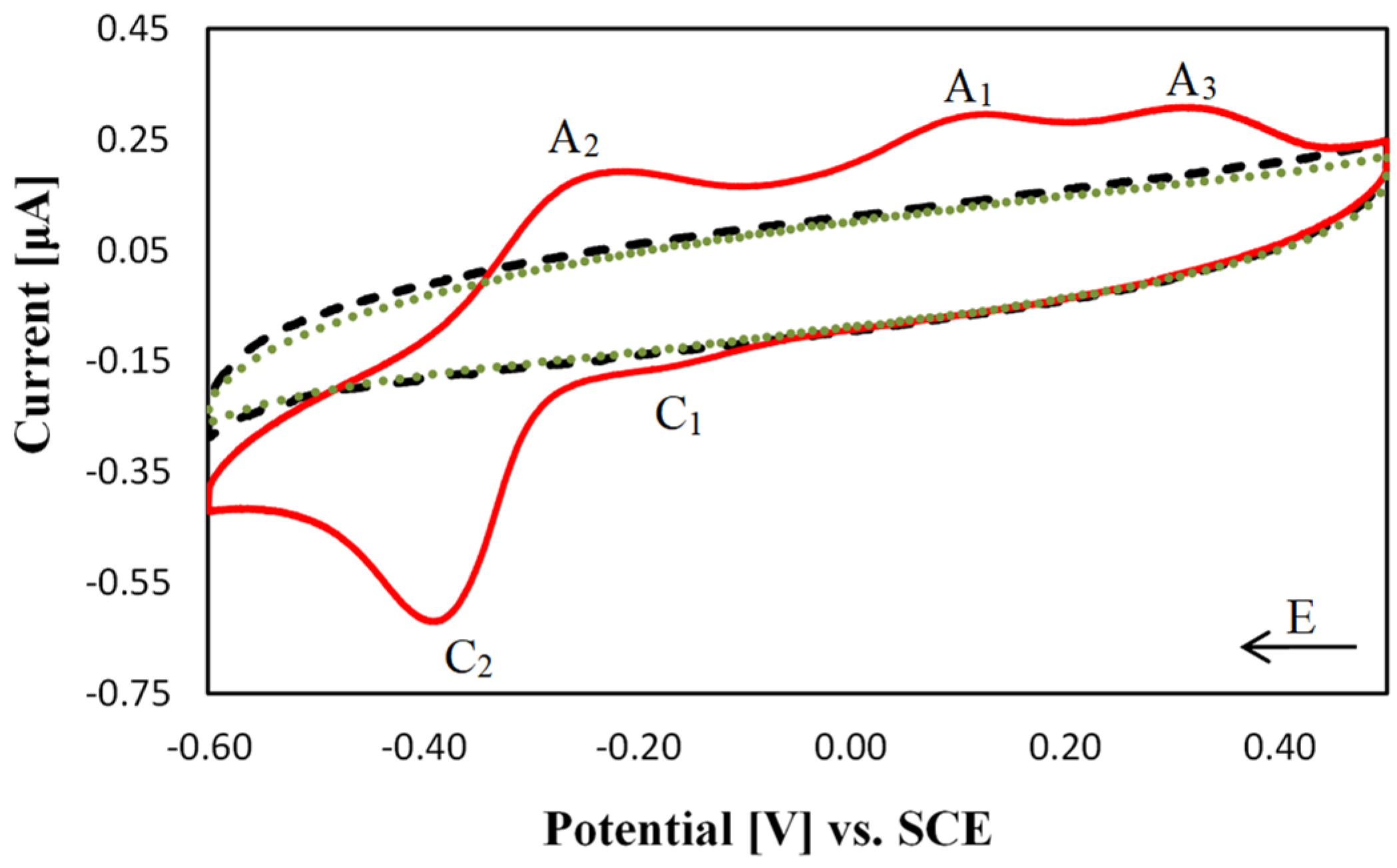
2.3. Electrochemical Characterisation of [Cu(TPMA)(N,N′)]2+ Complexes at the DNA Biosensor
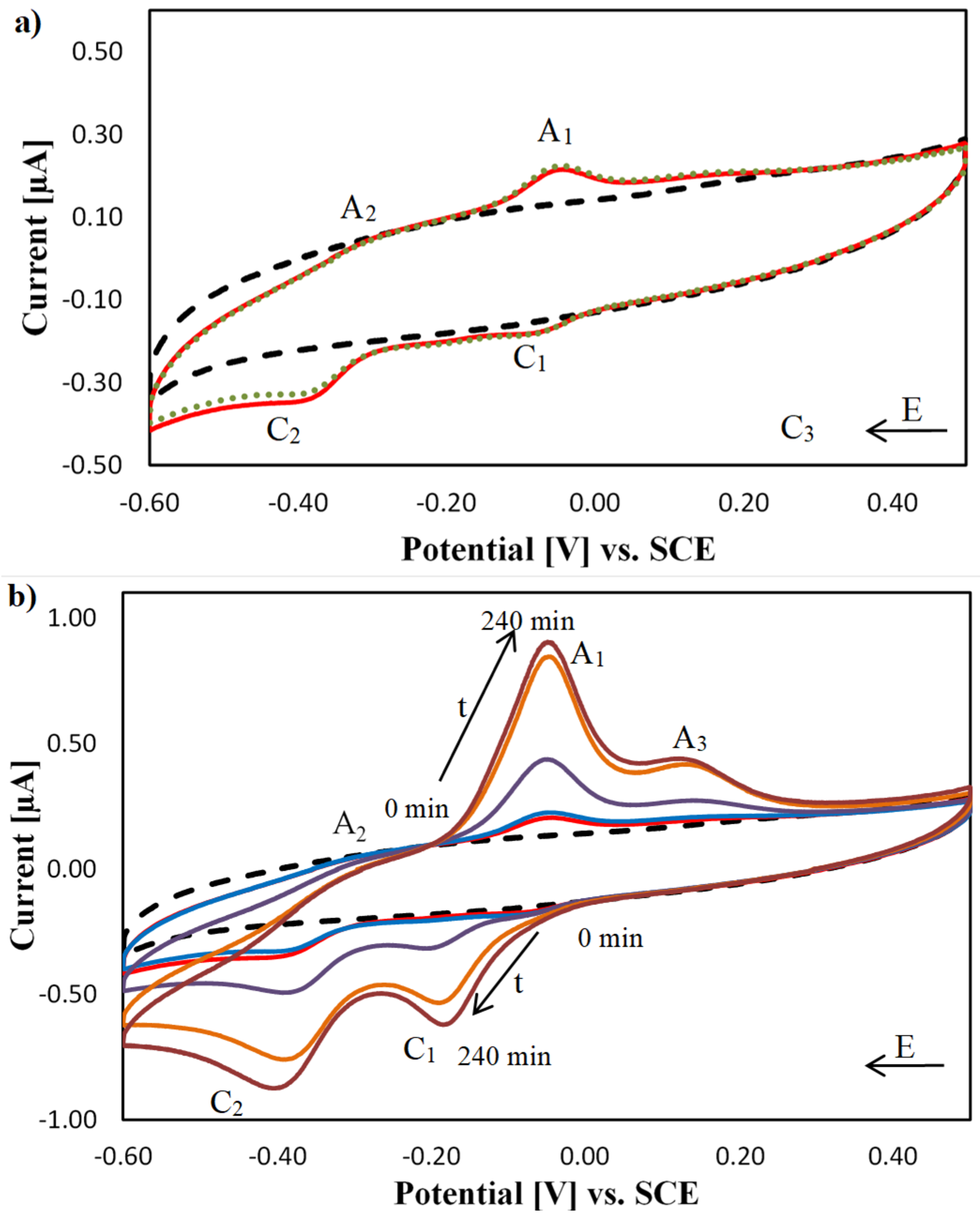
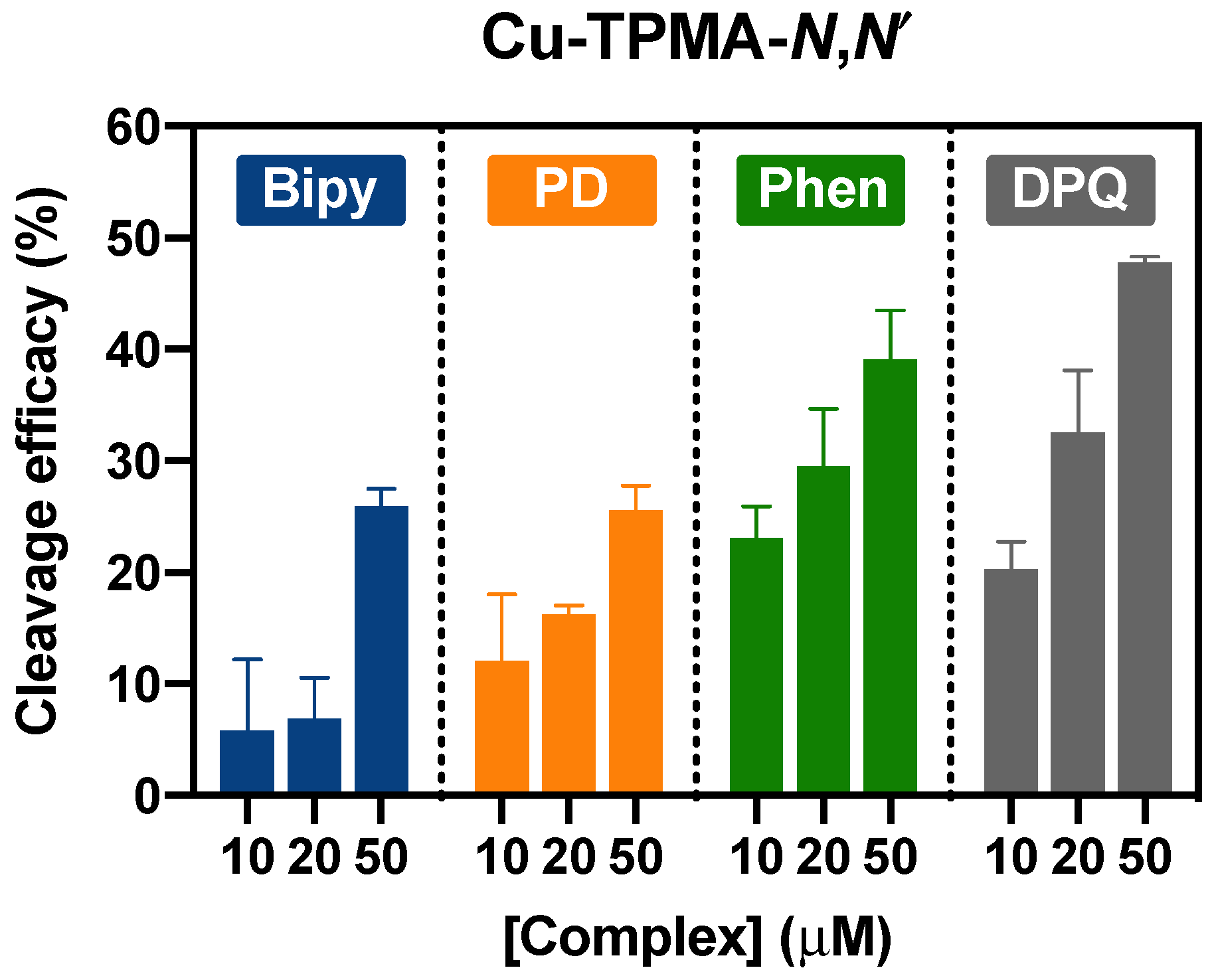
2.4. DNA Nuclease Efficacy of Copper Complexes
3. Materials and Methods
3.1. Materials
3.2. Equipment
3.3. Interactions between Immobilised DNA and Bioinorganic Compounds
3.4. Determination of DNA Surface Coverage
3.5. Electrochemical Measurements and Nuclease Assay Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brown, G.C. Living too long: The current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Rep. 2015, 16, 137–141. [Google Scholar] [CrossRef] [Green Version]
- WHO. Latest global cancer data: Cancer burden rises to 18.1 million new cases and 9.6 million cancer deaths in 2018. WHO: Geneva, Switzerland; pp. 1–3. Available online: https://www.iarc.who.int/wp-content/uploads/2018/09/pr263_E.pdf (accessed on 10 June 2018).
- Hennessy, J.; McGorman, B.; Molphy, Z.; Farrell, N.P.; Singleton, D.; Brown, T.; Kellett, A. A Click Chemistry Approach to Targeted DNA Crosslinking with cis-Platinum(II)-Modified Triplex-Forming Oligonucleotides. Angew. Chem. Int. Ed. 2021, 61, e202110455. [Google Scholar] [CrossRef]
- Kellett, A.; O’Connor, M.; McCann, M.; Howe, O.; Casey, A.; McCarron, P.; Kavanagh, K.; McNamara, M.; Kennedy, S.; May, D.D.; et al. Water-soluble bis(1,10-phenanthroline) octanedioate Cu2+ and Mn2+ complexes with unprecedented nano and picomolar in vitro cytotoxicity: Promising leads for chemotherapeutic drug development. MedChemComm 2011, 2, 579–584. [Google Scholar] [CrossRef] [Green Version]
- McGivern, T.; Afsharpour, S.; Marmion, C. Copper complexes as artificial DNA metallonucleases: From Sigman’s reagent to next generation anti-cancer agent? Inorganica Chim. Acta 2018, 472, 12–39. [Google Scholar] [CrossRef]
- Muhammad, N.; Guo, Z. Metal-based anticancer chemotherapeutic agents. Curr. Opin. Chem. Biol. 2014, 19, 144–153. [Google Scholar] [CrossRef]
- Thornton, L.; Dixit, V.; Assad, L.O.; Ribeiro, T.P.; Queiroz, D.; Kellett, A.; Casey, A.; Colleran, J.; Pereira, M.D.; Rochford, G.; et al. Water-soluble and photo-stable silver(I) dicarboxylate complexes containing 1,10-phenanthroline ligands: Antimicrobial and anticancer chemotherapeutic potential, DNA interactions and antioxidant activity. J. Inorg. Biochem. 2016, 159, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Fantoni, N.Z.; Brown, T.; Kellett, A. DNA-Targeted Metallodrugs: An Untapped Source of Artificial Gene Editing Technology. ChemBioChem 2021, 22, 2184–2205. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Garcia-Ramos, J.C.; Ruiz-Azuara, L.; Cheatham, T.E.; Cortes-Guzman, F. Intercalation processes of copper complexes in DNA. Nucleic Acids Res. 2015, 43, 5364–5376. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2014, 33, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzano, C.; Pellei, M.; Tisato, F.; Santini, C. Copper Complexes as Anticancer Agents. Anti-Cancer Agents Med. Chem. 2009, 9, 185–211. [Google Scholar] [CrossRef]
- McStay, N.; Slator, C.; Singh, V.; Gibney, A.; Westerlund, F.; Kellett, A. Click and Cut: A click chemistry approach to developing oxidative DNA damaging agents. Nucleic Acids Res. 2021, 49, 10289–10308. [Google Scholar] [CrossRef]
- Sangeetha Gowda, K.R.; Mathew, B.B.; Sudhamani, C.N.; Naik, H.S.B. Mechanism of DNA Binding and Cleavage. Biomed.Biotechnol. 2014, 2, 1–9. [Google Scholar] [CrossRef]
- Que, B.G.; Downey, K.M.; So, A.G. Degradation of deoxyribonucleic acid by a 1,10-phenanthroline-copper complex: The role of hydroxyl radicals. Biochemistry 1980, 19, 5987–5991. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.A.; Marshall, L.E.; Graham, D.R.; Sigman, D.S. Cleavage of DNA by the 1,10-phenanthroline-copper ion complex. Superoxide mediates the reaction dependent on NADH and hydrogen peroxide. J. Am. Chem. Soc. 1981, 103, 3582–3584. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; Molphy, Z.; Slator, C.; Menounou, G.; Toniolo, G.; Mitrikas, G.; McKee, V.; Chatgilialoglu, C.; Kellett, A. Polypyridyl-Based Copper Phenanthrene Complexes: A New Type of Stabilized Artificial Chemical Nuclease. Chem.–A Eur. J. 2018, 25, 221–237. [Google Scholar] [CrossRef]
- Abreu, F.; Goulart, M.; Brett, A.O. Detection of the damage caused to DNA by niclosamide using an electrochemical DNA-biosensor. Biosens. Bioelectron. 2002, 17, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Kellett, A.; Molphy, Z.; McKee, V.; Slator, C. CHAPTER 4. Recent Advances in Anticancer Copper Compounds. Met. Based Anticancer Agents 2019, 14, 91–119. [Google Scholar] [CrossRef]
- Toniolo, G.; Louka, M.; Menounou, G.; Fantoni, N.Z.; Mitrikas, G.; Efthimiadou, E.K.; Masi, A.; Bortolotti, M.; Polito, L.; Bolognesi, A.; et al. [Cu(TPMA)(Phen)](ClO4)2: Metallodrug Nanocontainer Delivery and Membrane Lipidomics of a Neuroblastoma Cell Line Coupled with a Liposome Biomimetic Model Focusing on Fatty Acid Reactivity. ACS Omega 2018, 3, 15952–15965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigman, D.S.; Graham, D.R.; D’Aurora, V.; Stern, A.M. Oxygen-dependent cleavage of DNA by the 1,10-phenanthroline. cuprous complex. Inhibition of Escherichia coli DNA polymerase I. J. Biol. Chem. 1979, 254, 12269–12272. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2013, 114, 815–862. [Google Scholar] [CrossRef]
- Fantoni, N.Z.; McGorman, B.; Molphy, Z.; Singleton, D.; Walsh, S.; El-Sagheer, A.H.; McKee, V.; Brown, T.; Kellett, A. Development of Gene-Targeted Polypyridyl Triplex-Forming Oligonucleotide Hybrids. ChemBioChem 2020, 21, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Fantoni, N.Z.; Molphy, Z.; O’Carroll, S.; Menounou, G.; Mitrikas, G.; Krokidis, M.G.; Chatgilialoglu, C.; Colleran, J.; Banasiak, A.; Clynes, M.; et al. Polypyridyl-Based Copper Phenanthrene Complexes: Combining Stability with Enhanced DNA Recognition. Chem.-A Eur. J. 2020, 27, 971–983. [Google Scholar] [CrossRef]
- Lauria, T.; Slator, C.; McKee, V.; Müller, M.; Stazzoni, S.; Crisp, A.L.; Carell, T.; Kellett, A. A Click Chemistry Approach to Developing Molecularly Targeted DNA Scissors. Chem.–A Eur. J. 2020, 26, 16782–16792. [Google Scholar] [CrossRef]
- Panattoni, A.; El-Sagheer, A.H.; Brown, T.; Kellett, A.; Hocek, M. Oxidative DNA Cleavage with Clip-Phenanthroline Triplex-Forming Oligonucleotide Hybrids. ChemBioChem 2019, 21, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Fantoni, N.Z.; El-Sagheer, A.H.; Brown, T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 2021, 121, 7122–7154. [Google Scholar] [CrossRef] [PubMed]
- Larragy, R.; Fitzgerald, J.; Prisecaru, A.; McKee, V.; Leonard, P.; Kellett, A. Protein engineering with artificial chemical nucleases. Chem. Commun. 2015, 51, 12908–12911. [Google Scholar] [CrossRef] [PubMed]
- Fojta, M.; Kubičárová, T.; Paleček, E. Cleavage of Supercoiled DNA by Deoxyribonuclease I in Solution and at the Electrode Surface. Electroanalysis 1999, 11, 1005–1012. [Google Scholar] [CrossRef]
- Ghosh, K.; Kumar, P.; Tyagi, N.; Singh, U.P.; Goel, N. Synthesis, structural characterization and DNA interaction studies on a mononuclear copper complex: Nuclease activity via self-activation. Inorg. Chem. Commun. 2011, 14, 489–492. [Google Scholar] [CrossRef]
- Hirohama, T.; Kuranuki, Y.; Ebina, E.; Sugizaki, T.; Arii, H.; Chikira, M.; Selvi, P.T.; Palaniandavar, M. Copper(II) complexes of 1,10-phenanthroline-derived ligands: Studies on DNA binding properties and nuclease activity. J. Inorg. Biochem. 2005, 99, 1205–1219. [Google Scholar] [CrossRef]
- Molphy, Z.; Prisecaru, A.; Slator, C.; Barron, N.; McCann, M.; Colleran, J.; Chandran, D.; Gathergood, N.; Kellett, A. Copper Phenanthrene Oxidative Chemical Nucleases. Inorg. Chem. 2014, 53, 5392–5404. [Google Scholar] [CrossRef]
- Kellett, A.; Molphy, Z.; Slator, C.; McKee, V.; Farrell, N.P. Molecular methods for assessment of non-covalent metallodrug–DNA interactions. Chem. Soc. Rev. 2019, 48, 971–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fojta, M.; Kubičárová, T.; Paleček, E. Electrode potential-modulated cleavage of surface-confined DNA by hydroxyl radicals detected by an electrochemical biosensor. Biosens. Bioelectron. 2000, 15, 107–115. [Google Scholar] [CrossRef]
- Labuda, J.; Bučková, M.; Vaníčková, M.; Mattusch, J.; Wennrich, R. Voltammetric Detection of the DNA Interaction with Copper Complex Compounds and Damage to DNA. Electroanalysis 1999, 11, 101–107. [Google Scholar] [CrossRef]
- Paleček, E.; Fojta, M.; Tomschik, M.; Wang, J. Electrochemical biosensors for DNA hybridization and DNA damage. Biosens. Bioelectron. 1998, 13, 621–628. [Google Scholar] [CrossRef]
- Banasiak, A.; Cassidy, J.; Colleran, J. A novel quantitative electrochemical method to monitor DNA double-strand breaks caused by a DNA cleavage agent at a DNA sensor. Biosens. Bioelectron. 2018, 117, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Jopp, M.; Becker, J.; Becker, S.; Miska, A.; Gandin, V.; Marzano, C.; Schindler, S. Anticancer activity of a series of copper(II) complexes with tripodal ligands. Eur. J. Med. Chem. 2017, 132, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Molphy, Z.; Slator, C.; Chatgilialoglu, C.; Kellett, A. DNA oxidation profiles of copper phenanthrene chemical nucleases. Front. Chem. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, K.J.; Johnson, A.E.; Karlin, K.D.; Rokita, S.E. Oxidative strand scission of nucleic acids by a multinuclear copper(II) complex. JBIC J. Biol. Inorg. Chem. 2002, 7, 835–842. [Google Scholar] [CrossRef]
- Humphreys, K.J.; Karlin, K.D.; Rokita, S.E. Efficient and Specific Strand Scission of DNA by a Dinuclear Copper Complex: Comparative Reactivity of Complexes with Linked Tris(2-pyridylmethyl)amine Moieties. J. Am. Chem. Soc. 2002, 124, 6009–6019. [Google Scholar] [CrossRef]
- Kraft, S.S.N.; Bischof, C.; Loos, A.; Braun, S.; Jafarova, N.; Schatzschneider, U. A [4+2] mixed ligand approach to ruthenium DNA metallointercalators [Ru(tpa)(N–N)](PF6)2 using a tris(2-pyridylmethyl)amine (tpa) capping ligand. J. Inorg. Biochem. 2009, 103, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Louka, F.R.; Doulain, P.E.; Landry, C.A.; Mautner, F.A.; Massoud, S.S. Hydrolytic cleavage of DNA promoted by cobalt(III)–tetraamine complexes: Synthesis and characterization of carbonatobis[2-(2-pyridylethyl)]-(2-pyridylmethyl)aminecobalt(III) perchlorate. Polyhedron 2009, 28, 1221–1228. [Google Scholar] [CrossRef]
- Prisecaru, A.; McKee, V.; Howe, O.; Rochford, G.; McCann, M.; Colleran, J.; Pour, M.; Barron, N.; Gathergood, N.; Kellett, A. Regulating Bioactivity of Cu2+ Bis-1,10-phenanthroline Artificial Metallonucleases with Sterically Functionalized Pendant Carboxylates. J. Med. Chem. 2013, 56, 8599–8615. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wu, J.; Xi, C.; Lehnert, N.; Major, T.; Bartlett, H.R.; Meyerhoff, M.E. Electrochemically Modulated Nitric Oxide (NO) Releasing Biomedical Devices via Copper(II)-Tri(2-pyridylmethyl)amine Mediated Reduction of Nitrite. ACS Appl. Mater. Interfaces 2014, 6, 3779–3783. [Google Scholar] [CrossRef]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- Evans, D.H.; Griffith, D.A. Effect of metal ions on the electrochemical reduction of some heterocyclic quinones. J. Electroanal. Chem. Interfacial Electrochem. 1982, 136, 149–157. [Google Scholar] [CrossRef]
- Kou, Y.-Y.; Xu, G.-J.; Gu, W.; Tian, J.-L.; Yan, S.-P. Synthesis and pH-sensitive redox properties of 1,10-phenanthroline-5,6-dione complexes. J. Co-ord. Chem. 2008, 61, 3147–3157. [Google Scholar] [CrossRef]
- Cory, R.M.; McKnight, D.M. Fluorescence spectroscopy reveals ubiquitous presence of oxidized and reduced quinones in dissolved organic matter. Environ. Sci. Technol. 2005, 39, 8142–8149. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, J.M.; Molero, L.; Cañete, A.; Del Rio, R.; Del Valle, A.M.; Mallavia, R.; Armijo, F. Electrochemical and spectroscopic properties of indolizino[1,2-B] quinole derivates. J. Chil. Chem. Soc. 2013, 58, 1976–1979. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, S.; Palaniandavar, M. Spectroscopic and Voltammetric Studies on Copper Complexes of 2,9-Dimethyl-1,10-phenanthrolines Bound to Calf Thymus DNA. Inorg. Chem. 1998, 37, 693–700. [Google Scholar] [CrossRef]
- Lucio, A.J.; Shaw, S.K. Pyridine and Pyridinium Electrochemistry on Polycrystalline Gold Electrodes and Implications for CO2 Reduction. J. Phys. Chem. C 2015, 119, 12523–12530. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics, 95th ed.; Haynes, W.M., Lide, D.R., Bruno, T.J., Eds.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Carter, M.T.; Rodriguez, M.; Bard, A.J. Voltammetric studies of the interaction of metal chelates with DNA. 2. Tris-chelated complexes of cobalt(III) and iron(II) with 1,10-phenanthroline and 2,2′-bipyridine. J. Am. Chem. Soc. 1989, 111, 8901–8911. [Google Scholar] [CrossRef]
- Chikira, M.; Ng, C.H.; Palaniandavar, M. Interaction of DNA with Simple and Mixed Ligand Copper(II) Complexes of 1,10-Phenanthrolines as Studied by DNA-Fiber EPR Spectroscopy. Int. J. Mol. Sci. 2015, 16, 22754–22780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draksharapu, A.; Boersma, A.J.; Leising, M.; Meetsma, A.; Browne, W.R.; Roelfes, G. Binding of copper(II) polypyridyl complexes to DNA and consequences for DNA-based asymmetric catalysis. Dalton Trans. 2014, 44, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.-W.; Abruña, H.D. Micromethod for the Investigation of the Interactions between DNA and Redox-Active Molecules. Anal. Chem. 1998, 70, 3162–3169. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2016, 30, 13–37. [Google Scholar] [CrossRef]
- Pinto, A.V.; De Castro, S.L. The Trypanocidal Activity of Naphthoquinones: A Review. Molecules 2009, 14, 4570–4590. [Google Scholar] [CrossRef] [PubMed]
- Steel, A.B.; Herne, T.M.; Tarlov, M.J. Electrochemical Quantitation of DNA Immobilized on Gold. Anal. Chem. 1998, 70, 4670–4677. [Google Scholar] [CrossRef] [PubMed]
- Santra, B.K.; Reddy, P.A.; Neelakanta, G.; Mahadevan, S.; Nethaji, M.; Chakravarty, A.R. Oxidative cleavage of DNA by a dipyridoquinoxaline copper(II) complex in the presence of ascorbic acid. J. Inorg. Biochem. 2002, 89, 191–196. [Google Scholar] [CrossRef]
- Marshall, L.E.; Graham, D.R.; Reich, K.A.; Sigman, D.S. Cleavage of deoxyribonucleic acid by the 1,10-phenanthroline-cuprous complex. Hydrogen peroxide requirement and primary and secondary structure specificity. Biochemistry 1981, 20, 244–250. [Google Scholar] [CrossRef]
- Ausubel, F.M. Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Smith, J.A., Seidman, J.G., Struhl, K., Eds.; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Pividori, M.I.; Merkoçi, A.; Alegret, S. Electrochemical genosensor design: Immobilisation of oligonucleotides onto transducer surfaces and detection methods. Biosens. Bioelectron. 2000, 15, 291–303. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Ep,C1/Ep,A1 [V] | ΔEp,C1/A1 [mV] | Ep,C2/Ep,A2 [V] | ΔEp,C2/A2 [mV] |
|---|---|---|---|---|
| [Cu(TPMA)(Phen)]2+ | −0.18/+0.30 | 480 | −0.46/+0.30 | 760 |
| [Cu(TPMA)(DPQ)]2+ | −0.21/+0.23 | 440 | −0.46/+0.23 | 590 |
| [Cu(TPMA)(Bipy)]2+ | −0.19/+0.35 | 540 | −0.45/+0.35 | 800 |
| [Cu(TPMA)(PD)]2+ | −0.07/−0.04 | 30 | −0.43/+0.27 | 700 |
| [Cu(TPMA)]2+ | −0.21/+0.01 | 220 | −0.42/+0.22 | 640 |
| ✻ [Cu(Phen)2]2+ | −0.20/−0.08 | 120 | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banasiak, A.; Zuin Fantoni, N.; Kellett, A.; Colleran, J. Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors. Molecules 2022, 27, 645. https://doi.org/10.3390/molecules27030645
Banasiak A, Zuin Fantoni N, Kellett A, Colleran J. Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors. Molecules. 2022; 27(3):645. https://doi.org/10.3390/molecules27030645
Chicago/Turabian StyleBanasiak, Anna, Nicolò Zuin Fantoni, Andrew Kellett, and John Colleran. 2022. "Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors" Molecules 27, no. 3: 645. https://doi.org/10.3390/molecules27030645
APA StyleBanasiak, A., Zuin Fantoni, N., Kellett, A., & Colleran, J. (2022). Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors. Molecules, 27(3), 645. https://doi.org/10.3390/molecules27030645