
Synthesis of Polysubstituted Ferrocenesulfoxides

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
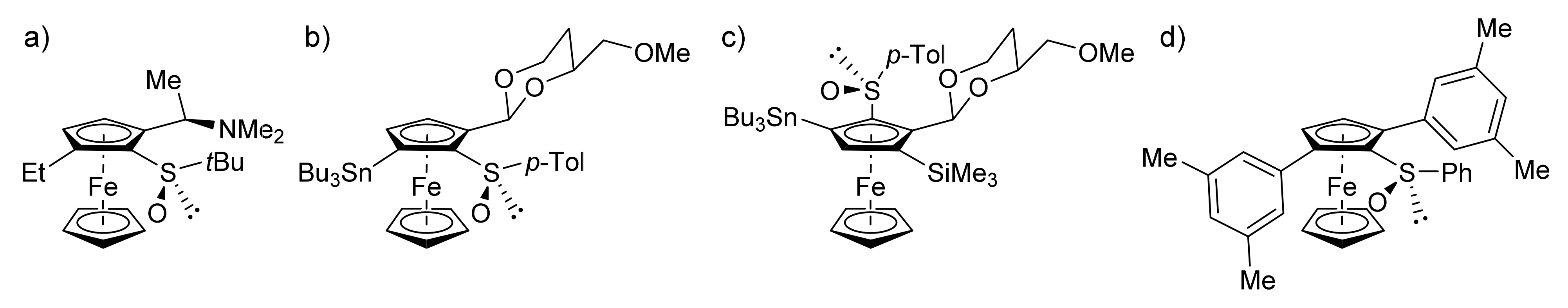
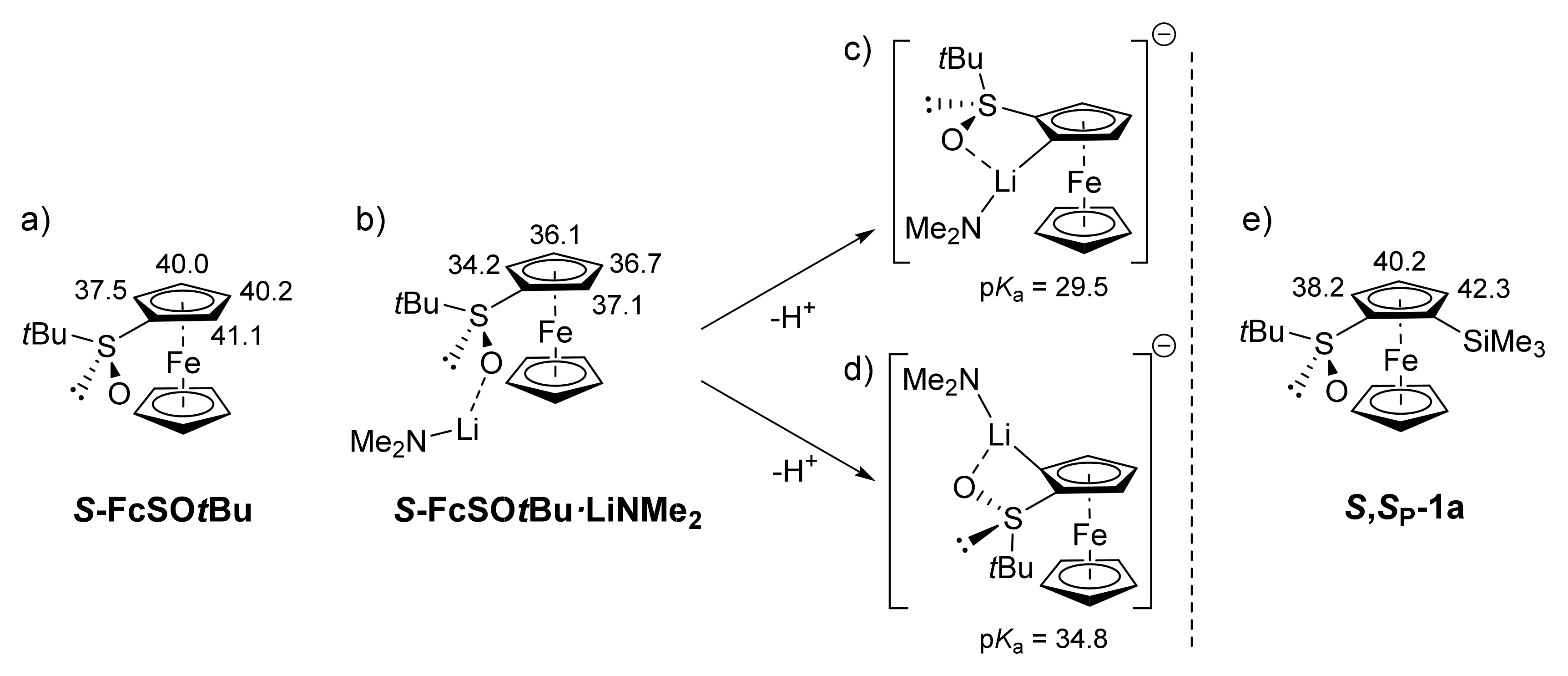
2.1. In Search of an Effective Protecting Group to Functionalize the Unfavorable Position Adjacent to the Sulfoxide
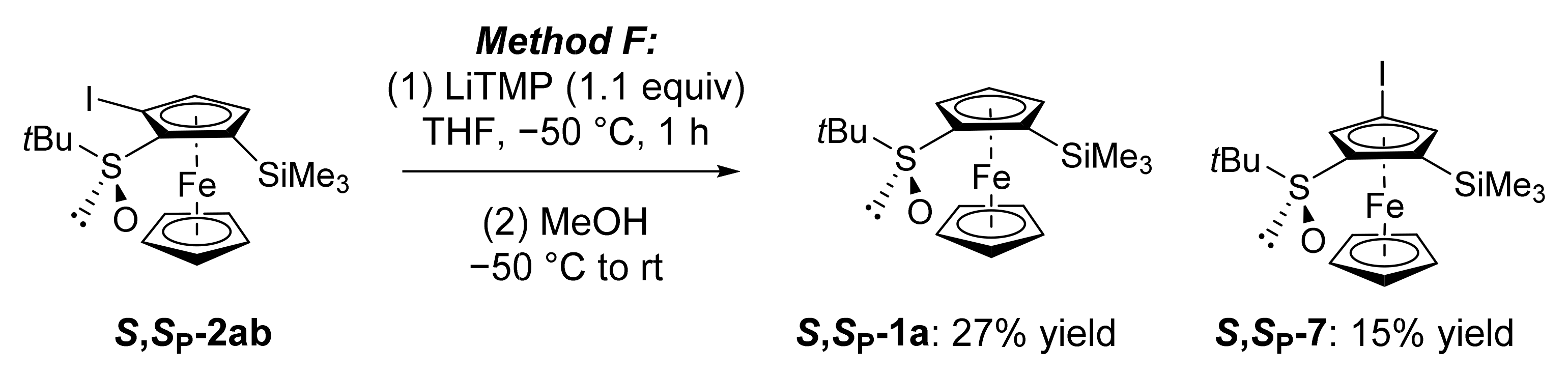
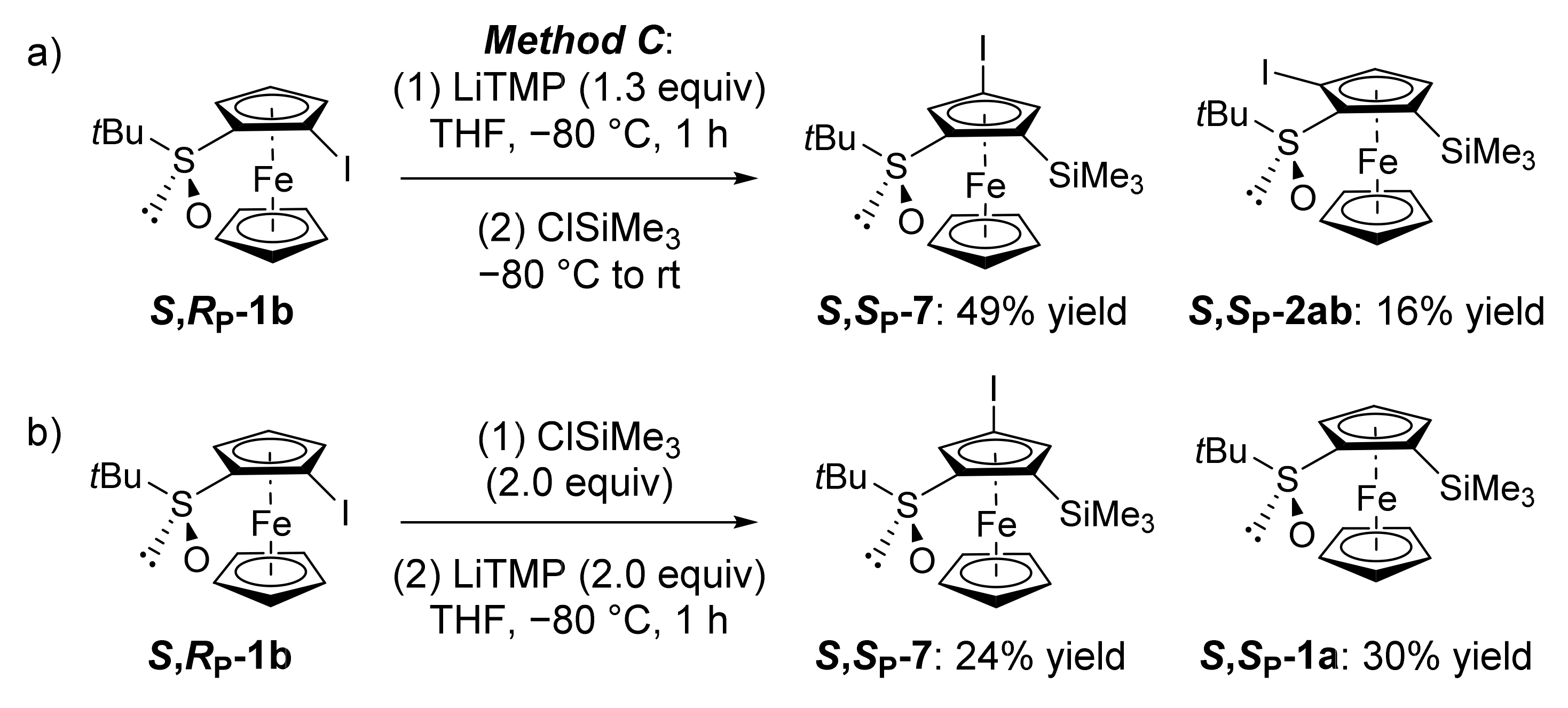
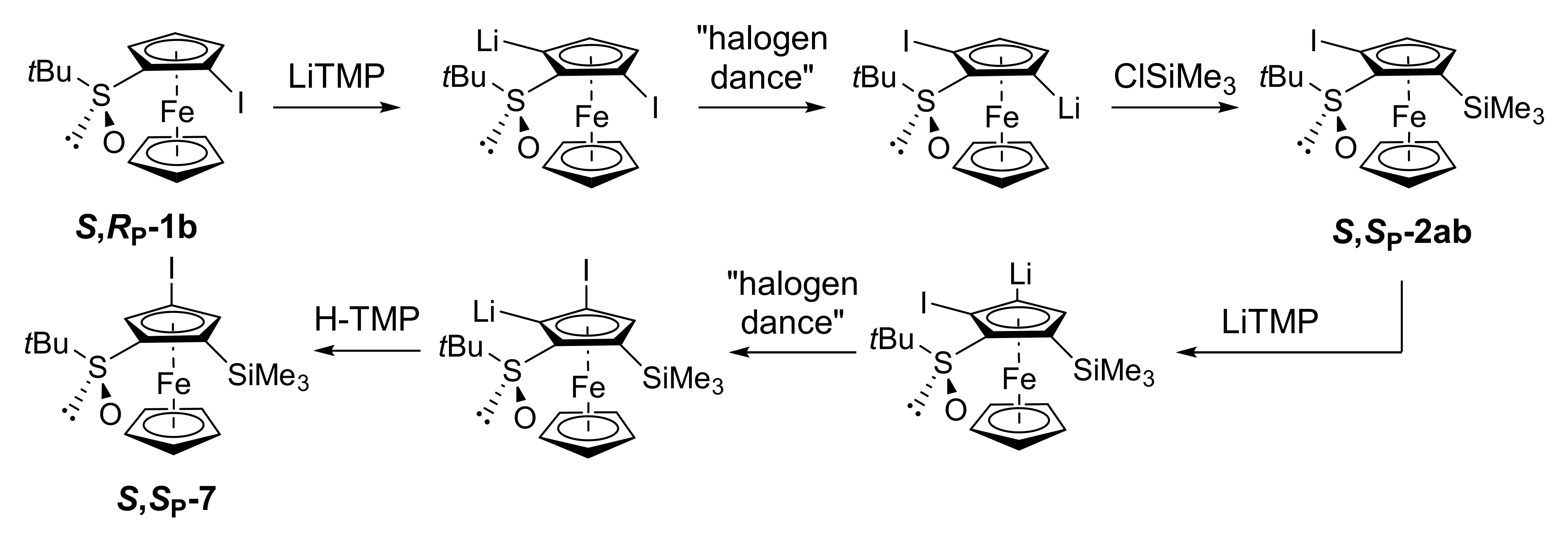
2.2. Attempts to Apply the “Halogen Dance” Reaction to the Ferrocenesulfoxide Series
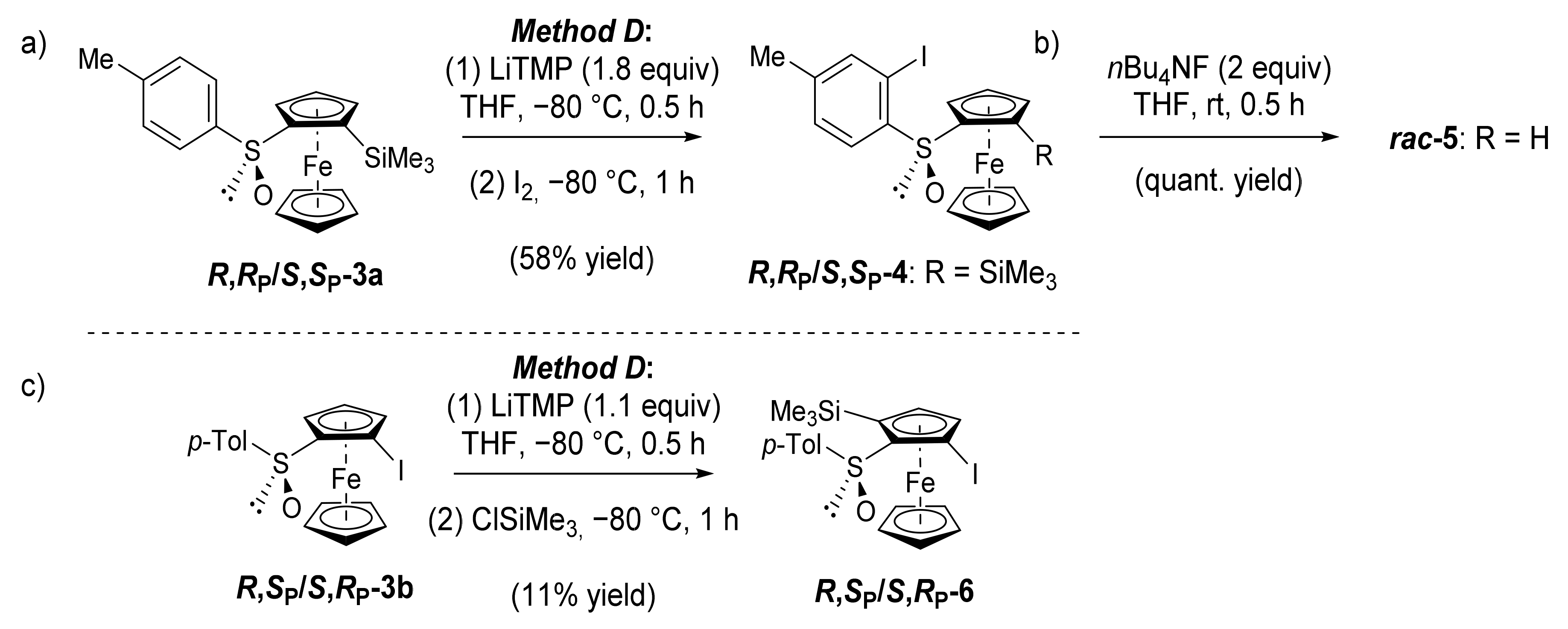
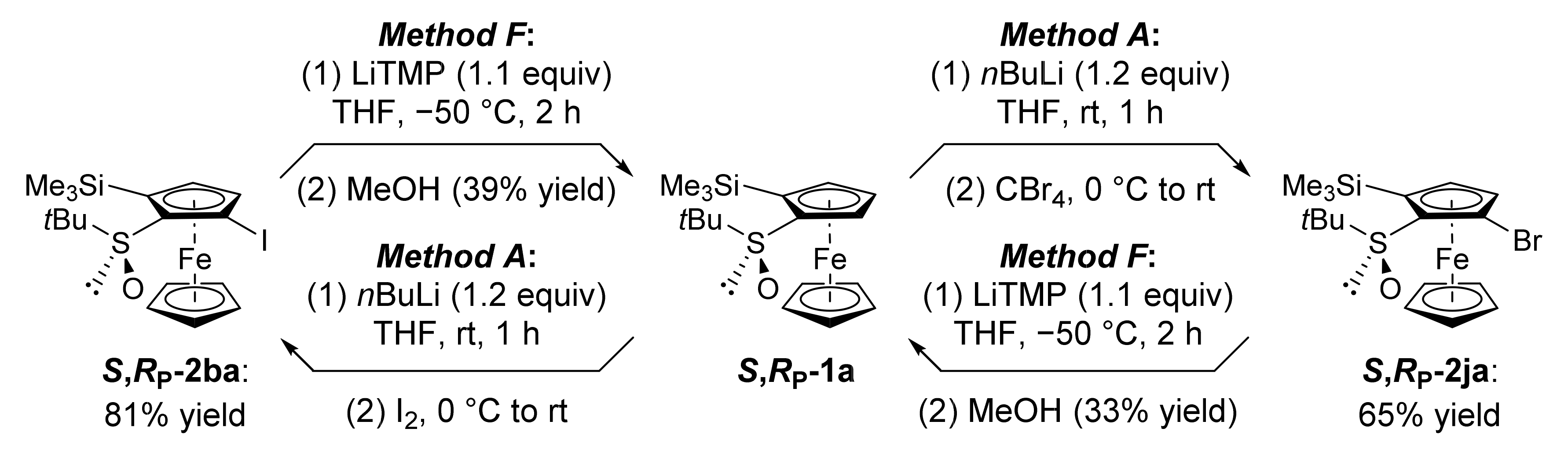
2.3. On the Way to Polysubstituted Ferrocenesulfoxides
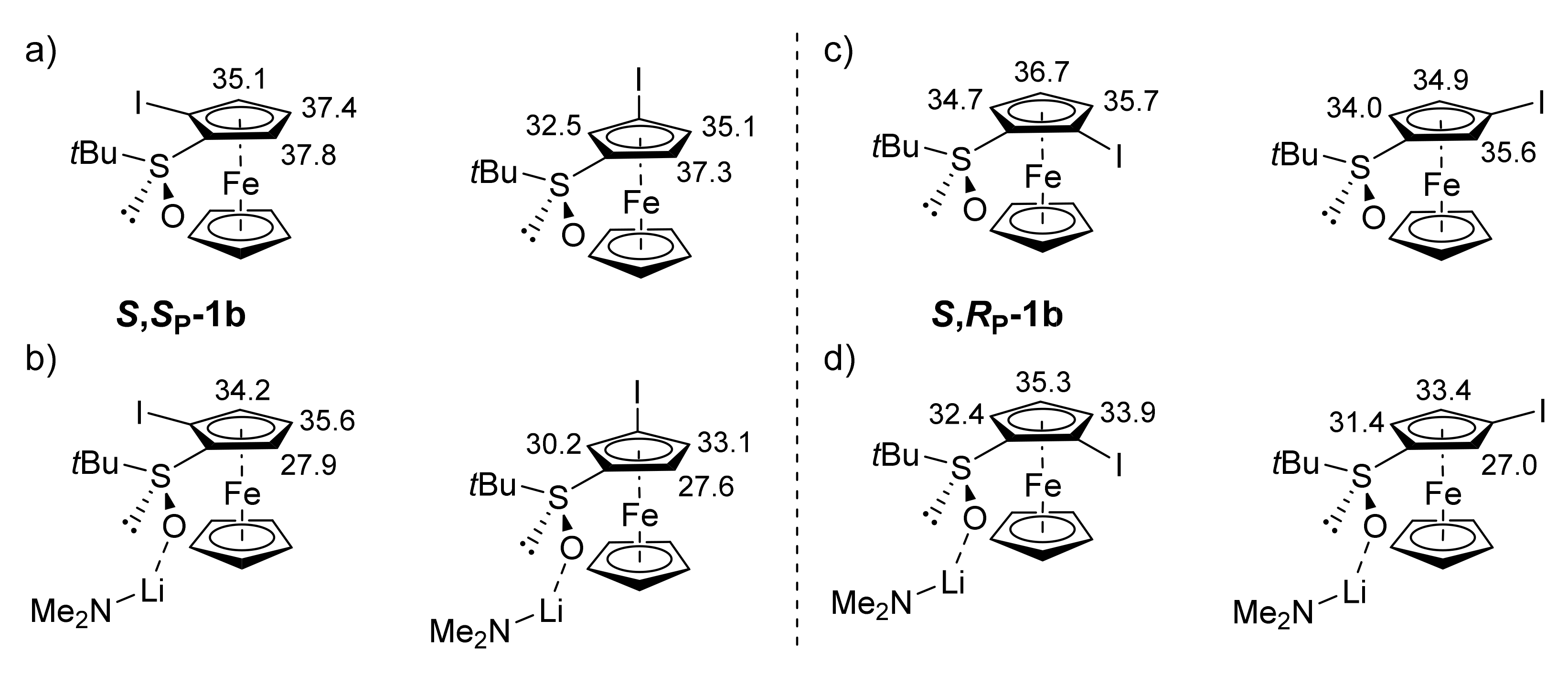
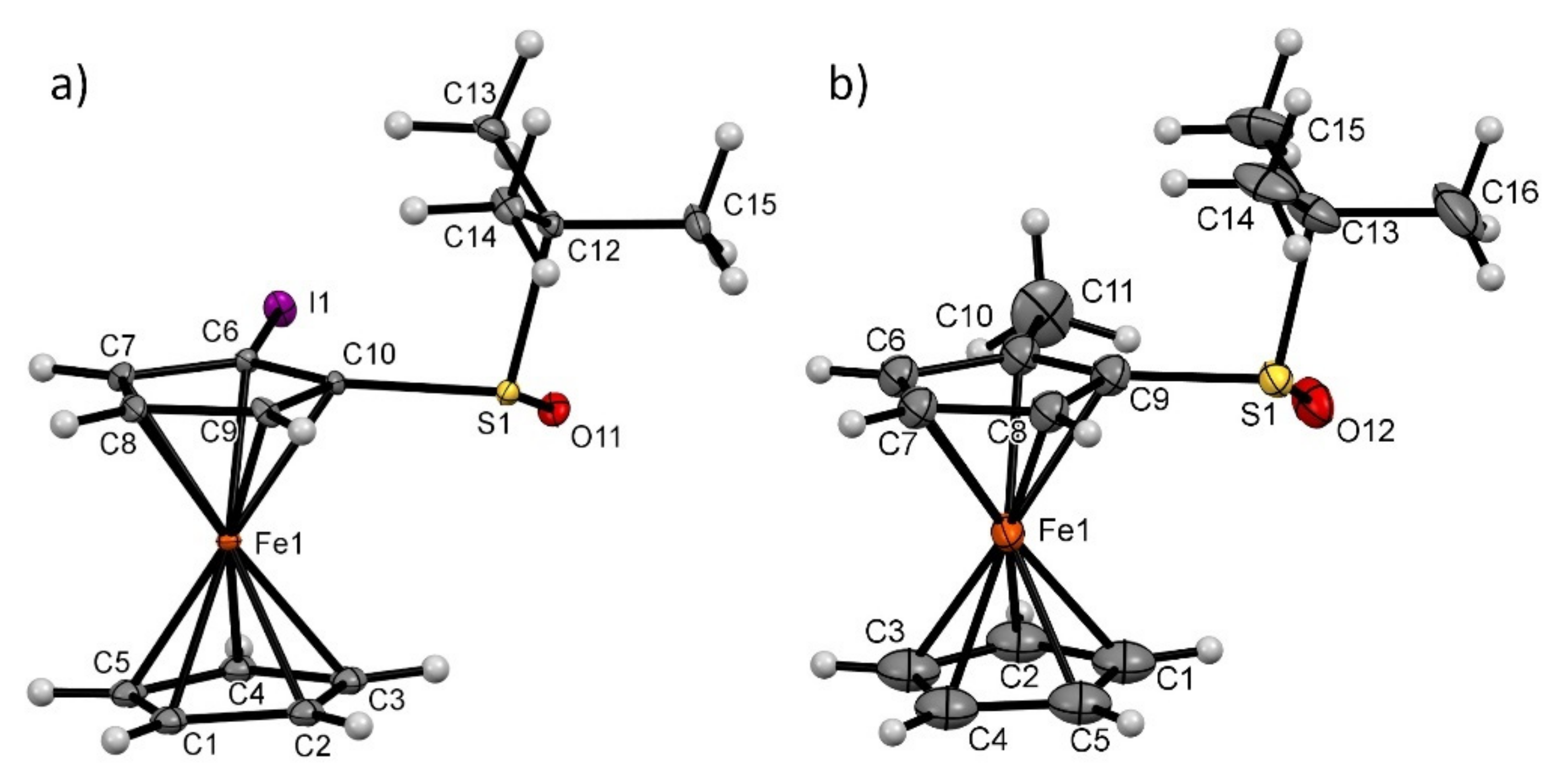
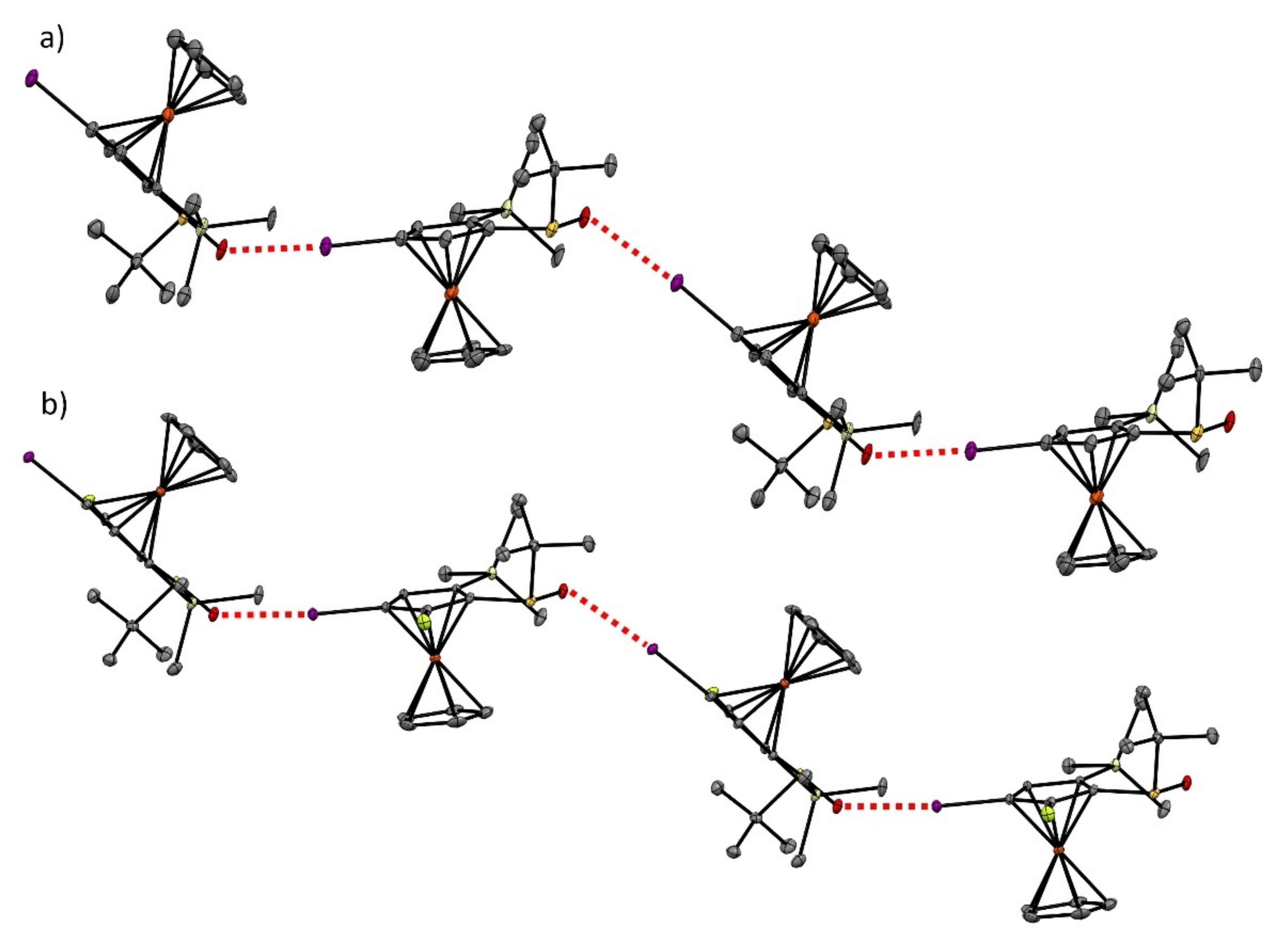
2.4. Specific Solid-State Structures of Some Ferrocenesulfoxides
3. Materials and Methods
3.1. General Information
3.2. Crystallography
3.3. Safety Considerations
3.4. Starting Materials
3.4.1. (tert-Butylthio)ferrocene
3.4.2. Racemic S-tert-Butylferrocenesulfoxide (rac-FcSOtBu)
3.4.3. (4-Tolylthio)ferrocene
3.4.4. Racemic S-(4-Tolyl)ferrocenesulfoxide (rac-FcSO-p-Tol)
3.5. General Procedure A: Deprotolithiation of S-tert-Butylferrocenesulfoxides Using nBuLi Followed by Electrophilic Trapping
3.5.1. (S,SP)-S-tert-Butyl-2-(trimethylsilyl)ferrocenesulfoxide (S,SP-1a)
3.5.2. (R,RP)-S-tert-Butyl-2-(trimethylsilyl)ferrocenesulfoxide (R,RP-1a)
3.5.3. (R,RP)- and (S,SP)-S-tert-Butyl-2-(trimethylsilyl)ferrocenesulfoxide (R,RP/S,SP-1a)
3.5.4. (S,RP)-S-tert-Butyl-2-iodoferrocenesulfoxide (S,RP-1b)
3.5.5. (S,RP)- and (R,SP)-S-tert-Butyl-2-iodoferrocenesulfoxide (S,RP/R,SP-1b)
3.5.6. (S,SP)-S-tert-Butyl-2-deuterioferrocenesulfoxide (S,SP-1c)
3.5.7. (S,SP)-S-tert-Butyl-2-methylferrocenesulfoxide (S,SP-1d)
3.5.8. (S)-S-tert-Butyl-2,5-bis(trimethylsilyl)ferrocenesulfoxide (S-2aa)
3.5.9. (R)- and (S)-S-tert-Butyl-2,5-bis(trimethylsilyl)ferrocenesulfoxide (rac-2aa)
3.5.10. (S,SP)-S-tert-Butyl-2-deuterio-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ac)
3.5.11. (S,SP)-S-tert-Butyl-2-iodo-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ab)
3.5.12. (R,RP)-S-tert-Butyl-2-iodo-5-(trimethylsilyl)ferrocenesulfoxide (R,RP-2ab)
3.5.13. (S,SP)-S-tert-Butyl-2-(diphenylphosphinyl)-5-iodoferrocenesulfoxide (S,SP-2eb)
3.5.14. (S,RP)-2-Bromo-S-tert-butyl-5-(trimethylsilyl)ferrocenesulfoxide (S,RP-2ja)
3.5.15. (S,RP)-S-tert-Butyl-2-iodo-5-(trimethylsilyl)ferrocenesulfoxide (S,RP-2ba)
3.6. General Procedure B: Deprotolithiation of Enantiopure S-tert-Butylferrocenesulfoxides Using tBuLi Followed by Electrophilic Trapping
3.6.1. (S,SP)-S-tert-Butyl-2-(diphenylphosphino)ferrocenesulfoxide (S,SP-1e)
3.6.2. (S,RP)-S-tert-Butyl-2-(tributylstannyl)ferrocenesulfoxide (S,RP-1f)
3.6.3. (S,SP)-S-tert-Butyl-2-[(α,α-diphenyl)hydroxymethyl]-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ag)
3.7. General Procedure C: Deprotolithiation of S-tert-Butylferrocenesulfoxides Using LiTMP Followed by Electrophilic Trapping
3.7.1. (S,SP)-S-tert-Butyl-2-(diphenylphosphino)-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ea)
3.7.2. (S,SP)-S-tert-Butyl-4-iodo-2-(trimethylsilyl)ferrocenesulfoxide (S,SP-7)
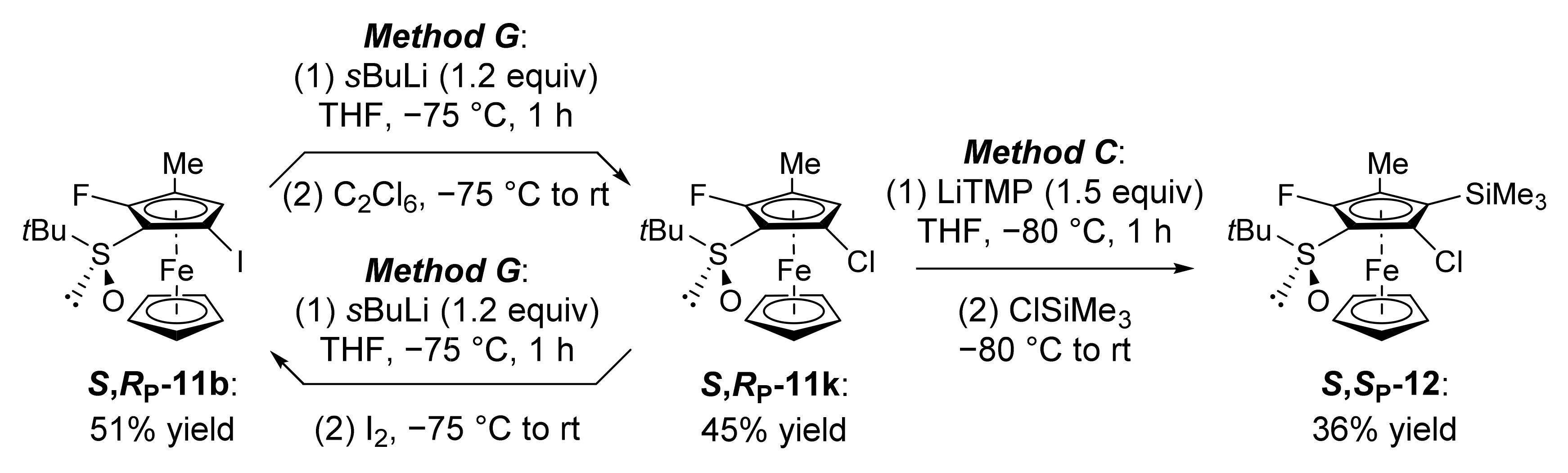
3.7.3. (S,SP)-S-tert-Butyl-2-chloro-5-fluoro-4-methyl-3-(trimethylsilyl)ferrocenesulfoxide (S,SP-12)
3.8. General Procedure D: Deprotolithiation of S-(4-tolyl)ferrocenesulfoxides Using LiTMP Followed by Electrophilic Trapping
3.8.1. (R,RP)- and (S,SP)-S-(4-Tolyl)-2-(trimethylsilyl)ferrocenesulfoxide (R,RP/S,SP-3a)
3.8.2. (R,SP)- and (S,RP)-2-Iodo-S-(4-tolyl)ferrocenesulfoxide (R,SP/S,RP-3b)
3.8.3. (S,SP)-2-Deuterio-S-(4-tolyl)ferrocenesulfoxide (S,SP-3c)
3.8.4. (R,RP)- and (S,SP)-S-(2-Iodo-4-tolyl)-2-(trimethylsilyl)ferrocenesulfoxide (R,RP/S,SP-4)
3.8.5. (R,SP)- and (S,RP)-2-Iodo-S-(4-tolyl)-5-(trimethylsilyl)ferrocenesulfoxide (R,SP/S,RP-6)
3.9. General Procedure E: One-Pot Deprotolithiation-Trimethylsilylation-Deprotolithiation-Trapping of S-tert-Butylferrocenesulfoxides
3.9.1. (S,SP)-S-tert-Butyl-2-iodoferrocenesulfoxide (S,SP-2ab)
3.9.2. (R,RP)-S-tert-Butyl-2-iodo-5-(trimethylsilyl)ferrocenesulfoxide (R,RP-2ab)
3.9.3. (S,SP)-S-tert-Butyl-2-methyl-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ad)
3.9.4. (S,RP)-S-tert-Butyl-2-(diphenylphosphino)-5-(trimethylsilyl)ferrocenesulfoxide (S,RP-2ae)
3.9.5. (S,SP)-S-tert-Butyl-2-[(α,α-diphenyl)hydroxymethyl]-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ag)
3.9.6. (S,SP)-S-tert-Butyl-2-(dimethylaminomethyl)-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ah)
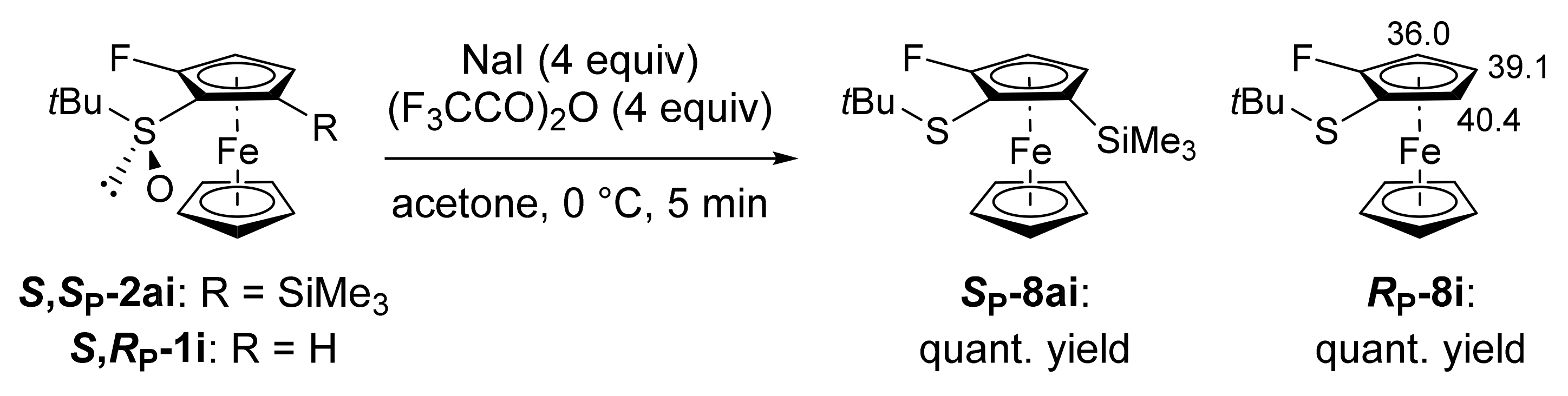
3.9.7. (S,SP)-S-tert-Butyl-2-fluoro-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-2ai)
3.10. General Procedure F: Attempted “Halogen Dance” Using LiTMP
3.10.1. From (S,RP)-S-tert-Butyl-2-iodo-5-(trimethylsilyl)ferrocenesulfoxide (S,RP-2ba)
3.10.2. From (S,SP)-2-Bromo-S-tert-butyl-5-(trimethylsilyl)ferrocenesulfoxide (S,RP-2ja)
3.11. General Procedure G: Deprotolithiation of Enantiopure Ferrocenes using sBuLi Followed by Electrophilic Trapping
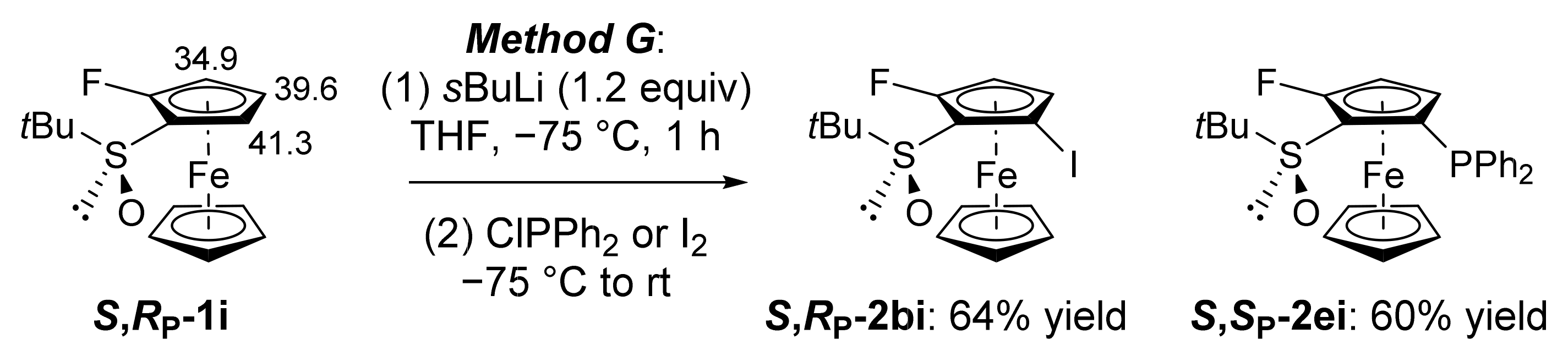
3.11.1. (S,RP)-S-tert-Butyl-2-fluoro-5-iodoferrocenesulfoxide (S,RP-2bi)
3.11.2. (S,SP)-S-tert-Butyl-2-(diphenylphosphino)-5-fluoroferrocenesulfoxide (S,SP-2ei)
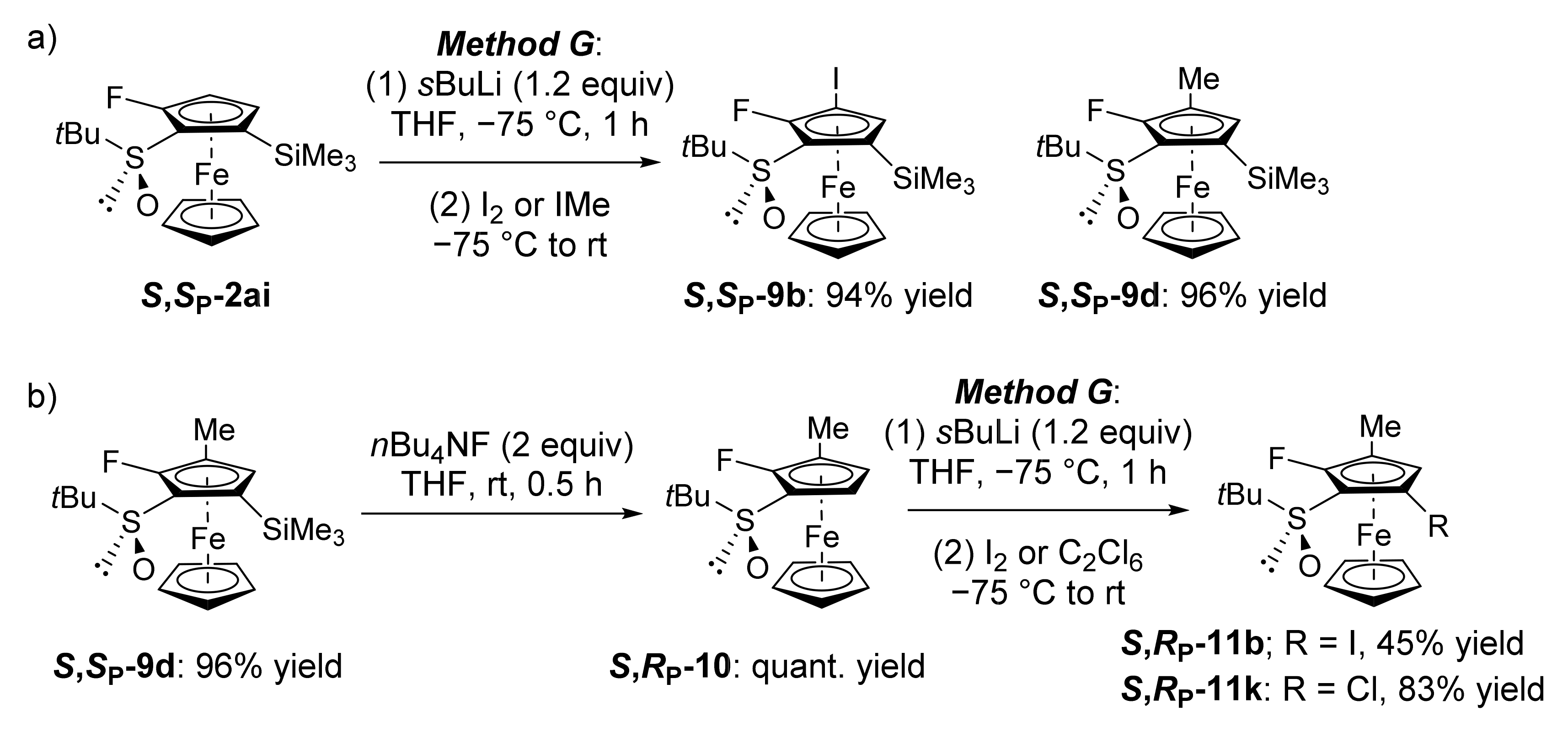
3.11.3. (S,SP)-S-tert-Butyl-2-fluoro-3-iodo-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-9b)
3.11.4. (S,SP)-S-tert-Butyl-2-fluoro-3-methyl-5-(trimethylsilyl)ferrocenesulfoxide (S,SP-9d)
3.11.5. (S,RP)-S-tert-Butyl-2-fluoro-5-iodo-3-methylferrocenesulfoxide (S,RP-11b)
3.11.6. (S,RP)-S-tert-Butyl-5-chloro-2-fluoro-3-methylferrocenesulfoxide (S,RP-11k)
3.12. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References and Note
- Durst, T. Comprehensive Organic Chemistry; Barton, D., Ollis, W.D., Eds.; Pergamon Press: Oxford, UK, 1979; Volume 3. [Google Scholar]
- Patai, S.; Rappoport, Z. (Eds.) Syntheses of Sulphones, Sulphoxides and Cyclic Sulphides; John Wiley & Sons: Hoboken, NJ, USA, 1995. [Google Scholar] [CrossRef]
- Delouvrié, B.; Fensterbank, L.; Nájera, F.; Malacria, M. The chemistry of C2-symmetric bis(sulfoxides): A new approach in asymmetric synthesis. Eur. J. Org. Chem. 2002, 3507–3525. [Google Scholar] [CrossRef]
- Fernández, I.; Khiar, N. Recent developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003, 103, 3651–3705. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Use of chiral sulfoxides in asymmetric synthesis. Tetrahedron 2006, 62, 5559–5601. [Google Scholar] [CrossRef]
- Flemming, J.P.; Berry, M.B.; Brown, J.M. Sequential ortho-lithiations; the sulfoxide group as a relay to enable meta-substitution. Org. Biomol. Chem. 2008, 6, 1215–1221. [Google Scholar] [CrossRef]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-forming and -breaking reactions at sulfur(IV): Sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef] [Green Version]
- Kupwade, R.V. A concise review on synthesis of sulfoxides and sulfones with special reference to oxidation of sulfides. J. Chem. Rev. 2019, 1, 99–113. [Google Scholar] [CrossRef]
- Li, G.; Nieves-Quinones, Y.; Zhang, H.; Liang, Q.; Su, S.; Liu, Q.; Kozlowski, M.C.; Jia, T. Transition-metal-free formal cross-coupling of aryl methyl sulfoxides and alcohols via nucleophilic activation of C-S bond. Nat. Commun. 2020, 11, 2890. [Google Scholar] [CrossRef]
- Anselmi, S.; Aggarwal, N.; Moody, T.S.; Castagnolo, D. Unconventional biocatalytic approaches to the synthesis of chiral sulfoxides. ChemBioChem 2021, 22, 298–307. [Google Scholar] [CrossRef]
- Wojaczyńska, E.; Wojaczyński, J. Modern stereoselective synthesis of chiral sulfinyl compounds. Chem. Rev. 2020, 120, 4578–4611. [Google Scholar] [CrossRef]
- Carreño, M.C. Applications of sulfoxides to asymmetric synthesis of biologically active compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Prilezhaeva, E.N. Sulfones and sulfoxides in the total synthesis of biologically active natural compounds. Russ. Chem. Rev. 2000, 69, 367–408. [Google Scholar] [CrossRef]
- Wojaczyńska, E.; Wojaczyński, J. Enantioselective synthesis of sulfoxides: 2000–2009. Chem. Rev. 2010, 110, 4303–4356. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Rao, M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem. Int. Ed. 2015, 54, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otocka, S.; Kwiatkowska, M.; Madalińska, L.; Kiełbasiński, P. Chiral organosulfur ligands/catalysts with a stereogenic sulfur atom: Applications in asymmetric synthesis. Chem. Rev. 2017, 117, 4147–4181. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Jia, T.; Wang, M.; Liao, J. Chiral sulfoxide ligands in asymmetric catalysis. Top. Curr. Chem. 2019, 377, 399–427. [Google Scholar] [CrossRef]
- Kealy, T.J.; Pauson, P.L. A new type of organo-iron compound. Nature 1951, 168, 1039–1040. [Google Scholar] [CrossRef]
- Miller, S.A.; Tebboth, J.A.; Tremaine, J.F. Dicyclopentadienyliron. J. Chem. Soc. 1952, 1952, 632–635. [Google Scholar] [CrossRef]
- Astruc, D. Why is ferrocene so exceptional? Eur. J. Inorg. Chem. 2017, 2017, 6–29. [Google Scholar] [CrossRef]
- Colacot, T.J. A concise update on the applications of chiral ferrocenyl phosphines in homogeneous catalysis leading to organic synthesis. Chem. Rev. 2003, 103, 3101–3118. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Butt, N.A.; Liu, D.; Zhang, W. The design and synthesis of planar chiral ligands and their application to asymmetric catalysis. Synlett 2014, 25, 615–630. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [Green Version]
- Ong, Y.C.; Roy, S.; Andrews, P.C.; Gasser, G. Metal compounds against neglected tropical diseases. Chem. Rev. 2019, 119, 730–796. [Google Scholar] [CrossRef]
- Gibson, V.C.; Long, N.J.; Oxford, P.J.; White, A.J.P.; Williams, D.J. Ferrocene-substituted bis(imino)pyridine iron and cobalt complexes: Toward redox-active catalysts for the polymerization of ethylene. Organometallics 2006, 25, 1932–1939. [Google Scholar] [CrossRef]
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Drozd, V.N.; Sazonova, V.A.; Nesmeyanov, A.N. Ferrocenyl sulfones. Ferrocenyl mesityl sulfone under the conditions of the smiles rearrangement. Dokl. Akad. Nauk SSSR 1964, 159, 591–594. [Google Scholar]
- Herrmann, R.; Hübener, G.; Ugi, I. Chiral sulfoxides from (R)-α-dimethylaminoethylferrocene. Tetrahedron 1985, 41, 941–947. [Google Scholar] [CrossRef]
- Schlögl, K. Stereochemistry of metallocenes. Top. Stereochem. 1967, 1, 39–91. [Google Scholar]
- Rebière, F.; Riant, O.; Ricard, L.; Kagan, H.B. Asymmetric synthesis and highly diastereoselective ortho-lithiation of ferrocenyl sulfoxides. Application in the synthesis of planar chiral ferrocenyl derivatives. Angew. Chem. Int. Ed. Engl. 1993, 32, 568–570. [Google Scholar] [CrossRef]
- Ferber, B.; Kagan, H.B. Metallocene sulfoxides as precursors of metallocenes with planar chirality. Adv. Synth. Catal. 2007, 349, 493–507. [Google Scholar] [CrossRef]
- Hua, D.H.; Lagneau, N.M.; Chen, Y.; Robben, P.M.; Clapham, G.; Robinson, P.D. Enantioselective synthesis of sulfur-containing 1,2-disubstituted ferrocenes. J. Org. Chem. 1996, 61, 4508–4509. [Google Scholar] [CrossRef]
- Riant, O.; Argouarch, G.; Guillaneux, D.; Samuel, O.; Kagan, H.B. A straightforward asymmetric synthesis of enantiopure 1,2-disubstituted ferrocenes. J. Org. Chem. 1998, 63, 3511–3514. [Google Scholar] [CrossRef]
- Ferber, B.; Top, S.; Welter, R.; Jaouen, G. A new efficient route to chiral 1,3-disubstituted ferrocenes: Application to the syntheses of (RP)- and (SP)-17α-[(3’-formylferrocenyl)ethynyl]estradiol. Chem. Eur. J. 2006, 12, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, N.; Lambusta, D.; Morrone, R.; Nicolosi, G.; Secundo, F. Biocatalytic procedure for obtaining all four diastereoisomers of 1-(1-hydroxyethyl)-3-ethylferrocene: Synthons for chiral 1,3-disubstituted ferrocenes. Tetrahedron Asymmetry 2004, 15, 3835–3840. [Google Scholar] [CrossRef]
- Priego, J.; García Mancheño, O.; Cabrera, S.; Gómez Arrayás, R.; Llamas, T.; Carretero, J.C. 1-Phosphino-2-sulfenylferrocenes: Efficient ligands in enantioselective palladium-catalyzed allylic substitutions and ring opening of 7-oxabenzonorbornadienes. Chem. Commun. 2002, 38, 2512–2513. [Google Scholar] [CrossRef]
- Steurer, M.; Tiedl, K.; Wang, Y.; Weissensteiner, W. Stereoselective synthesis of chiral, non-racemic 1,2,3-tri- and 1,3-disubstituted ferrocene derivatives. Chem. Commun. 2005, 41, 4929–4931. [Google Scholar] [CrossRef]
- Lagneau, N.M.; Chen, Y.; Robben, P.M.; Sin, H.-S.; Takasu, K.; Chen, J.-S.; Robinson, P.D.; Hua, D.H. Chiral sulfur-containing 1,2-disubstituted ferrocenes. Tetrahedron 1998, 54, 7301–7334. [Google Scholar] [CrossRef]
- All along this paper, the absolute configuration of planar chirality was given according to Schlögl’s rules: Schlogl, K. Fortschr. Chem. Forsch. 1966, 6, 479–514. See also Ref. 33, especially Figure 10, left. In the compound names and numbers, it was given after the absolute configuration of central chirality.
- Sasamori, T.; Sakagami, M.; Niwa, M.; Sakai, H.; Furukawa, Y.; Tokitoh, N. Synthesis of a stable 1,2-bis(ferrocenyl)diphosphene. Chem. Commun. 2012, 48, 8562–8564. [Google Scholar] [CrossRef]
- Caniparoli, U.; Escofet, I.; Echavarren, A.M. Planar chiral 1,3-disubstituted ferrocenyl phosphine gold(I) catalysts. ACS Catal. 2022, 12, 3317–3322. [Google Scholar] [CrossRef]
- Wen, M.; Erb, W.; Mongin, F.; Blot, M.; Roisnel, T. Enantiopure ferrocene-1,2-disulfoxides: Synthesis and reactivity. Chem. Commun. 2022, 58, 2002–2005. [Google Scholar] [CrossRef] [PubMed]
- Rebière, F.; Samuel, O.; Kagan, H.B. A convenient method for the preparation of monolithioferrocene. Tetrahedron Lett. 1990, 31, 3121–3124. [Google Scholar] [CrossRef]
- Han, Z.S.; Meyer, A.M.; Xu, Y.; Zhang, Y.; Busch, R.; Shen, S.; Grinberg, N.; Lu, B.Z.; Krishnamurthy, D.; Senanayake, C.H. Enantioselective synthesis of diverse sulfinamides and sulfinylferrocenes from phenylglycine-derived chiral sulfinyl transfer agent. J. Org. Chem. 2011, 76, 5480–5484. [Google Scholar] [CrossRef] [PubMed]
- Gelat, F.; Lohier, J.-F.; Gaumont, A.-C.; Perrio, S. Tert-butyl sulfoxides: Key precursors for palladium-catalyzed arylation of sulfenate salts. Adv. Synth. Catal. 2015, 357, 2011–2016. [Google Scholar] [CrossRef]
- Škvorcová, A.; Šebesta, R. Computational study of diastereoselective ortho-lithiations of chiral ferrocenes. Org. Biomol. Chem. 2014, 12, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Tazi, M.; Erb, W.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Mongin, F. From 2- to 3-substituted ferrocene carboxamides or how to apply halogen “dance” to the ferrocene series. Organometallics 2017, 36, 4770–4778. [Google Scholar] [CrossRef]
- Tazi, M.; Hedidi, M.; Erb, W.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Bentabed-Ababsa, G.; Mongin, F. Fluoro- and chloroferrocene: From 2- to 3-substituted derivatives. Organometallics 2018, 37, 2207–2211. [Google Scholar] [CrossRef]
- Erb, W.; Wen, M.; Hurvois, J.P.; Mongin, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T. O-isopropylferrocenesulfonate: Synthesis of polysubstituted derivatives and electrochemical study. Eur. J. Inorg. Chem. 2021, 2021, 3165–3176. [Google Scholar] [CrossRef]
- Wen, M.; Erb, W.; Mongin, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V. Functionalization of N,N-dialkylferrocenesulfonamides toward substituted derivatives. Organometallics 2021, 40, 1129–1147. [Google Scholar] [CrossRef]
- Hedidi, M.; Maillard, J.; Erb, W.; Lassagne, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T.; Dorcet, V.; Hamzé, M.; et al. Fused systems based on 2-aminopyrimidines: Synthesis combining deprotolithiation-in situ zincation with N-arylation reactions and biological properties. Eur. J. Org. Chem. 2017, 2017, 5903–5915. [Google Scholar] [CrossRef]
- Gómez Arrayás, R.; Alonso, I.; Familiar, O.; Carretero, J.C. Synthesis of enantiopure planar chiral bisferrocenes bearing sulfur or nitrogen substituents. Organometallics 2004, 23, 1991–1996. [Google Scholar] [CrossRef]
- García Mancheño, O.; Priego, J.; Cabrera, S.; Gómez Arrayás, R.; Llamas, T.; Carlos Carretero, J. 1-Phosphino-2-sulfenylferrocenes as planar chiral ligands in enantioselective palladium-catalyzed allylic substitutions. J. Org. Chem. 2003, 68, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Chiffre, J.; Coppel, Y.; Balavoine, G.G.A.; Daran, J.-C.; Manoury, E. Selective functionalization of chiral ferrocenyl acetals. Easy access to various tri- and tetrasubstituted ferrocenes with controlled geometry. Organometallics 2002, 21, 4552–4555. [Google Scholar] [CrossRef]
- Mokhtari Brikci-Nigassa, N.; Bentabed-Ababsa, G.; Erb, W.; Mongin, F. In situ ‘trans-metal trapping’: An efficient way to extend the scope of aromatic deprotometalation. Synthesis 2018, 50, 3615–3633. [Google Scholar] [CrossRef]
- Zirakzadeh, A.; Herlein, A.; Gross, M.A.; Mereiter, K.; Wang, Y.; Weissensteiner, W. Halide-mediated ortho-deprotonation reactions applied to the synthesis of 1,2- and 1,3-disubstituted ferrocene derivatives. Organometallics 2015, 34, 3820–3832. [Google Scholar] [CrossRef]
- Nayyar, B.; Koop, S.; Lutter, M.; Jurkschat, K. Ferrocene-based, potentially D,C,D-coordinating (D = O, S), pincer-type proligands and their organotin derivatives. Eur. J. Inorg. Chem. 2017, 2017, 3233–3238. [Google Scholar] [CrossRef]
- Nayyar, B.; Kapoor, R.; Lutter, M.; Alnasr, H.; Jurkschat, K. It’s getting tight: Highly substituted intramolecularly p:O→sn coordinated ferrocene derivatives. Eur. J. Inorg. Chem. 2017, 2017, 3967–3978. [Google Scholar] [CrossRef]
- Fraser, R.R.; Mansour, T.S.; Savard, S. Acidity measurements on pyridines in tetrahydrofuran using lithiated silylamines. J. Org. Chem. 1985, 50, 3232–3234. [Google Scholar] [CrossRef]
- Tazi, M.; Erb, W.; Roisnel, T.; Dorcet, V.; Mongin, F.; Low, P.J. From ferrocene to fluorine-containing penta-substituted derivatives and all points in-between; or, how to increase the available chemical space. Org. Biomol. Chem. 2019, 17, 9352–9359. [Google Scholar] [CrossRef] [PubMed]
- Gronowitz, S. Recent advances in the chemistry of thiophenes. Adv. Heterocycl. Chem. 1963, 14, 1–124. [Google Scholar] [CrossRef] [PubMed]
- Queguiner, G.; Marsais, F.; Snieckus, V.; Epsztajn, J. Directed metalation of pi-deficient azaaromatics: Strategies of functionalization of pyridines, quinolines, and diazines. Adv. Heterocycl. Chem. 1991, 52, 187–304. [Google Scholar] [CrossRef]
- Fröhlich, J. Substituted heterocyclic compounds by selective control of halogen-dance reactions. Prog. Heterocycl. Chem. 1994, 6, 1–35. [Google Scholar] [CrossRef]
- Schlosser, M. The organometallic approach to molecular diversity—halogens as helpers. Eur. J. Org. Chem. 2001, 2001, 3975–3984. [Google Scholar] [CrossRef]
- Schlosser, M. The 2 × 3 toolbox of organometallic methods for regiochemically exhaustive functionalization. Angew. Chem. Int. Ed. 2005, 44, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-F.; Zhang, Z.-B. Recent progress of halogen-dance reactions in heterocycles. Heterocycles 2005, 65, 2005–2012. [Google Scholar] [CrossRef]
- Schlosser, M.; Mongin, F. Pyridine elaboration through organometallic intermediates: Regiochemical control and completeness. Chem. Soc. Rev. 2007, 36, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Schnürch, M. Recent progress on the halogen dance reaction on heterocycles. Top. Heterocycl. Chem. 2012, 27, 185–218. [Google Scholar] [CrossRef]
- Erb, W.; Mongin, F. Halogen ‘dance’: A way to extend the boundaries of arene deprotolithiation. Tetrahedron 2016, 72, 4973–4988. [Google Scholar] [CrossRef]
- Dayaker, G.; Sreeshailam, A.; Chevallier, F.; Roisnel, T.; Radha Krishna, P.; Mongin, F. Deprotonative metallation of ferrocenes using mixed lithium-zinc and lithium-cadmium combinations. Chem. Commun. 2010, 46, 2862–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb, W.; Roisnel, T. Asymmetric synthesis of hetero-1,2,3,4,5-pentasubstituted ferrocenes. Chem. Commun. 2019, 55, 9132–9135. [Google Scholar] [CrossRef]
- Blockhaus, T.; Bernhartzeder, S.; Kempinger, W.; Klein-Heβling, C.; Weigand, S.; Sünkel, K. Evidence for “halogen-dance” and ring-exchange reactions in chloro-methylthio-ferrocenes. Eur. J. Org. Chem. 2020, 2020, 6576–6587. [Google Scholar] [CrossRef]
- Erb, W.; Kadari, L.; Al-Mekhlafi, K.; Roisnel, T.; Dorcet, V.; Radha Krishna, P.; Mongin, F. Functionalization of 3-iodo-N,N-diisopropylferrocenecarboxamide, a pivotal substrate to open the chemical space to 1,3-disubstituted ferrocenes. Adv. Synth. Catal. 2020, 362, 832–850. [Google Scholar] [CrossRef] [Green Version]
- Erb, W.; Roisnel, T. The chemistry of ferrocenesulfonyl fluoride revealed. Dalton Trans. 2021, 50, 16483–16487. [Google Scholar] [CrossRef]
- Mongin, F.; Marzi, E.; Schlosser, M. Extensive halogen scrambling and buttressing effects encountered upon treatment of oligobromoarenes with bases. Eur. J. Org. Chem. 2001, 2001, 2771–2777. [Google Scholar] [CrossRef]
- Bridges, A.J.; Lee, A.; Maduakor, E.C.; Schwartz, C.E. Fluorine as an ortho-directing group in aromatic metalation: Generality of the reaction and the high position of fluorine in the dir-met potency scale. Tetrahedron Lett. 1992, 33, 7495–7498. [Google Scholar] [CrossRef]
- Mongin, F.; Curty, C.; Marzi, E.; Leroux, F.R.; Schlosser, M. Substituent effects on the relative rates and free energies of ortho-lithiation reactions: Families of fluorobenzenes as the substrates. ARKIVOC 2015, 2015, 48–65. [Google Scholar] [CrossRef] [Green Version]
- Bonini, B.F.; Fochi, M.; Comes-Franchini, M.; Ricci, A.; Thijs, L.; Zwanenburg, B. Synthesis of ferrocenyl-oxazolines by ring expansion of n-ferrocenoyl-aziridine-2-carboxylic esters. Tetrahedron Asymmetry 2003, 14, 3321–3327. [Google Scholar] [CrossRef]
- Pichon, C.; Odell, B.; Brown, J.M. A direct meta-lithiation route to 1,3-disubstituted ferrocenes. Chem. Commun. 2004, 40, 598–599. [Google Scholar] [CrossRef]
- Marsh, B.; Frost, C.; Pearce, D. 1,1′-[[(Substituted Alkyl)Imino]Bis(Alkylene)]Bis-Ferrocenes and Their Use in i Electrochemical Assays by Labelling Substrates of Interest. WO 2013/190328 A1, 27 December 2013. [Google Scholar]
- Huffman, J.W.; Keith, L.H.; Asbury, R.L. Some reactions of chloroferrocene with organolithium compounds. J. Org. Chem. 1965, 30, 1600–1604. [Google Scholar] [CrossRef]
- Hedberg, F.L.; Rosenberg, H. Preparation and reactions of decachloroferrocene and decachlororuthenocene. J. Am. Chem. Soc. 1973, 95, 870–875. [Google Scholar] [CrossRef]
- Romanov, A.S.; Mulroy, J.M.; Khrustalev, V.N.; Antipin, M.Y.; Timofeeva, T.V. Monohalogenated ferrocenes C5H5FeC5H4X (X = Cl, Br and I) and a second polymorph of C5H5FeC5H4I. Acta Crystallogr. Sect. C 2009, 65, m426–m430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahimova, N.Z.; Jafarov, G.M.; Taghiev, D.B.; Lyatifov, I.U. Crystal structure of 1,2,4,1′,2′,4′-hexamethylferrocene. Russ. J. Coord. Chem. 2020, 46, 53–57. [Google Scholar] [CrossRef]
- Mamane, V.; Peluso, P.; Aubert, E.; Weiss, R.; Wenger, E.; Cossu, S.; Pale, P. Disubstituted ferrocenyl iodo- and chalcogenoalkynes as chiral halogen and chalcogen bond donors. Organometallics 2020, 39, 3936–3950. [Google Scholar] [CrossRef]
- Bartashevich, E.; Mukhitdinova, S.; Yushina, I.; Tsirelson, V. Electronic criterion for categorizing the chalcogen and halogen bonds: Sulfur-iodine interactions in crystals. Acta Crystallogr. Sect. B 2019, 75, 117–126. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Burchat, A.F.; Chong, J.M.; Nielsen, N. Titration of alkyllithiums with a simple reagent to a blue endpoint. J. Organomet. Chem. 1997, 542, 281–283. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Solladié, G.; Hutt, J.; Girardin, A. Improved preparation of optically active methyl p-tolyl sulfoxide. Synthesis 1987, 19, 173. [Google Scholar] [CrossRef]
- Rausch, M.D. Convenient synthesis of ferrocenyl aryl sulfides. J. Org. Chem. 1961, 26, 3579–3580. [Google Scholar] [CrossRef]
- Butler, I.R.; Drew, M.G.B. 1,2-dibromoferrocenes: Synthesis and structure. Inorg. Chem. Commun. 1999, 2, 234–237. [Google Scholar] [CrossRef]
- Minière, S.; Reboul, V.; Metzner, P. Synthesis of planar chiral ferrocenyl sulfides and evaluation as catalysts for the asymmetric epoxidation of aldehydes. ARKIVOC 2005, 2005, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Espino, G.; Xiao, L.; Puchberger, M.; Mereiter, K.; Spindler, F.; Manzano, B.R.; Jalón, F.A.; Weissensteiner, W. Synthesis, coordination behavior, structural features and use in asymmetric hydrogenations of bifep-type biferrocenes. Dalton Trans. 2009, 38, 2751–2763. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the coulomb-attenuating method (cam-b3lyp. Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Fraser, R.R.; Mansour, T.S.; Savard, S. Acidity measurements in THF. V. Heteroaromatic compounds containing 5-membered rings. Can. J. Chem. 1985, 63, 3505–3509. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Substrate | Method | Electrophile 1 | Product 1, Yield (%) 2 | |
|---|---|---|---|---|---|
| 1 | S-FcSOtBu | A | ClSiMe3 | 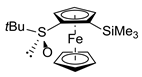 | S,SP-1a, 89 |
| 2 | rac-FcSOtBu | R,RP/S,SP-1a, 88 | |||
| 3 | S-FcSOtBu | A | I2 | 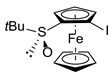 | S,RP-1b, 85 |
| 4 | rac-FcSOtBu | R,SP/S,RP-1b, 84 | |||
| 5 | S-FcSOtBu | A | D2O | 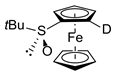 | S,SP-1c, 98 (95% D) |
| 6 | S-FcSOtBu | A | IMe | 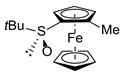 | S,SP-1d, 62 |
| 7 | S-FcSOtBu | B | ClPPh2 |  | S,SP-1e, 92 |
| 8 | S-FcSOtBu | B | ClSnBu3 | 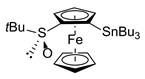 | S,RP-1f, 95 |

| Entry | Substrate 1 (E) | Method | Electrophile 1 | Product 2, Yield (%) 2 | |
|---|---|---|---|---|---|
| 1 | S,SP-1a (SiMe3) | A | ClSiMe3 | 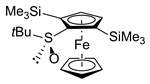 | S-2aa, 80 |
| 2 | R,RP/S,SP-1a (SiMe3) | rac-2aa, 63 | |||
| 3 | S,SP-1a (SiMe3) | A | I2 |  | S,SP-2ab, 78 |
| 4 | R,RP-1a (SiMe3) | 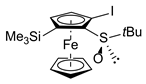 | R,RP-2ab, 77 | ||
| 5 | S,SP-1a (SiMe3) | A | D2O | 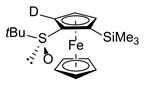 | S,SP-2ac, 98 (90% D) |
| 6 | S,SP-1a (SiMe3) | B | Ph2CO | 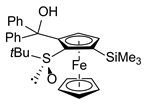 | S,SP-2ag, 35 3 |
| 7 | S,SP-1e (PPh2) | A | I2 |  | S,SP-2eb, 40 4 |
| 8 | S,SP-1e (PPh2) | C | ClSiMe3 | 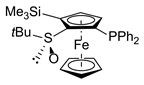 | S,SP-2ea, 70 |
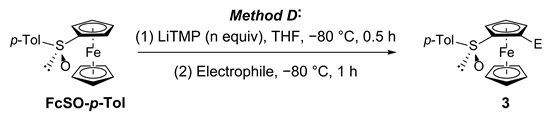
| Entry | Substrate | n Equiv | Electrophile 1 | Product 3, Yield (%) 2 | |
|---|---|---|---|---|---|
| 1 | rac-FcSO-p-Tol | 1.2 | ClSiMe3 |  | R,RP/S,SP-3a, 61 |
| 2 | rac-FcSO-p-Tol | 1.5 | ClSiMe3 | 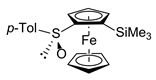 | R,RP/S,SP-3a, 43 |
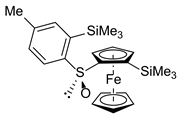 | R,RP/S,SP-3a’, 19 | ||||
| 3 | rac-FcSO-p-Tol | 1.1 | I2 | 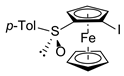 | R,SP/S,RP-3b, 51 3 |
| 4 | 1.5 | R,SP/S,RP-3b, 81 | |||
| 5 | S-FcSO-p-Tol | 2 | DCl/D2O |  | S,SP-3c, quant. (80% D) |

| Entry | Substrate | Electrophile 1 | Product 2, Yield (%) 2 | |
|---|---|---|---|---|
| 1 | S-FcSOtBu | I2 | 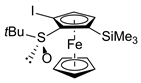 | S,SP-2ab, 83 3 |
| 2 | R-FcSOtBu |  | R,RP-2ab, 53 | |
| 3 | S-FcSOtBu | IMe | 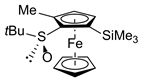 | S,SP-2ad, 74 |
| 4 | S-FcSOtBu | ClPPh2 | 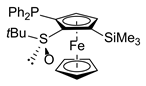 | S,RP-2ae, 48 |
| 5 | S-FcSOtBu | Ph2CO | 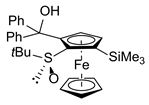 | S,SP-2ag, 51 |
| 6 | S-FcSOtBu | CH2=NMe2I | 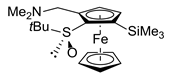 | S,SP-2ah, 66 4 |
| 7 | S-FcSOtBu | NFSI | 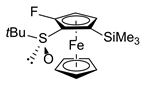 | S,SP-2ai, 67 |
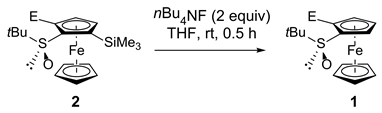
| Entry | Substrate 2 (E) | Product 1, Yield (%) 1 | |
|---|---|---|---|
| 1 | R,RP-2ab (I) |  | R,RP-1b, 83 |
| 2 | S,SP-2ac (D) |  | S,RP-1c, 60 |
| 3 | S,SP-2ad (Me) | 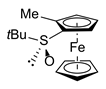 | S,RP-1d, quant. |
| 4 | S,SP-2ah (CH2NMe2) | 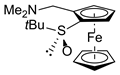 | S,RP-1h, quant. |
| 5 | S,SP-2ai (F) | 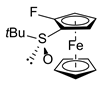 | S,RP-1i, quant. |
| 6 2 | S-2aa (SiMe3) | 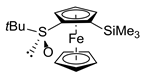 | S,SP-1a, 78 |
| 7 2 | rac-2aa (SiMe3) | S,SP/R,RP-1a, 67 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, M.; Erb, W.; Mongin, F.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Roisnel, T. Synthesis of Polysubstituted Ferrocenesulfoxides. Molecules 2022, 27, 1798. https://doi.org/10.3390/molecules27061798
Wen M, Erb W, Mongin F, Halauko YS, Ivashkevich OA, Matulis VE, Roisnel T. Synthesis of Polysubstituted Ferrocenesulfoxides. Molecules. 2022; 27(6):1798. https://doi.org/10.3390/molecules27061798
Chicago/Turabian StyleWen, Min, William Erb, Florence Mongin, Yury S. Halauko, Oleg A. Ivashkevich, Vadim E. Matulis, and Thierry Roisnel. 2022. "Synthesis of Polysubstituted Ferrocenesulfoxides" Molecules 27, no. 6: 1798. https://doi.org/10.3390/molecules27061798
APA StyleWen, M., Erb, W., Mongin, F., Halauko, Y. S., Ivashkevich, O. A., Matulis, V. E., & Roisnel, T. (2022). Synthesis of Polysubstituted Ferrocenesulfoxides. Molecules, 27(6), 1798. https://doi.org/10.3390/molecules27061798