
Important Requirements for the Selection of Internal Standards during the Development of Desorption/Ionization Assays for Drug Quantification in Biological Matrices—A Practical Example

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Choice of the MALDI Matrix for Analysis
2.3. Sample Preparation
2.4. Mass Spectrometric (MS) Analyses
2.5. Data Processing
3. Results
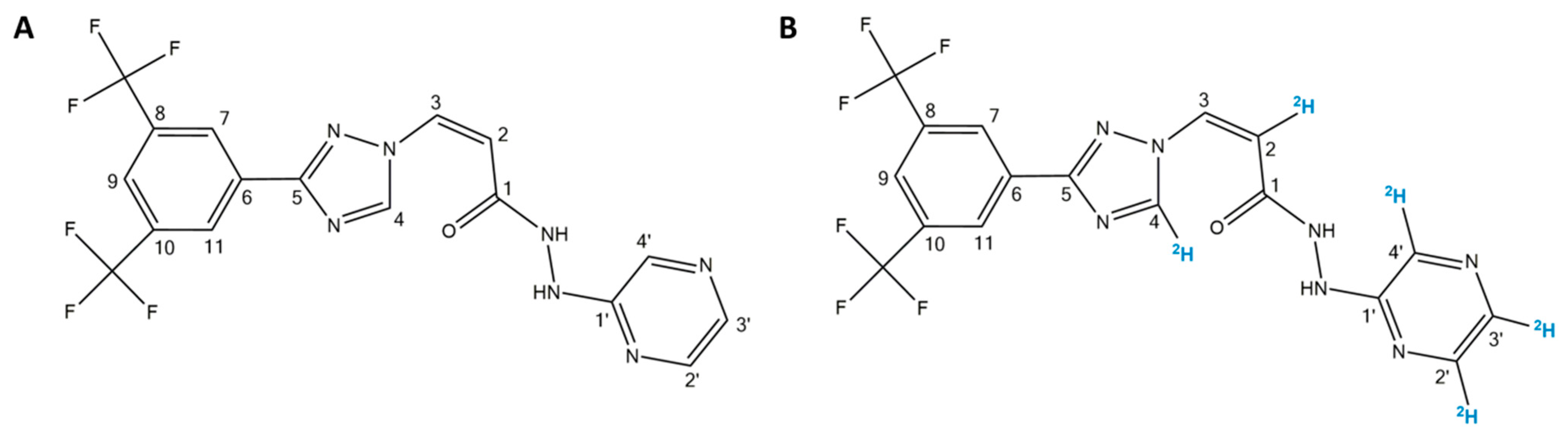
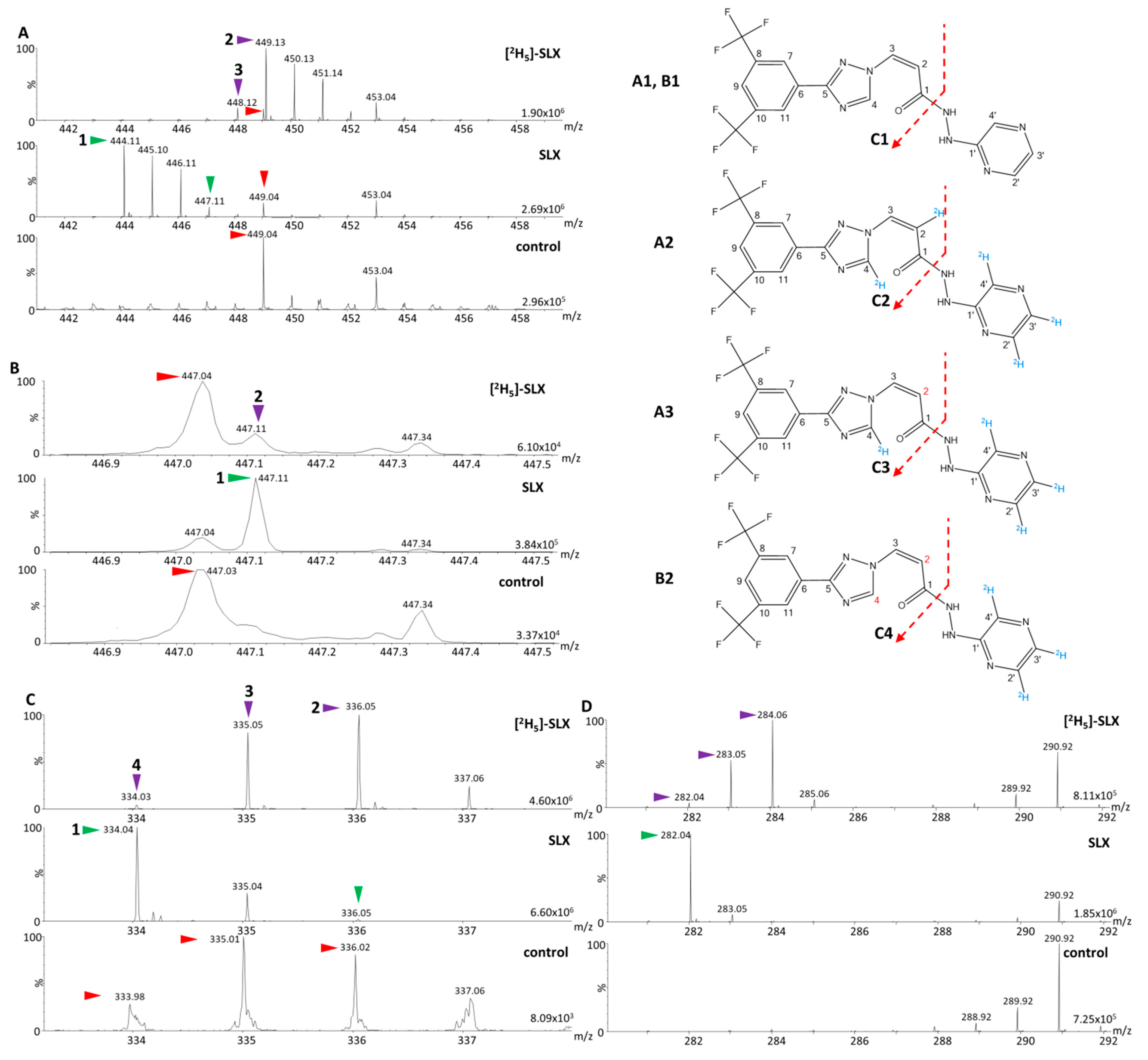
3.1. Determination of the Mass of Parent and Fragment Ions of Selinexor (SLX) and [2H5]-SLX
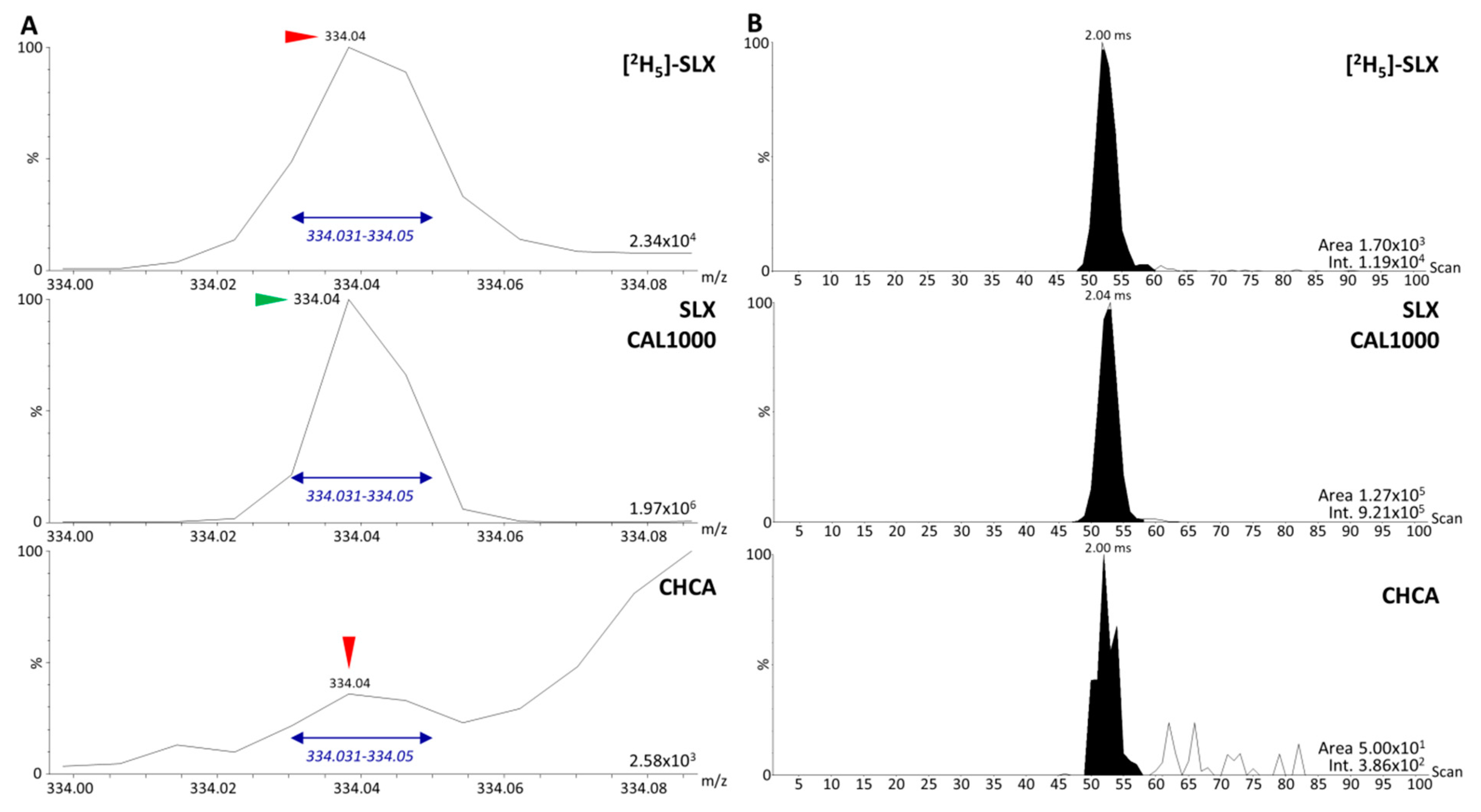
3.2. Analysis of Dilution Series of Selinexor in Biological Matrix and Calibration Using Selinexor Fragments
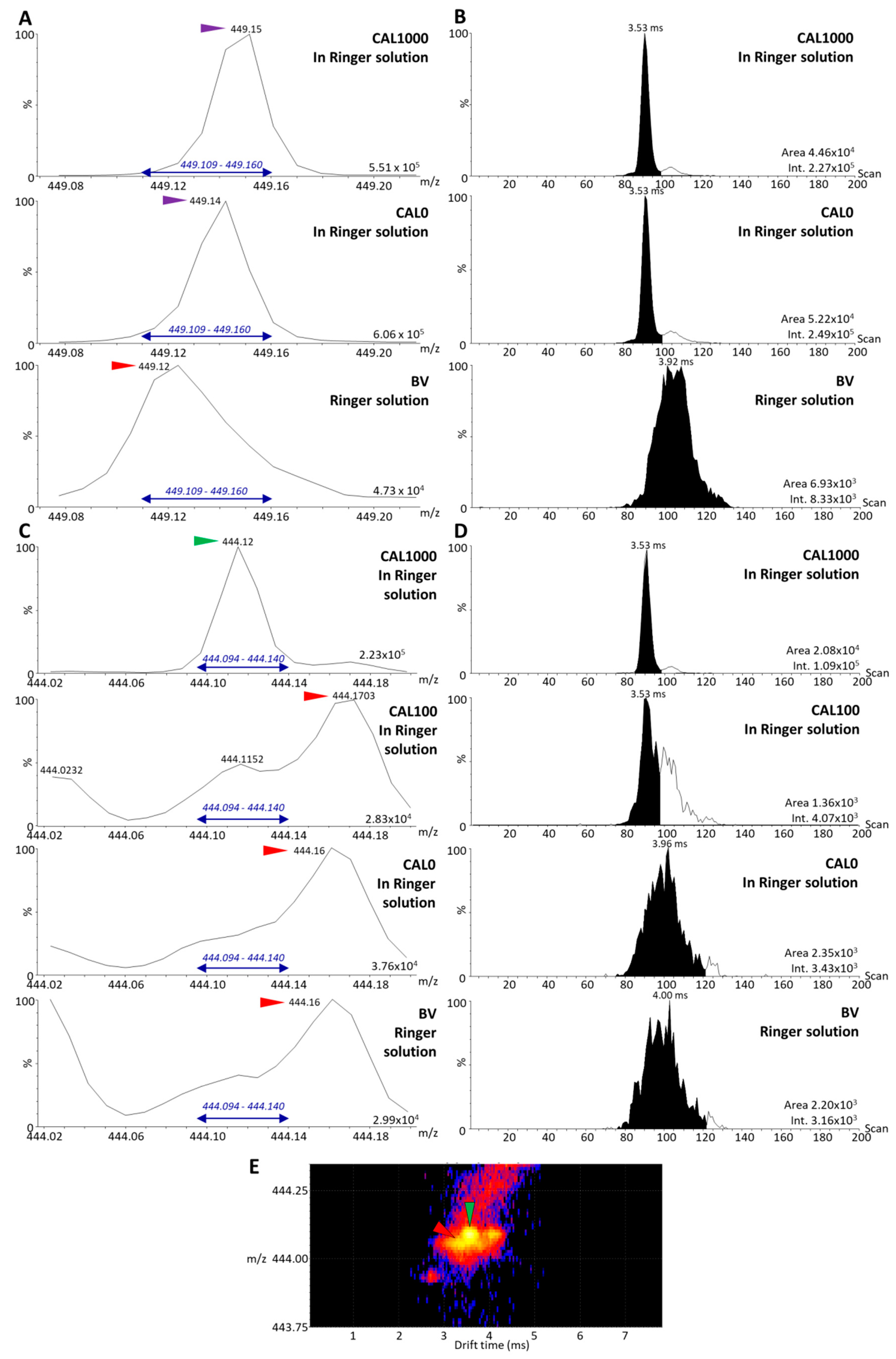
3.3. Analysis of the Dilution Series of Selinexor in Blank Matrix and Calibration Using the Parent Compounds
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fresnais, M.; Burhenne, J.; Haefeli, W.E.; Longuespée, R. Desorption/Ionization-MS Methods for Drug Quantification in Biological Matrices and Their Validation Following Regulatory Guidance. Anal. Chem. 2021, 93, 7152–7163. [Google Scholar] [CrossRef] [PubMed]
- Fresnais, M.; Yildirim, E.; Karabulut, S.; Jäger, D.; Zörnig, I.; Benzel, J.; Pajtler, K.; Pfister, S.; Burhenne, J.; Haefeli, W.; et al. Rapid MALDI-MS Assays for Drug Quantification in Biological Matrices: Lessons Learned, New Developments, and Future Perspectives. Molecules 2021, 26, 1281. [Google Scholar] [CrossRef] [PubMed]
- Mims, D.; Hercules, D. Quantification of bile acids directly from plasma by MALDI-TOF-MS. Anal. Bioanal. Chem. 2004, 378, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Müller, M.; Stokes, C.S.; Volmer, D.A. Rapid Quantification of 25-Hydroxyvitamin D3 in Human Serum by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, J.J.A.; Reedijk, M.L.; Burgers, P.C.; Dekker, L.J.M.; Hartwig, N.G.; Van Der Ende, I.E.; De Groot, R.; Osterhaus, A.D.M.E.; Burger, D.M.; Luider, T.M.; et al. Ultra-Fast Analysis of Plasma and Intracellular Levels of HIV Protease Inhibitors in Children: A Clinical Application of MALDI Mass Spectrometry. PLoS ONE 2010, 5, e11409. [Google Scholar] [CrossRef]
- Cabrales-Rico, A.; de la Torre, B.G.; Garay, H.E.; Machado, Y.; Gómez, J.A.; Audain, E.; Morales, O.; Besada, V.; Marcelo, J.L.; Reyes, V.; et al. Bio-analytical method based on MALDI-MS analysis for the quantification of CIGB-300 anti-tumor peptide in human plasma. J. Pharm. Biomed. Anal. 2015, 105, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Meesters, R.J.; den Boer, E.; Mathot, R.A.; de Jonge, R.; van Klaveren, R.J.; Lindemans, J.; Luider, T.M. Ultrafast selective quantification of methotrexate in human plasma by high-throughput MALDI-isotope dilution mass spectrometry. Bioanalysis 2011, 3, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Castrati, L.; Colombo, P.; Flammini, L.; Barocelli, E.; Bettini, R.; Elviri, L. Development and validation of a DESI-HRMS/MS method for the fast profiling of esomeprazole and its metabolites in rat plasma: A pharmacokinetic study. Drug Test. Anal. 2015, 8, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Fresnais, M.; Muck, A.; Majewsky, M.; Statz, B.; Krausert, S.; Benzel, J.; Castel, D.; Le Dret, L.; Pfister, S.; Haefeli, W.E.; et al. Rapid and Sensitive Drug Quantification in Tissue Sections Using Matrix Assisted Laser Desorption Ionization-Ion Mobility-Mass Spectrometry Profiling. J. Am. Soc. Mass Spectrom. 2020, 31, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.; Becker, M.; Groseclose, M.R.; Schadt, S.; Hopf, C. Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr. Opin. Biotechnol. 2018, 55, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Fresnais, M.; Haefeli, W.E.; Burhenne, J.; Longuespée, R. Rapid drug detection in whole blood droplets using a desorption electrospray ionization static profiling approach—A proof-of-concept. Rapid Commun. Mass Spectrom. 2019, 34, e8614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresnais, M.; Roth, A.; Foerster, K.I.; Jäger, D.; Pfister, S.M.; Haefeli, W.E.; Burhenne, J.; Longuespée, R. Rapid and Sensitive Quantification of Osimertinib in Human Plasma Using a Fully Validated MALDI–IM–MS/MS Assay. Cancers 2020, 12, 1897. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M.; Foerster, K.I.; Benzel, J.; Pfister, S.; Pajtler, K.W.; Haefeli, W.E.; Burhenne, J. Bioanalysis of selinexor in mouse plasma micro-samples utilizing UPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1176, 122781. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH Harmonised Guidelines M10—Bioanalytical Method Validation (Draft Version). Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf (accessed on 7 December 2021).
- FDA. US Food and Drug Administration, Bioanalytical Method Validation—Guidance for Industry. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 7 December 2021).
- EMA. European Medicines Agency, Guideline of Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 7 December 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calibration Point | Concentration in Solution (ng/mL) | Concentration in Biological Matrix (ng/mL) |
|---|---|---|
| CAL1000 | 2000 | 1000 |
| CAL500 | 1000 | 500 |
| CAL200 | 400 | 200 |
| CAL100 | 200 | 100 |
| CAL50 | 100 | 50 |
| CAL20 | 40 | 20 |
| CAL10 | 20 | 10 |
| CAL5 | 10 | 5 |
| CAL0 | 0 | 0 |
| Blind value (BV) | 0 | 0 |
| Method | Quadrupole | Collision Energy (eV) | Ion Mobility | Target Ion SLX (m/z) | Internal Standard | Target Ion IS (m/z) |
|---|---|---|---|---|---|---|
| 3 | m/z 444 | 0 | ✓ | 444.11 | SLX-d5 | 449.13 |
| 4 | m/z 444 | 32 | ✓ | 444.11 | SLX-d5 | 449.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresnais, M.; Karabulut, S.; Abou Zeed, Y.; Ungermann, J.; Benzel, J.; Pajtler, K.W.; Pfister, S.M.; Haefeli, W.E.; Burhenne, J.; Longuespée, R. Important Requirements for the Selection of Internal Standards during the Development of Desorption/Ionization Assays for Drug Quantification in Biological Matrices—A Practical Example. Molecules 2022, 27, 690. https://doi.org/10.3390/molecules27030690
Fresnais M, Karabulut S, Abou Zeed Y, Ungermann J, Benzel J, Pajtler KW, Pfister SM, Haefeli WE, Burhenne J, Longuespée R. Important Requirements for the Selection of Internal Standards during the Development of Desorption/Ionization Assays for Drug Quantification in Biological Matrices—A Practical Example. Molecules. 2022; 27(3):690. https://doi.org/10.3390/molecules27030690
Chicago/Turabian StyleFresnais, Margaux, Seda Karabulut, Yasmin Abou Zeed, Johannes Ungermann, Julia Benzel, Kristian W. Pajtler, Stefan M. Pfister, Walter E. Haefeli, Jürgen Burhenne, and Rémi Longuespée. 2022. "Important Requirements for the Selection of Internal Standards during the Development of Desorption/Ionization Assays for Drug Quantification in Biological Matrices—A Practical Example" Molecules 27, no. 3: 690. https://doi.org/10.3390/molecules27030690