3. Materials and Methods
3.1. General Information
All air- and/or water-sensitive reactions were carried out under an argon atmosphere. THF, CH2Cl2 and toluene were dried over alumina columns in a solvent purification apparatus (Innovative Technology, Oldham, UK). Methanol, isopropanol, chlorobenzene, trifluoromethylbenzene, ethyl acetate and acetonitrile from Sigma-Aldrich (Darmstadt, Germany) were used without further purification. Hexafluoroisopropanol (HFIP) was purchased from TCI and was used without further purification. Formic acid/triethylamine (1:1) mixture was purchased from Fluka or Alfa Aesar and was used without further purification. Reactions were monitored by thin layer chromatography carried out on precoated silica gel plates (Merck 60F254, Darmstadt, Germany) and revealed with either an ultraviolet lamp (λ = 254 nm) or a potassium permanganate solution. Proton nuclear magnetic resonance (1H-NMR) spectra were recorded using a Bruker AC 400 (400 MHz). The chemical shifts are expressed in parts per million (ppm) referenced to residual chloroform (7.26 ppm). Data are reported as follows: chemical shifts (δ), multiplicity (recorded as s, singlet; d, doublet; t, triplet; q, quadruplet; quint, quintuplet; sext, sextuplet; hept, heptuplet; m, multiplet and br, broad), coupling constants and integration. Carbon-13 nuclear magnetic resonance (13C-NMR) spectra were recorded using a Bruker AC 400 (101 MHz). The chemical shifts are expressed in parts per million (ppm) relative to the centre line of the triplet at 77.16 ppm for CDCl3. Melting points (m. p.) were determined on a Köfler melting point apparatus. Optical rotations were measured on a Jasco P-1010 polarimeter. High resolution mass spectrometric (HRMS) analyses were measured on LTQ-Orbitrap (Thermo Fisher Scientific, Waltham, Massachusetts, US) at Sorbonne Université.
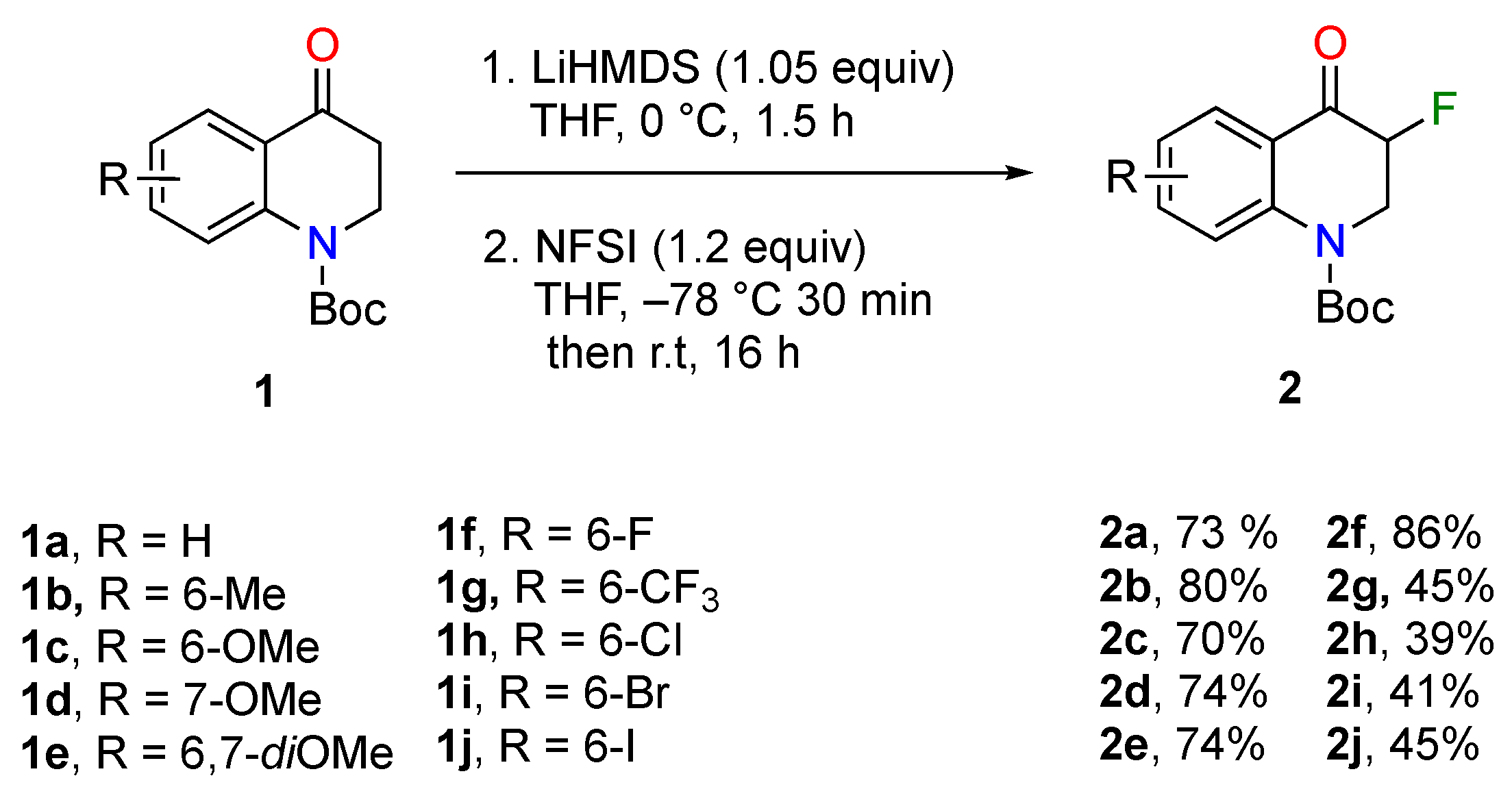
3.2. Method for the Synthesis of Compounds 2
To a 0 °C solution of LiHMDS (1.0 M in THF, 1.05 equiv) in a round-bottom tube set under argon, was slowly added a solution of the ketone (1.2 mmol, 1.0 equiv) in THF (2 mL) over 5 min. The mixture was stirred at 0 °C for 1.5 h. The resulting solution was then added dropwise over 5 min via a cannula to a −78 °C solution of NFSI (1.4 mmol, 1.2 equiv) in THF (4 mL). The reaction mixture was stirred at −78 °C for 30 min and then allowed to come to room temperature overnight. The reaction was diluted with 3 mL of CH
2Cl
2, quenched with 5 mL of saturated NH
4Cl aqueous solution and extracted with CH
2Cl
2 (3 × 10 mL). The combined organic layers were washed with brine, dried over MgSO
4, filtered, and concentrated under reduced pressure [
25]. The pure products were isolated by flash column chromatography on silica gel (see
Supplementary Materials for NMR spectra).
tert-Butyl 3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2a). 2.0 g of tert-butyl 4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (8.09 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 petroleum ether/ethyl acetate) yielded 1.56 g of 2a as a white solid (73%). m.p. 96–100 °C. 1H-NMR (400 MHz, Chloroform-d) δ 8.03 (dd, J = 8.0, 1.6 Hz, 1H), 7.81 (dd, J = 8.5, 1.0 Hz, 1H), 7.54 (ddd, J = 8.7, 7.2, 1.7 Hz, 1H), 7.20 (td, J = 7.6, 1.0 Hz, 1H), 5.10 (ddd, J = 46.9, 10.5, 4.7 Hz, 1H), 4.63 (td, J = 13.0, 4.7 Hz, 1H), 4.04 (ddd, J = 13.3, 10.6, 4.8 Hz, 1H), 1.57 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −198.77. 13C-NMR (101 MHz, Chloroform-d) δ 189.7 (C-F, 2JCF = 14.1 Hz), 189.6 (C-F, 2JCF = 14.1 Hz), 152.8, 144.1, 135.0, 128.1, 124.6, 123.8, 123.2, 88.1 (C-F, 1JCF = 190.9 Hz), 86.2 (C-F, 1JCF = 190.9 Hz), 83.2, 48.8 (C-F, 2JCF = 27.3 Hz), 48.5 (C-F, 2JCF = 27.3 Hz), 28.3. HRMS (APCI): m/z [M + H]+ calcd. for C14H17FNO3 266.1187, found 266.1186.
tert-Butyl 3-fluoro-6-methyl-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2b). 550 mg of tert-butyl 6-methyl-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2.10 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 petroleum ether/ethyl acetate) yielded 470 mg of 2b as a white solid (80%). m.p. 122–126 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.75 (s, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.28 (dd, J = 9.8, 3.4 Hz, 1H), 5.01 (ddd, J = 46.9, 10.6, 4.7 Hz, 1H), 4.55 (tdd, J = 13.2, 4.7, 0.9 Hz, 1H), 3.93 (ddd, J = 13.5, 10.6, 4.8 Hz, 1H), 2.28 (s, 3H), 1.49 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ –198.75. 13C-NMR (101 MHz, Chloroform-d) δ 189.9 (C-F, 2JCF = 14.1 Hz), 189.8 (C-F, 2JCF = 14.1 Hz), 152.9, 141.8, 136.0, 134.5, 127.8, 123.7, 123.0, 88.2 (C-F, 1JCF = 190.9 Hz), 86.3 (C-F, 1JCF = 190.9 Hz), 83.0, 48.8 (C-F, 2JCF = 27.3 Hz), 48.5 (C-F, 2JCF = 27.3 Hz), 28.4, 20.7. HRMS (APCI): m/z [M − Boc + H]+ calcd. for C10H11FNO 180.0825, found 180.0819.
tert-Butyl 3-fluoro-6-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2c). 700 mg of tert-butyl 6-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2.52 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 to 85:15 petroleum ether/ethyl acetate) yielded 487 mg of 2c as a pale-yellow solid (70%). m.p. 136–140 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.69 (dd, J = 9.7, 2.9 Hz, 1H), 7.43 (dd, J = 7.0, 4.2 Hz, 1H), 7.11 (dt, J = 12.5, 8.2 Hz, 1H), 5.08 (ddd, J = 46.8, 17.6, 13.6 Hz, 1H), 4.60 (td, J = 13.2, 8.9 Hz, 1H), 4.01 (ddt, J = 15.3, 10.6, 3.2 Hz, 1H), 3.82 (s, 3H), 1.54 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ –198.57. 13C-NMR (101 MHz, Chloroform-d) δ 189.7 (C-F, 2JCF = 15.2 Hz), 189.5 (C-F, 2JCF = 15.2 Hz), 156.5, 152.9, 137.9, 125.5, 124.0, 123.5, 108.9, 88.7 (C-F, 1JCF = 190.9 Hz), 86.4 (C-F, 1JCF = 190.9 Hz), 82.9, 55.8, 48.9 (C-F, 2JCF = 27.3 Hz), 48.7 (C-F, 2JCF = 27.3 Hz), 28.3. HRMS (APCI): m/z [M − Boc + H]+ calcd. for C10H11FNO2 196.0774, found 196.0769.
tert-Butyl 3-fluoro-7-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2d). 313 mg of tert-butyl 7-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (1.13 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 to 85:15 petroleum ether/ethyl acetate) yielded 247 mg of 2d as a pale-yellow solid (74%). m.p. 93–97 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.98 (dd, J = 9.0, 1.3 Hz, 1H), 7.37 (d, J = 2.4 Hz, 1H), 6.74 (dd, J = 8.9, 2.4 Hz, 1H), 5.04 (ddd, J = 47.0, 10.3, 4.6 Hz, 1H), 4.58 (td, J = 13.4, 4.6 Hz, 1H), 4.03 (ddd, J = 13.2, 10.3, 5.2 Hz, 1H), 3.88 (s, 3H), 1.58 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ –199.06. 13C-NMR (101 MHz, Chloroform-d) δ 188.3 (C-F, 2JCF = 15.2 Hz), 188.1 (C-F, 2JCF = 15.2 Hz), 165.1, 152.7, 146.0, 130.2, 116.9, 112.0, 107.7, 87.8 (C-F, 1JCF = 189.9 Hz), 85.9 (C-F, 1JCF = 189.9 Hz), 83.2, 55.9, 48.9 (C-F, 2JCF = 27.3 Hz), 48.7 (C-F, 2JCF = 27.3 Hz), 28.4. HRMS (APCI): m/z [M + H]+ calcd. for C15H19FNO4 296.1293, found 296.1295.
tert-Butyl 3-fluoro-6,7-dimethoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2e). 330 mg of tert-butyl 6,7-dimethoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (1.07 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (90:10 to 80:20 petroleum ether/ethyl acetate) yielded 257 mg of 2e as a pale-yellow solid (74%). m.p. 168–172 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.46–7.36 (m, 2H), 5.05 (ddd, J = 47.0, 10.1, 4.5 Hz, 1H), 4.56 (ddd, J = 14.3, 13.2, 4.5 Hz, 1H), 4.06 (ddd, J = 13.3, 10.1, 5.4 Hz, 1H), 3.96 (s, 3H), 3.91 (s, 3H), 1.57 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −198.64. 13C-NMR (101 MHz, Chloroform-d) δ 188.3 (C-F, 2JCF = 15.2 Hz), 188.1 (C-F, 2JCF = 15.2 Hz), 154.8, 152.8, 146.6, 140.0, 116.2, 108.0, 106.3, 87.9 (C-F, 1JCF = 188.9 Hz), 86.0 (C-F, 1JCF = 188.9 Hz), 83.0, 56.4, 56.3, 49.4 (C-F, 2JCF = 27.3 Hz), 49.1 (C-F, 2JCF = 27.3 Hz), 28.4. HRMS (APCI): m/z [M + H]+ calcd. for C16H21FNO5 326.1398, found 326.1399.
tert-Butyl 3,6-difluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2f). 500 mg of tert-butyl 6-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (1.88 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 to 85:15 petroleum ether/ethyl acetate) yielded 474 mg of 2f as a pale-yellow solid (86%). m.p. 89–93 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.81 (ddd, J = 9.7, 4.5, 2.3 Hz, 1H), 7.65 (dt, J = 8.3, 4.3 Hz, 1H), 7.31–7.20 (m, 1H), 5.10 (ddd, J = 46.7, 10.0, 2.7 Hz, 1H), 4.59 (td, J = 13.4, 3.8 Hz, 1H), 4.06 (ddd, J = 12.1, 5.1, 2.5 Hz, 1H), 1.55 (s, 9H). m.p. 89–93 °C. 19F-NMR (376 MHz, Chloroform-d) δ −116.65, −198.88. 13C-NMR (101 MHz, Chloroform-d) δ 188.8 (C-F, 2JCF = 15.2 Hz), 188.6 (C-F, 2JCF = 15.2 Hz), 160.5 (C-F, 1JCF = 248.5 Hz), 158.1 (C-F, 1JCF = 248.5 Hz), 152.7, 140.4, 126.1 (C-F, 3JCF = 7.1 Hz), 126.1 (C-F, 3JCF = 7.1 Hz), 124.6 (C-F, 3JCF = 6.1 Hz), 124.6 (C-F, 3JCF = 6.1 Hz), 122.5 (C-F, 2JCF = 23.2 Hz), 122.3 (C-F, 2JCF = 23.2 Hz), 113.5 (C-F, 2JCF = 23.2 Hz), 113.3 (C-F, 2JCF = 23.2 Hz), 88.0 (C-F, 1JCF = 191.9 Hz), 86.1 (C-F, 1JCF = 191.9 Hz), 83.4, 48.8 (C-F, 2JCF = 26.3 Hz), 48.5 (C-F, 2JCF = 26.3 Hz), 28.3. HRMS (APCI): m/z [M − Boc + H]+ calcd. for C9H8F2NO 184.0574, found 184.0569.
tert-Butyl 3-fluoro-4-oxo-6-(trifluoromethyl)-3,4-dihydroquinoline-1(2H)-carboxylate (2g). 700 mg of tert-butyl 4-oxo-6-(trifluoromethyl)-3,4-dihydroquinoline-1(2H)-carboxylate (2.22 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 to 85:15 petroleum ether/ethyl acetate) yielded 335 mg of 2g as a white solid (45%). m.p. 101–105 °C. 1H-NMR (400 MHz, Chloroform-d) δ 8.29 (d, J = 2.3 Hz, 1H), 8.03 (dd, J = 8.9, 0.6 Hz, 1H), 7.75 (dd, J = 7.1, 2.8 Hz, 1H), 5.13 (ddd, J = 46.7, 10.3, 4.6 Hz, 1H), 4.62 (ddd, J = 14.3, 13.6, 4.6 Hz, 1H), 4.12 (ddd, J = 13.5, 10.3, 5.1 Hz, 1H), 1.58 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −62.76, −199.13. 13C-NMR (101 MHz, Chloroform-d) δ 188.4 (C-F, 2JCF = 15.2 Hz), 188.2 (C-F, 2JCF = 15.2 Hz), 152.3, 146.6, 131.3 (C-F, 3JCF = 4.0 Hz), 131.2 (C-F, 3JCF = 4.0 Hz), 127.6 (C-F, 1JCF3 = 272.7 Hz), 127.2 (C-F, 2JCF3 = 33.3 Hz), 126.9 (C-F, 2JCF3 = 33.3 Hz), 126.6 (C-F, 2JCF3 = 33.3 Hz), 126.2 (C-F, 2JCF3 = 33.3 Hz), 125.6, 124.9 (C-F, 1JCF3 = 272.7 Hz), 124.2, 122.7, 122.2 (C-F, 1JCF3 = 272.7 Hz), 119.5 (C-F, 1JCF3 = 272.7 Hz), 87.6 (C-F, 1JCF = 190.9 Hz), 85.7 (C-F, 1JCF = 190.9 Hz), 84.1, 48.5 (C-F, 2JCF = 27.3 Hz), 48.3 (C-F, 2JCF = 27.3 Hz), 28.3. HRMS (APCI): m/z [M − Boc + H]+ calcd. for C10H8F4NO 234.0542, found 234.0537.
tert-Butyl 6-chloro-3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2h). 260 mg of tert-butyl 4-oxo-6-chloro-3,4-dihydroquinoline-1(2H)-carboxylate (0.92 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 petroleum ether/ethyl acetate) yielded 107 mg of 2h as a yellow solid (39%). m.p. 117–121 °C. 1H-NMR (400 MHz, Chloroform-d) δ 7.97 (d, J = 2.6 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.48 (dd, J = 9.0, 2.6 Hz, 1H), 5.09 (ddd, J = 46.8, 10.4, 4.7 Hz, 1H), 4.60 (td, J = 13.5, 4.6 Hz, 1H), 4.06 (ddd, J = 13.4, 10.4, 4.9 Hz, 1H), 1.56 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −198.92. 13C-NMR (101 MHz, Chloroform-d) δ 188.5 (C-F, 2JCF = 15.2 Hz), 188.4 (C-F, 2JCF = 15.2 Hz), 152.51, 142.5, 134.8, 130.4, 127.4, 125.3, 124.1, 87.8 (C-F, 1JCF = 191.9 Hz), 85.9 (C-F, 1JCF = 191.9 Hz), 83.6, 48.6 (C-F, 2JCF = 26.3 Hz), 48.4 (C-F, 2JCF = 26.3 Hz), 28.3. HRMS (APCI): m/z [M + H]+ calcd. for C14H16ClFNO3 300.0797, found 300.0799.
tert-Butyl 6-bromo-3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2i). 380 mg of tert-butyl 4-oxo-6-bromo-3,4-dihydroquinoline-1(2H)-carboxylate (1.16 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (95:5 petroleum ether/ethyl acetate) yielded 165 mg of 2i as a white solid (41%). m.p. 111–114 °C. 1H-NMR (400 MHz, Chloroform-d) δ 8.12 (d, J = 2.4 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.62 (dd, J = 9.0, 2.5 Hz, 1H), 5.09 (ddd, J = 46.8, 10.4, 4.7 Hz, 1H), 4.59 (td, J = 13.5, 4.6 Hz, 1H), 4.05 (ddd, J = 13.4, 10.4, 5.0 Hz, 1H), 1.56 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −198.90. 13C-NMR (101 MHz, Chloroform-d) δ 188.4 (C-F, 2JCF = 15.2 Hz), 188.3 (C-F, 2JCF = 15.2 Hz), 152.4, 143.0, 137.7, 130.5, 125.5, 124.4, 117.8, 87.8 (C-F, 1JCF = 191.9 Hz), 85.9 (C-F, 1JCF = 191.9 Hz), 83.7, 48.6 (C-F, 2JCF = 27.3 Hz), 48.3 (C-F, 2JCF = 27.3 Hz), 28.3. HRMS (APCI): m/z [M − Boc + H]+ calcd. for C9H8BrFNO 243.9773, found 243.9768.
tert-Butyl 3-fluoro-6-iodo-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2j). 339 mg of tert-butyl 4-oxo-6-iodo-3,4-dihydroquinoline-1(2H)-carboxylate (0.91 mmol, 1.0 equiv) were used following the described procedure. Purification via flash column chromatography on silica gel (97:3 petroleum ether/ethyl acetate) yielded 160 mg of 2j as a white solid (45%). m.p. 113–117 °C. 1H-NMR (400 MHz, Chloroform-d) δ 8.31 (d, J = 2.2 Hz, 1H), 7.80 (dd, J = 8.9, 2.3 Hz, 1H), 7.61 (d, J = 8.9 Hz, 1H), 5.08 (ddd, J = 46.8, 10.4, 4.7 Hz, 1H), 4.59 (td, J = 13.5, 4.7 Hz, 1H), 4.04 (ddd, J = 13.4, 10.4, 5.0 Hz, 1H), 1.56 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −198.84. 13C-NMR (101 MHz, Chloroform-d) δ 188.3 (C-F, 2JCF = 15.2 Hz), 188.2 (C-F, 2JCF = 15.2 Hz), 152.4, 143.6, 143.4, 136.7, 125.6, 124.5, 88.0, 87.6 (C-F, 1JCF = 190.9 Hz), 85.8 (C-F, 1JCF = 190.9 Hz), 83.7, 48.5 (C-F, 2JCF = 24.2 Hz), 48.3 (C-F, 2JCF = 24.2 Hz), 28.3. HRMS (APCI): m/z [M + H]+ calcd. for C14H16FINO3 392.0153, found 392.0155.
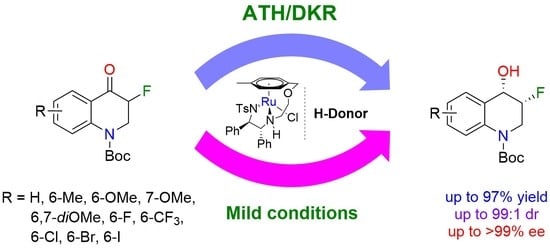
3.3. General Procedure for the ATH of tert-Butyl 3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylates
In a round-bottom tube charged with the corresponding 3-fluoro-4-oxo-3,4-dihydroquinoline-1(2
H)-carboxylate (1.0 equiv) set under argon, the necessary volume of a solution of (
R,R)-A in acetonitrile (1.0 mL of a 1.24 mg/mL solution; 0.005 equiv) was added. The mixture was stirred for one minute before adding, by syringe, the necessary volume of formic acid/triethylamine (1:1) mixture (6.0 equiv). The reaction mixture was stirred at 40 °C for the time needed and then quenched with 3 mL of saturated NaHCO
3 aqueous solution. The media was extracted with CH
2Cl
2 (2 × 4 mL) and the organic layers were dried over MgSO
4, filtered, and concentrated under vacuum. The diastereoisomeric ratio was determined by
1H-NMR analysis of the crude product. The product was purified with a flash column chromatography on silica gel (petroleum ether/EtOAc) and the enantiomeric excess was determined by SFC analysis (CHIRALPAK IE or IF column) (see
Supplementary Materials for NMR and SFC spectra).
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-3,4-dihydroquinoline-1(2H)-carboxylate (3a). 100 mg of tert-butyl 3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2a) (0.38 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 93 mg of a beige/white solid (93%). m.p. 142–144 °C dr (cis/trans) = 98:2, eecis = > 99%. [α]D30 = −5 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.70 (d, J = 8.3 Hz, 1H), 7.51 (dt, J = 7.6, 1.4 Hz, 1H), 7.34–7.24 (m, 1H), 7.14 (td, J = 7.5, 1.2 Hz, 1H), 5.05 (ddt, J = 50.8, 4.8, 3.5 Hz, 1H), 4.78 (dd, J = 21.5, 5.9 Hz, 1H), 4.10 (ddd, J = 14.9, 14.2, 4.8 Hz, 1H), 3.94 (ddd, J = 29.9, 14.2, 3.7 Hz, 1H), 2.38 (d, J = 8.3 Hz, 1H), 1.53 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.34. 13C-NMR (101 MHz, Chloroform-d) δ 153.8, 136.8, 128.2, 127.9, 127.3, 124.3, 123.5, 89.6 (C-F, 1JCF = 176.8 Hz), 87.9 (C-F, 1JCF = 176.8 Hz), 81.9, 68.0 (C-F, 2JCF = 19.2 Hz), 67.8 (C-F, 2JCF = 19.2 Hz), 46.3 (C-F, 2JCF = 23.2 Hz), 46.1 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + H]+ calcd. for C14H19FNO3 268.1343, found 268.1345. SFC: Chiralpak IE, scCO2/EtOH 96/4, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 11.04 min, tR [trans] = 12.85 min, tR [cis-(3R,4S)] = 14.54 min (major), tR [cis-(3S,4R)] = 15.43 min.
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-methyl-3,4-dihydroquinoline-1(2H)-carboxylate (3b). 100 mg of tert-butyl 3-fluoro-6-methyl-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2b) (0.36 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 91 mg of a colourless oil (91%). dr (cis/trans) = 98:2, eecis = > 99%. [α]D22 = −8.4 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.56 (d, J = 8.4 Hz, 1H), 7.34–7.28 (m, 1H), 7.08 (dd, J = 8.4, 2.1 Hz, 1H), 5.00 (dq, J = 50.9, 4.2 Hz, 1H), 4.72 (dd, J = 22.0, 7.0 Hz, 1H), 4.10–3.84 (m, 2H), 2.53 (s, 1H), 2.32 (s, 3H), 1.52 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.17. 13C-NMR (101 MHz, Chloroform-d) δ 153.8, 134.2, 133.9, 128.9, 127.6, 123.4, 89.7, 88.0 (C-F, 1JCF = 176.8 Hz), 81.7 (C-F, 1JCF = 176.8 Hz), 67.9 (C-F, 2JCF = 20.2 Hz), 67.7 (C-F, 2JCF = 20.2 Hz), 46.2 (C-F, 2JCF = 23.2 Hz), 46.0 (C-F, 2JCF = 23.2 Hz), 28.4, 20.9. HRMS (APCI): m/z [M + H]+ calcd. for C15H21FNO3 282.1500, found 282.1502. SFC: Chiralpak IE, scCO2/EtOH 97.5/2.5, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 27.71 min, tR [cis-(3R,4S)] = 32.43 min (major), tR [cis-(3S,4R)] = 37.48 min, tR [trans] = 42.41 min.
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-methoxy-3,4-dihydroquinoline-1(2H)-carboxylate (3c). 100 mg of tert-butyl 3-fluoro-6-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2c) (0.34 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 5 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 95 mg of a white/red solid (95%). m.p. 114–118 °C dr (cis/trans) = 98:2, eecis = > 99%. [α]D22 = −13.3 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.57 (d, J = 9.1 Hz, 1H), 7.05 (dd, J = 3.0, 1.0 Hz, 1H), 6.83 (dd, J = 9.1, 3.0 Hz, 1H), 5.06 (dd, J = 51.1, 3.7 Hz, 1H), 4.74 (ddd, J = 22.7, 9.4, 3.2 Hz, 1H), 4.08 (ddd, J = 15.9, 14.4, 4.4 Hz, 1H), 3.92 (ddd, J = 31.4, 14.5, 3.7 Hz, 1H), 3.81 (s, 3H), 2.37 (d, J = 9.4 Hz, 1H), 1.52 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.32. 13C-NMR (101 MHz, Chloroform-d) δ 156.5, 153.9, 129.8, 129.4, 125.0, 114.3, 111.4, 89.9 (C-F, 1JCF = 176.8 Hz), 88.1 (C-F, 1JCF = 176.8 Hz), 81.6, 68.2 (C-F, 2JCF = 20.2 Hz), 68.0 (C-F, 2JCF = 20.2 Hz), 55.7, 46.6 (C-F, 2JCF = 23.2 Hz), 46.3 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + Na]+ calcd. for C15H20FNO4Na 320.1269, found 320.1273. SFC: Chiralpak IF, scCO2/EtOH 95.5/45, 2.0 mL/min, P = 100 bar, λ = 254 nm, tR [trans] = 15.71 min, tR [cis-(3S,4R)] = 17.11 min, tR [trans] = 18.69 min, tR [cis-(3R,4S)] = 22.79 min (major)
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-7-methoxy-3,4-dihydroquinoline-1(2H)-carboxylate (3d). 100 mg of tert-butyl 3-fluoro-7-methoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2d) (0.34 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 5 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 95 mg of a beige/white solid (95%). m.p. 92–96 °C dr(cis/trans) = 95:5, eecis = >99%. [α]D22 = −9.2 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.42–7.31 (m, 2H), 6.70 (dt, J = 8.6, 2.3 Hz, 1H), 4.98 (dd, J = 50.0, 5.9 Hz, 1H), 4.76 (d, J = 19.1 Hz, 1H), 4.16–4.04 (m, 1H), 4.01–3.85 (m, 1H), 3.80 (s, 3H), 2.33 (s, 1H), 1.54 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −201.08. 13C-NMR (101 MHz, Chloroform-d) δ 159.7, 153.6, 138.0, 128.9, 119.8, 110.8, 108.6, 89.1 (C-F, 1JCF = 177.8 Hz), 87.3 (C-F, 1JCF = 177.8 Hz), 82.0, 67.6 (C-F, 2JCF = 19.2 Hz), 67.4 (C-F, 2JCF = 19.2 Hz), 55.5, 45.7 (C-F, 2JCF = 25.3 Hz), 45.5 (C-F, 2JCF = 25.3 Hz), 28.5. HRMS (APCI): m/z [M + NH4]+ calcd. for C15H24FN2O4 315.1715, found 315.1718. SFC: Chiralpak IE, scCO2/EtOH 95/5, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 11.87 min, tR [trans] = 13.19 min, tR [cis-(3R,4S)] = 16.63 min (major), tR [cis-(3S,4R)] = 18.62 min.
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6,7-dimethoxy-3,4-dihydroquinoline-1(2H)-carboxylate (3e). 100 mg of tert-butyl 3-fluoro-6,7-dimethoxy-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2e) (0.31 mmol; 1.0 equiv) were used according to the described procedure with 1.0 mol% of catalyst (R,R)-D (reaction time of 24 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 60:40) and yielded 90 mg of a colourless oil (90%). dr(cis/trans) = 94:6, eecis = 99%. [α]D22 = −11.8 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.32 (s, 1H), 6.97 (d, J = 0.8 Hz, 1H), 4.99 (ddt, J = 50.2, 5.6, 3.4 Hz, 1H), 4.74 (dq, J = 19.9, 3.5 Hz, 1H), 4.20–4.11 (m, 1H), 3.94–3.80 (m, 7H), 2.38 (d, J = 8.4 Hz, 1H), 1.53 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −202.03. 13C-NMR (101 MHz, Chloroform-d) δ 153.8, 148.5, 146.2, 130.4, 119.6, 110.0, 107.5, 89.1 (C-F, 1JCF = 177.8 Hz), 87.3 (C-F, 1JCF = 177.8 Hz), 81.7, 67.7 (C-F, 2JCF = 20.2 Hz), 67.5 (C-F, 2JCF = 20.2 Hz), 56.2, 56.1, 46.0 (C-F, 2JCF = 24.2 Hz), 45.8 (C-F, 2JCF = 24.2 Hz), 28.5. HRMS (APCI): m/z [M + Na]+ calcd. for C16H22FNO5Na 350.1374, found 320.1377. SFC: Chiralpak IF, scCO2/EtOH 95/5, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 14.75 min, tR [trans] = 15.89 min, tR [cis-(3R,4S)] = 17.45 min (major), tR [cis-(3S,4R)] = 19.07 min.
tert-Butyl (3R,4S)-3,6-difluoro-4-hydroxy-3,4-dihydroquinoline-1(2H)-carboxylate (3f). 100 mg of tert-butyl 3,6-difluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2f) (0.35 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 24 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 70 mg of a white solid (70%). m.p. 78–82 °C dr(cis/trans) = 98:2, eecis = > 99%. [α]D22 = −11.7 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.64 (dd, J = 9.3, 5.1 Hz, 1H), 7.23 (ddd, J = 8.8, 3.0, 1.0 Hz, 1H), 6.97 (td, J = 8.9, 8.1, 3.1 Hz, 1H), 5.08 (dq, J = 51.2, 3.6 Hz, 1H), 4.71 (ddd, J = 23.6, 9.5, 3.1 Hz, 1H), 4.09 (ddd, J = 16.6, 14.6, 3.9 Hz, 1H), 3.91 (ddd, J = 33.1, 14.6, 3.6 Hz, 1H), 2.48 (dd, J = 9.7, 2.0 Hz, 1H), 1.52 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −118.05, −200.38. 13C-NMR (101 MHz, Chloroform-d) δ 160.8 (C-F, 1JCF = 244.4 Hz), 158.4 (C-F, 1JCF = 244.4 Hz), 153.7, 132.6 (C-F, 4JCF = 2.0 Hz), 132.6 (C-F, 4JCF = 2.0 Hz), 130.3 6 (C-F, 3JCF = 6.1 Hz), 130.3 (C-F, 3JCF = 6.1 Hz), 125.4 (C-F, 3JCF = 7.1 Hz), 125.3 (C-F, 3JCF = 7.1 Hz), 115.1 (C-F, 2JCF = 22.2 Hz), 114.9 (C-F, 2JCF = 22.2 Hz), 113.5 (C-F, 2JCF = 22.2 Hz), 113.3 (C-F, 2JCF = 22.2 Hz), 89.7 (C-F, 1JCF = 176.8 Hz), 87.9 (C-F, 1JCF = 176.8 Hz), 82.1, 67.9 (C-F, 2JCF = 20.2 Hz), 67.7 (C-F, 2JCF = 20.2 Hz), 46.8 (C-F, 2JCF = 23.2 Hz), 46.6 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + H]+ calcd. for C14H18F2NO3 286.1249, found 282.1248. SFC: Chiralpak IE, scCO2/MeOH 94/6, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 4.50 min, tR [trans] = 4.96 min, tR [cis-(3S,4R)] = 6.44 min, tR [cis-(3R,4S)] = 7.00 min (major).
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-(trifluoromethyl)-3,4-dihydroquinoline-1(2H)-carboxylate (3g). 100 mg of tert-butyl 3-fluoro-6-trifluoromethyl-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2g) (0.30 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 97 mg of a beige/white solid (97%). m.p. 152–156 °C dr (cis/trans) = 98:2, eecis = > 99%. [α]D22 = −0.9 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.88 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 2.2 Hz, 1H), 7.51 (ddt, J = 8.7, 2.2, 0.7 Hz, 1H), 5.10 (dddd, J = 50.9, 7.7, 3.7, 1.4 Hz, 1H), 4.79 (dd, J = 23.1, 6.9 Hz, 1H), 4.17 (tdd, J = 15.5, 4.1, 1.1 Hz, 1H), 3.91 (dd, J = 33.7, 3.6 Hz, 1H), 2.45 (s, 1H), 1.54 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −62.20, −200.84. 13C-NMR (101 MHz, Chloroform-d) δ 153.4, 139.7, 128.3 (C-F, 1JCF3 = 272.7 Hz), 128.2, 128.1, 126.5 (C-F, 2JCF3 = 33.3 Hz), 126.2 (C-F, 2JCF3 = 33.3 Hz), 125.9 (C-F, 2JCF3 = 33.3 Hz), 125.6 (C-F, 1JCF3 = 272.7 Hz + C-F, 2JCF3 = 33.3 Hz), 125.1 (C-F, 3JCF3 = 4.0 Hz), 125.1 (C-F, 3JCF3 = 4.0 Hz), 124.4 (C-F, 3JCF3 = 4.0 Hz), 124.4 (C-F, 3JCF3 = 4.0 Hz), 123.4, 122.9 (C-F, 1JCF3 = 272.7 Hz), 120.2 (C-F, 1JCF3 = 272.7 Hz), 89.1 (C-F, 1JCF = 176.8 Hz), 87.4 (C-F, 1JCF = 176.8 Hz), 82.7, 67.7 (C-F, 2JCF = 20.2 Hz), 67.5 (C-F, 2JCF = 20.2 Hz), 46.8 (C-F, 2JCF = 22.2 Hz), 46.6 (C-F, 2JCF = 22.2 Hz), 28.3. HRMS (APCI): m/z [M + NH4]+ calcd. for C15H21FN2O3 353.1483, found 353.1483. SFC: Chiralpak IF, scCO2/EtOH 96/4, 2.0 mL/min, P = 100 bar, λ = 254 nm, tR [cis-(3R,4S)] = 5.68 min (major), tR [cis-(3S,4R)] = 6.84 min, tR [trans] = 8.54 min.
tert-Butyl (3R,4S)-6-chloro-3-fluoro-4-hydroxy-3,4-dihydroquinoline-1(2H)-carboxylate (3h). 60 mg of tert-butyl 3-fluoro-6-chloro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2h) (0.20 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 97 mg of a beige/white solid (96%). m.p. 113–116 °C dr (cis/trans) = 99:1, eecis = > 99%. [α]D22 = −12.8 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.66 (d, J = 8.9 Hz, 1H), 7.51 (dd, J = 2.6, 1.1 Hz, 1H), 7.22 (d, J = 9.2 Hz, 1H), 5.07 (dt, J = 50.8, 3.9 Hz, 1H), 4.78–4.67 (m, 1H), 4.17–4.05 (m, 1H), 3.99–3.81 (m, 1H), 2.44 (br, 1H), 1.52 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −200.57. 13C-NMR (101 MHz, Chloroform-d) δ 153.5, 135.2, 129.6, 128.2, 126.9, 124.8, 89.4 (C-F, 1JCF = 176.8 Hz), 87.7 (C-F, 1JCF = 176.8 Hz), 82.3, 67.7 (C-F, 2JCF = 20.2 Hz), 67.5 (C-F, 2JCF = 20.2 Hz), 46.7 (C-F, 2JCF = 23.2 Hz), 46.5 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + H]+ calcd. for C14H18ClFNO3 302.0954, found 302.0955. SFC: Chiralpak IE, scCO2/MeOH 94/6, 2.0 mL/min, P = 100 bar, λ = 254 nm, tR [trans] = 6.95 min, tR [trans] = 7.49 min, tR [cis-(3S,4R)] = 10.31 min, tR [cis-(3R,4S)] = 11.09 min (major).
tert-Butyl (3R,4S)-6-bromo-3-fluoro-4-hydroxy-3,4-dihydroquinoline-1(2H)-carboxylate (3i). 80 mg of tert-butyl 3-fluoro-6-bromo-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2i) (0.24 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 97 mg of a beige/white solid (95%). m.p. 128–131 °C dr (cis/trans) = 98:2, eecis = > 99%. [α]D22 = −15.6 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.68–7.65 (m, 1H), 7.59 (d, J = 8.9 Hz, 1H), 7.37 (dd, J = 8.9, 2.4 Hz, 1H), 5.05 (dq, J = 51.0, 3.6 Hz, 1H), 4.71 (ddd, J = 23.2, 9.3, 3.1 Hz, 1H), 4.10 (ddd, J = 15.7, 14.4, 4.1 Hz, 1H), 3.88 (ddd, J = 33.1, 14.5, 3.5 Hz, 1H), 2.52 (d, J = 9.4 Hz, 1H), 1.52 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.49. 13C-NMR (101 MHz, Chloroform-d) δ 153.5, 135.7, 131.1, 130.0, 129.9, 125.1, 117.2, 89.4 (C-F, 1JCF = 176.8 Hz), 87.6 (C-F, 1JCF = 176.8 Hz), 82.3, 67.7 (C-F, 2JCF = 20.2 Hz), 67.5 (C-F, 2JCF = 20.2 Hz), 46.7 (C-F, 2JCF = 23.2 Hz), 46.4 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + NH4]+ calcd. for C14H21BrFN2O3 363.0714, found 363.0714. SFC: Chiralpak IE, scCO2/MeOH 93/7, 2.0 mL/min, P = 100 bar, λ = 254 nm, tR [trans] = 7.72 min, tR [cis-(3S,4R)] = 11.07 min, tR [cis-(3R,4S)] = 11.96 min (major).
tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-iodo-3,4-dihydroquinoline-1(2H)-carboxylate (3j). 80 mg of tert-butyl 3-fluoro-6-iodo-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2j) (0.20 mmol; 1.0 equiv) were used according to the described procedure (reaction time of 3 h). The product was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate 75:25) and yielded 97 mg of a beige/white solid (94%). m.p. 125–128 °C dr (cis/trans) = 98:2, eecis = > 99%. [α]D22 = −19.6 (c 1.0, CHCl3). 1H-NMR (400 MHz, Chloroform-d) δ 7.84 (d, J = 0.8 Hz, 1H), 7.56 (dt, J = 8.7, 1.6 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 5.04 (dt, J = 50.9, 3.7 Hz, 1H), 4.71 (dd, J = 23.3, 8.8 Hz, 1H), 4.10 (ddt, J = 18.6, 15.1, 3.6 Hz, 1H), 3.97–3.78 (m, 1H), 2.46 (s, 1H), 1.52 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.57. 13C-NMR (101 MHz, Chloroform-d) δ 153.4, 137.0, 136.6, 135.9, 130.1, 125.3, 89.3 (C-F, 1JCF = 176.8 Hz), 87.8 (C-F, 1JCF = 176.8 Hz), 87.5, 82.4, 67.5 (C-F, 2JCF = 20.2 Hz), 67.3 (C-F, 2JCF = 20.2 Hz), 46.6 (C-F, 2JCF = 23.2 Hz), 46.3 (C-F, 2JCF = 23.2 Hz), 28.4. HRMS (APCI): m/z [M + NH4]+ calcd. for C14H21FIN2O3 411.0575, found 411.0577. SFC: Chiralpak IE, scCO2/MeOH 93/7, 2.0 mL/min, P = 100 bar, λ = 254 nm, tR [trans] = 10.92 min, tR [trans] = 11.41 min, tR [cis-(3S,4R)] = 15.73 min, tR [cis-(3R,4S)] = 17.25 min (major).
3.4. Gram-Scale Experiment
In a 30 mL-round-bottom tube charged with tert-butyl 3-fluoro-4-oxo-3,4-dihydroquinoline-1(2H)-carboxylate (2a, 1.0 g, 3.8 mmol; 1.0 equiv) and the catalyst (R,R)-D (12.4 mg, 12 μmol; 0.005 equiv) set under argon were added 10 mL of acetonitrile. The mixture was stirred for one minute before adding by syringe a (1:1) mixture of formic acid and triethylamine (3.40 mL, 16 mmol; 6.0 equiv). The reaction mixture was stirred at 40 °C for 24 h. Then it was cooled down and quenched with 30 mL of NaHCO3 aqueous solution. The mixture was extracted with CH2Cl2 (2 × 40 mL) and the organic layers dried over MgSO4, filtered, and concentrated under vacuum. The diastereoisomeric ratio was determined by 1H-NMR analysis of the crude product. The product was purified with a flash column chromatography on silica gel (petroleum ether/EtOAc 70:30). 960 mg (96%) of (3R,4R)- 3-fluorochroman-4-ol (3a) was obtained as a white solid. dr (cis/trans) 97:3, eecis = > 99%. 1H-NMR (400 MHz, Chloroform-d) δ 7.70 (d, J = 8.3 Hz, 1H), 7.51 (dt, J = 7.6, 1.4 Hz, 1H), 7.34–7.24 (m, 1H), 7.14 (td, J = 7.5, 1.2 Hz, 1H), 5.05 (ddt, J = 50.8, 4.8, 3.5 Hz, 1H), 4.78 (dd, J = 21.5, 5.9 Hz, 1H), 4.10 (ddd, J = 14.9, 14.2, 4.8 Hz, 1H), 3.94 (ddd, J = 29.9, 14.2, 3.7 Hz, 1H), 2.38 (d, J = 8.3 Hz, 1H), 1.53 (s, 9H). 19F-NMR (376 MHz, Chloroform-d) δ −200.34. 13C-NMR (101 MHz, Chloroform-d) δ 153.8, 136.8, 128.2, 127.9, 127.3, 124.3, 123.5, 89.6 (C-F, 1JCF = 176.8 Hz), 87.9 (C-F, 1JCF = 176.8 Hz), 81.9, 68.0 (C-F, 2JCF = 19.2 Hz), 67.8 (C-F, 2JCF = 19.2 Hz), 46.3 (C-F, 2JCF = 23.2 Hz), 46.1 (C-F, 2JCF = 23.2 Hz), 28.4. SFC: Chiralpak IE, scCO2/EtOH 96/4, 2.0 mL/min, P = 100 bar, λ = 215 nm, tR [trans] = 11.04 min, tR [trans] = 12.85 min, tR [cis-(3R,4S)] = 14.54 min (major), tR [cis-(3S,4R)] = 15.43 min.
3.5. Sonogashira Coupling Reaction
Tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-(phenylethynyl)-3,4-dihydroquinoline-1(2H)-carboxylate (4). Tert-Butyl (3R,4S)-3-fluoro-4-hydroxy-6-iodo-3,4-dihydroquinoline-1(2H)-carboxylate (3j) (77 mg; 0.2 mmol; 1.0 equiv) was introduced into a 10 mL round-bottom tube at open air with 1 mL of a 1:1 CH3CN/Et3N mixture. Next, phenylacetylene was added (31 µL; 0.3 mmol; 1.5 equiv) followed by CuI (2.8 mg; 10 µmol; 0.05 equiv) and PdCl2(PPh3)2 (6.4 mg; 6 µmol; 0.03 equiv). The mixture was stirred for 30 min at room temperature. It was then quenched with 2 mL of NH4Cl saturated solution and extracted with EtOAc (3 × 10 mL). The combined organic phases were dried with MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel, (petroleum ether/EtOAc/toluene 7:2:1) to afford 4 as a pale-yellow solid (49 mg; 68%). 1H-NMR (400 MHz, CDCl3) δ 7.76–7.69 (m, 2H), 7.55–7.49 (m, 2H), 7.44 (ddd, J = 8.6, 2.1, 0.6 Hz, 1H), 7.37–7.30 (m, 3H), 5.06 (ddt, J = 50.8, 4.5, 3.4 Hz, 1H), 4.76 (dd, J = 23.3, 6.0 Hz, 1H), 4.13 (ddd, J = 15.3, 14.3, 4.4 Hz, 1H), 3.91 (ddd, J = 32.0, 14.3, 3.5 Hz, 1H), 2.52 (d, J = 9.0 Hz, 1H), 1.54 (s, 9H). 19F-NMR (376 MHz, CDCl3) δ −200.41. 13C-NMR (101 MHz, CDCl3) δ 153.5, 136.7, 131.7, 131.3, 130.5, 128.5, 128.3, 127.9, 123.4, 123.2, 118.9, 89.4, 89.3 (C-F, 1JCF = 176.8 Hz), 89.1, 87.6 (C-F, 1JCF = 176.8 Hz), 82.3, 67.7 (C-F, 2JCF = 20.2 Hz), 67.5 (C-F, 2JCF = 20.2 Hz), 46.6 (C-F, 2JCF = 23.2 Hz), 46.4 (C-F, 2JCF = 23.2 Hz), 28.4. MS (ESI) [M − Boc + H]+ = 267.
3.6. N-Boc Deprotection
(3R,4S)-3-Fluoro-1,2,3,4-tetrahydroquinolin-4-ol (
5).
Tert-Butyl (3
R,4
S)-3-fluoro-4-hydroxy-3,4-dihydroquinoline-1(2
H)-carboxylate 3a (200 mg; 0.75 mmol; 1.0 equiv) was introduced in a 50 mL round-bottom tube set under argon. 3.0 mL of 1,4-dioxane were added followed by 5.0 mL of HPLC-grade H
2O. The mixture was heated to reflux for 34 h [
26]. It was then allowed to cool down to room temperature, and it was extracted with MTBE (3 × 20 mL). The combined organic phases were dried with MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (petroleum ether/EtOAc 8:2 to 7:3) to afford 5 as a pale-yellow solid (112 mg; 90%).
1H-NMR (400 MHz, CDCl
3) δ 7.35 (dt,
J = 7.8, 1.0 Hz, 1H), 7.11 (ddd,
J = 8.5, 7.5, 1.6 Hz, 1H), 6.75 (td,
J = 7.4, 1.2 Hz, 1H), 6.55 (dd,
J = 8.0, 1.1 Hz, 1H), 5.08–4.75 (m, 2H), 3.87 (s, 1H), 3.66 (dt,
J = 11.9, 7.9 Hz, 1H), 3.45 (dddd,
J = 22.2, 11.9, 3.3, 1.4 Hz, 1H), 2.26 (s, 1H).
19F-NMR (376 MHz, CDCl
3) δ –203.21.
13C-NMR (101 MHz, CDCl
3) δ 143.4, 129.7, 129.6, 120.0, 118.2, 114.2, 88.8 (C-F,
1JCF = 176.8 Hz), 87.1 (C-F,
1JCF = 176.8 Hz), 67.3 (C-F,
2JCF = 19.2 Hz), 67.1 (C-F,
2JCF = 19.2 Hz), 41.8 (C-F,
2JCF = 24.2 Hz), 41.6 (C-F,
2JCF = 24.2 Hz). HRMS (ESI)
m/
z: [M+H]
+ calcd. for C
9H
11FNO 168.0819. Found 168.0819.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


