
Computational Simulations Highlight the IL2Rα Binding Potential of Polyphenol Stilbenes from Fenugreek


 ,
,
Abstract
:1. Introduction
2. Material and Methods
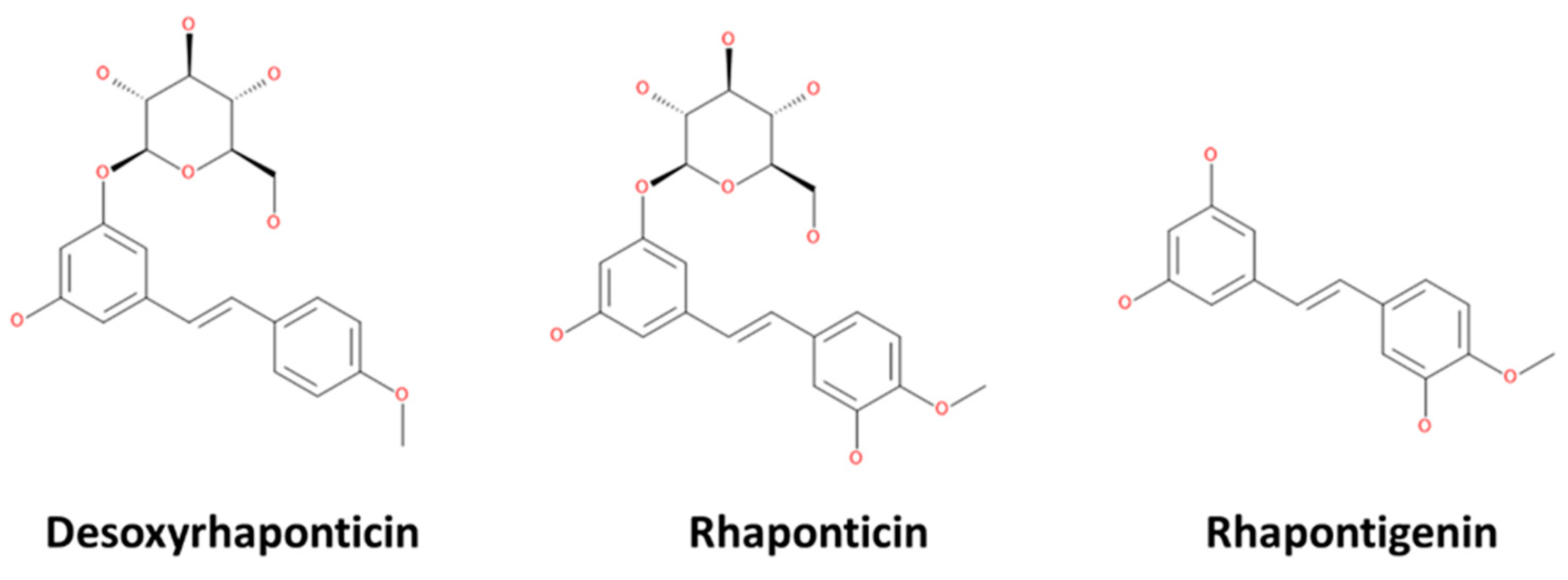
2.1. Receptor and Ligands Preparation
2.2. Molecular Docking
2.3. Molecular Dynamics (MD) Simulation
2.4. Binding Free Energy
2.5. Physiochemical Properties and ADMET Prediction
3. Results and Discussion
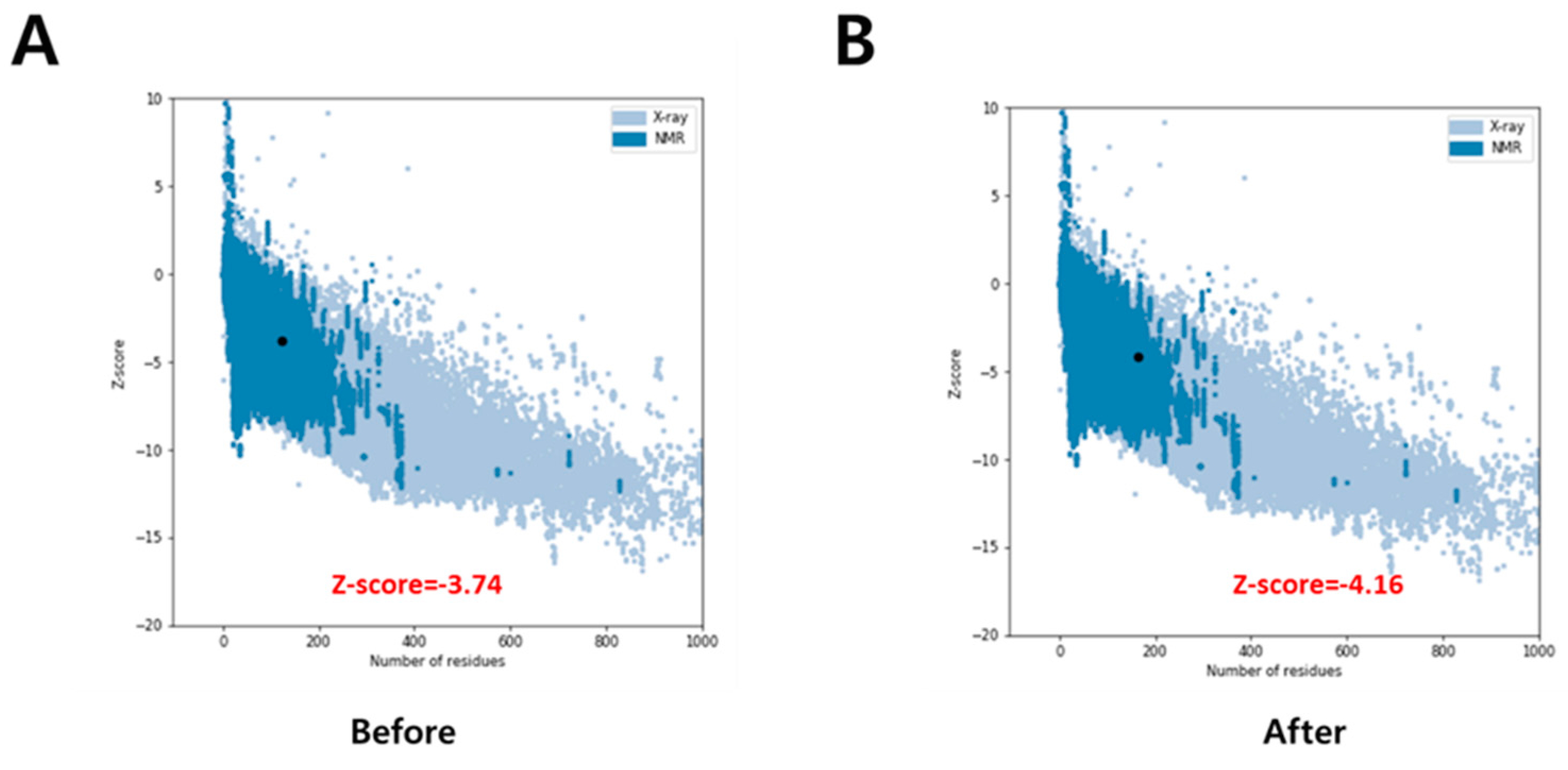
3.1. Receptor Preparation
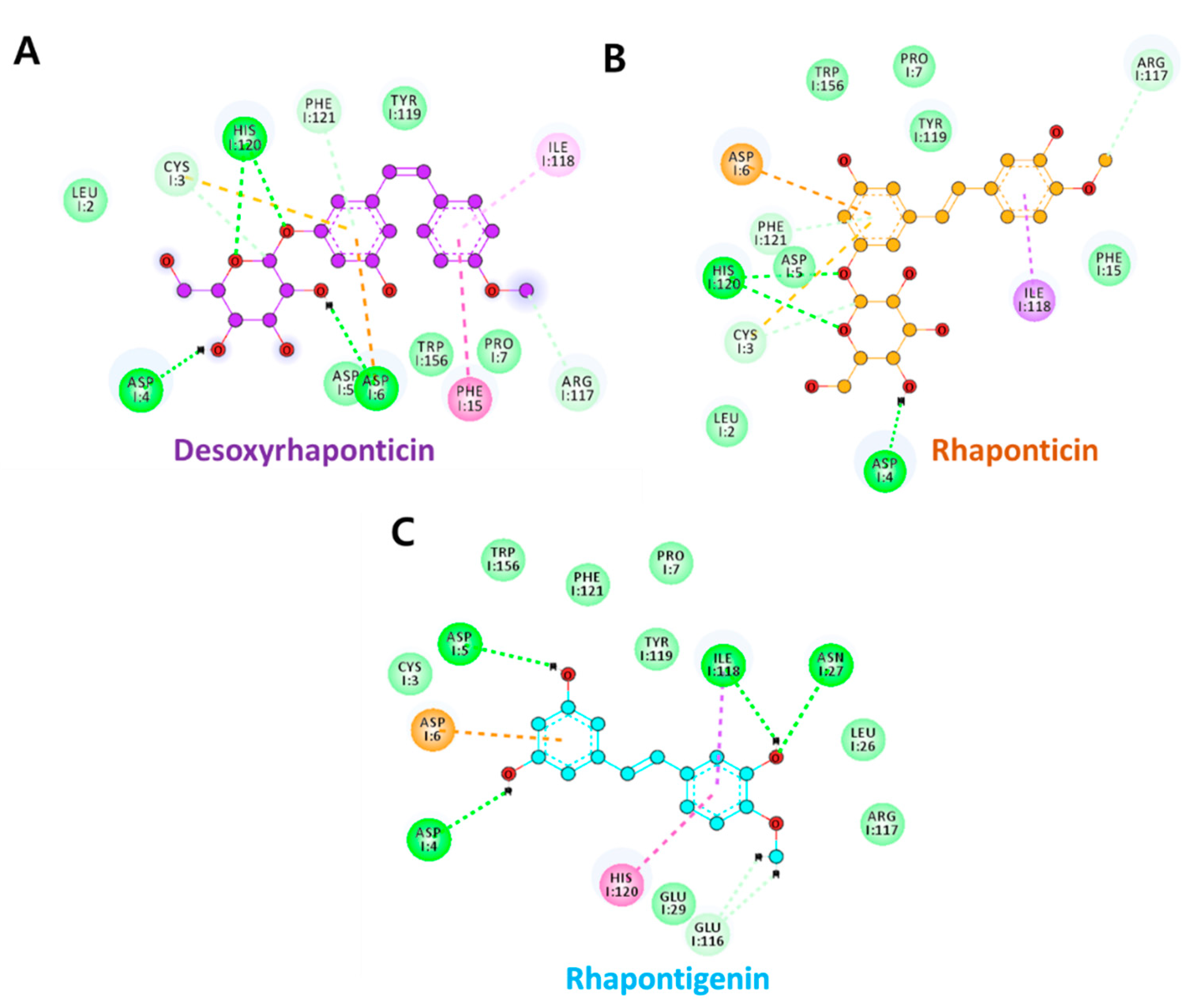
3.2. Molecular Docking Analysis
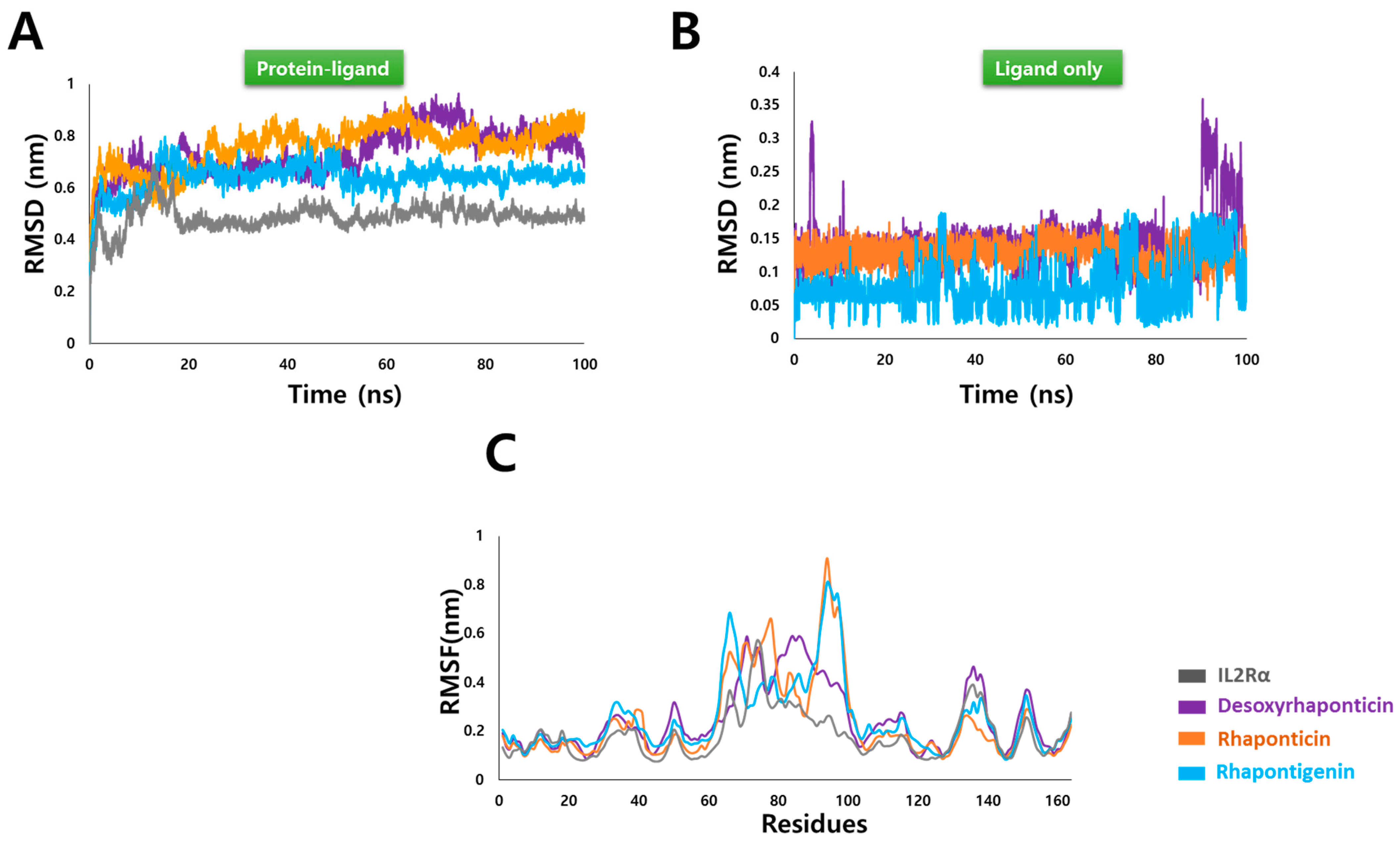
3.3. MD Simulation Analysis
3.3.1. Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF)
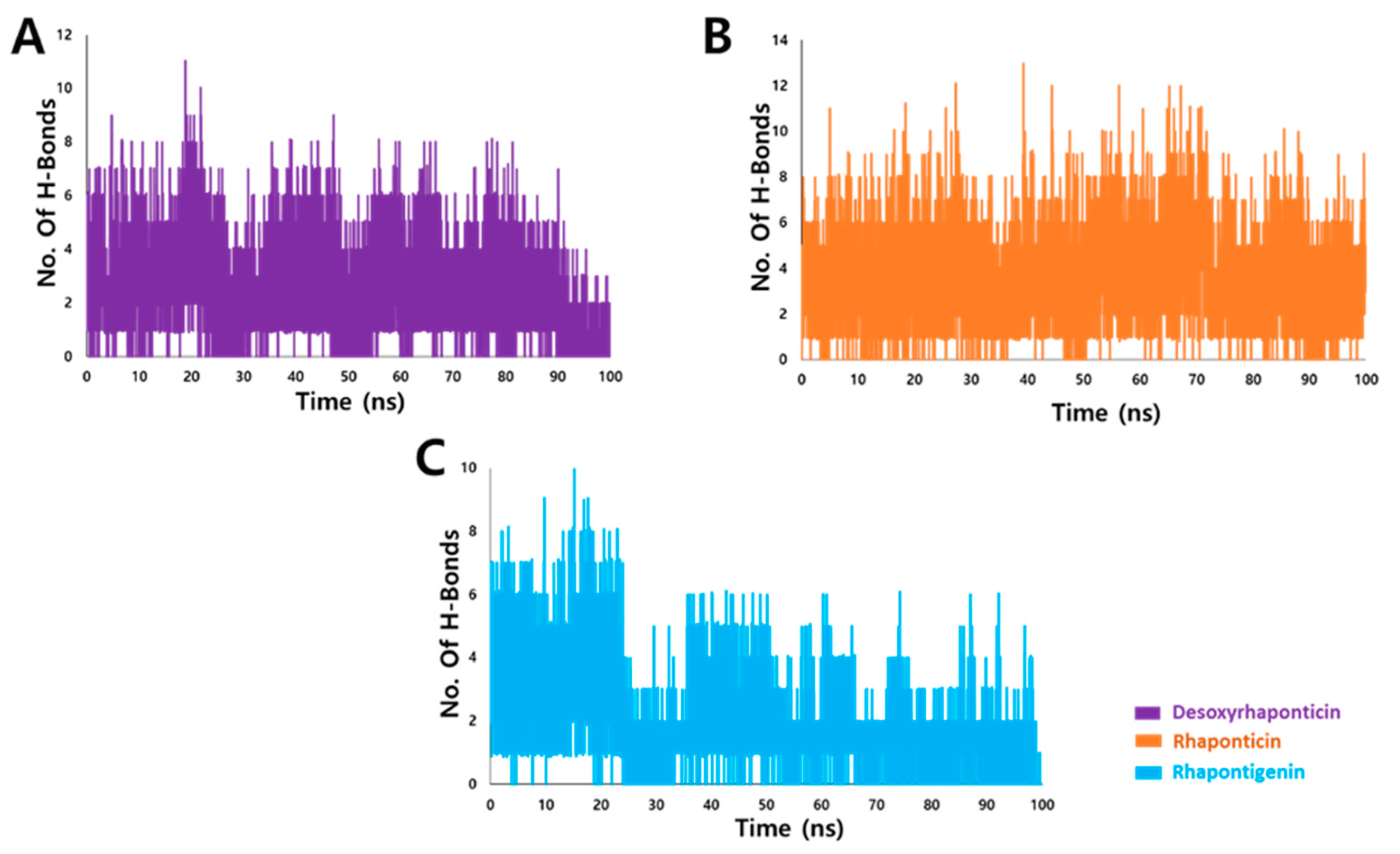
3.3.2. Hydrogen Bond Analysis
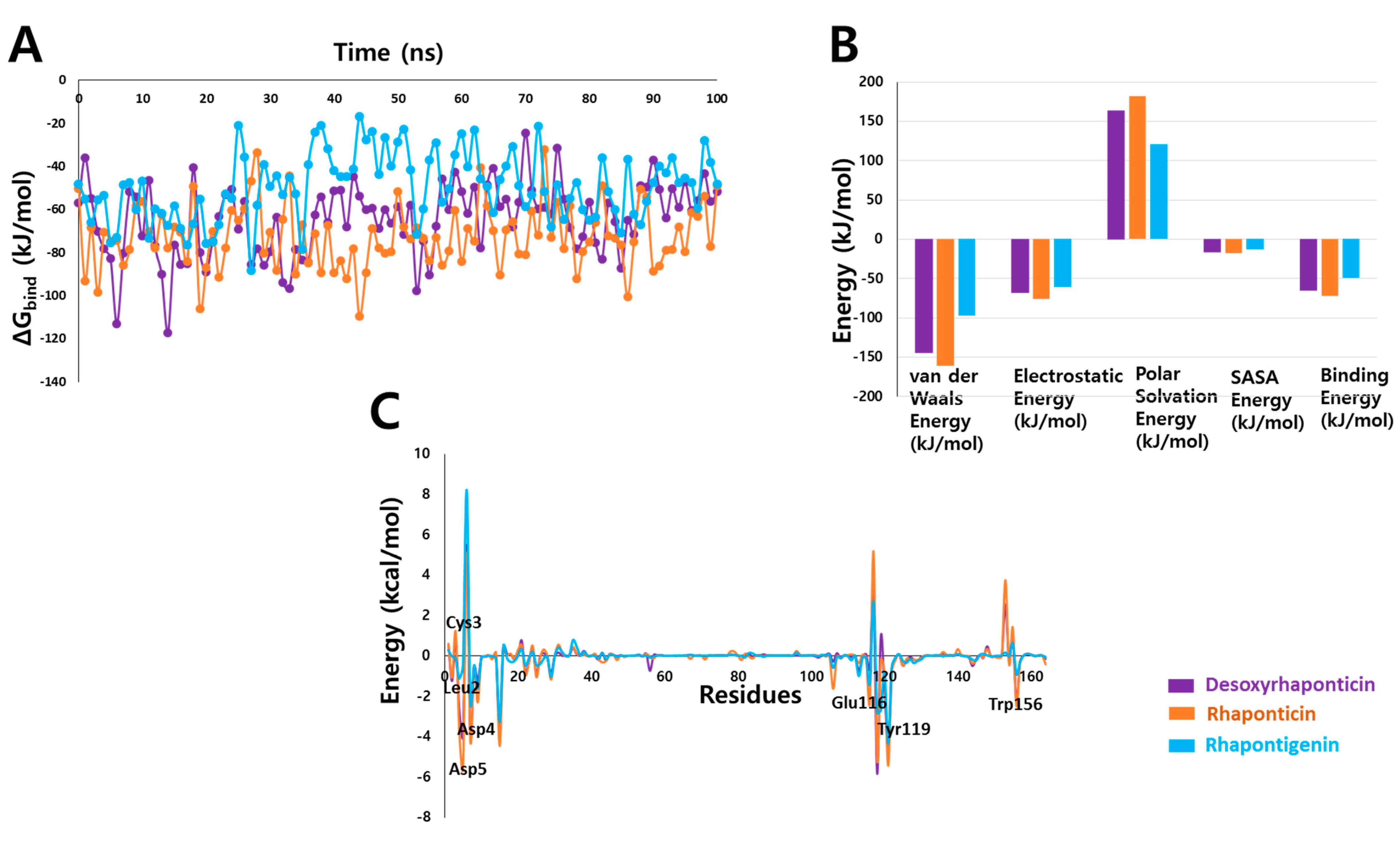
3.4. Binding Free Energy Analysis
3.5. Physicochemical Properties and ADMET Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Syed, Q.A.; Rashid, Z.; Ahmad, M.H.; Shukat, R.; Ishaq, A.; Muhammad, N.; Rahman, H.U. Nutritional and therapeutic properties of fenugreek (Trigonella foenum-graecum): A review. Int. J. Food. Prop. 2020, 23, 1777–1791. [Google Scholar] [CrossRef]
- Ozturk, M.; Hakeem, K.R.; Ashraf, M.; Ahmad, M.S.A. Global Perspectives on Underutilized Crops; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1–448. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, K.S.; Lee, E.K.; Park, N.C. Pd21-12 efficacy and safety of a mixed extract of trigonella foenum-graecum seed and lespedeza cuneata in the treatment of testosterone deficiency syndrome: A randomized, double-blind, placebo-controlled clinical trial. J. Urol. 2019, 201, e387. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ding, C.; Wang, X.; Wang, H.; Suo, Y. Using Response Surface Methodology to Optimize Countercurrent Chromatographic Separation of Polyphenol Compounds from Fenugreek (Trigonella foenumgraecum L.) Seeds. J. Liq. Chromatogr. Relat. Technol. 2014, 38, 29–35. [Google Scholar] [CrossRef]
- Li, G.; Luan, G.; He, Y.; Tie, F.; Wang, Z.; Suo, Y.; Ma, C.; Wang, H. Polyphenol stilbenes from fenugreek (Trigonella foenumgraecum L.) seeds improve insulin sensitivity and mitochondrial function in 3T3-L1 adipocytes. Oxid. Med. Cell. Longev. 2018, 2018, 7634362. [Google Scholar] [CrossRef]
- Kolodziejczyk-Czepas, J.; Czepas, J. Rhaponticin as an anti-inflammatory component of rhubarb: A minireview of the current state of the art and prospects for future research. Phytochem. Rev. 2019, 18, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Tian, W.; Wang, X.; Ma, X. Inhibitory effect of desoxyrhaponticin and rhaponticin, two natural stilbene glycosides from the Tibetan nutritional food Rheum tanguticum Maxim. ex Balf., on fatty acid synthase and human breast cancer cells. Food Funct. 2014, 5, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.A.; Iqbal, A.; Shahzad, S.A. Synthetic approaches toward stilbenes and their related structures. Mol. Divers. 2017, 21, 483–509. [Google Scholar] [CrossRef]
- De Filippis, B.; Ammazzalorso, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Anticancer Activity of Stilbene-Based Derivatives. ChemMedChem 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Pecyna, P.; Wargula, J.; Murias, M.; Kucinska, M. More Than Resveratrol: New Insights into Stilbene-Based Compounds. Biomolecules 2020, 10, 1111. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Dou, Q.P. The role of interleukin-2 receptor alpha in cancer. Front. Biosci. 2005, 10, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; He, J.; Zhou, W.; Shu, S.; Li, J.; Liu, W.; Deng, Y.; Lu, C.; Lin, S.; Ma, Y.; et al. High IL2RA mRNA expression is an independent adverse prognostic biomarker in core binding factor and intermediate-risk acute myeloid leukemia. J. Transl. Med. 2019, 17, 191. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.I.; Muroi, K.; Yamamoto, C.; Hatano, K.; Okazuka, K.; Sato, K.; Oh, I.; Ohmine, K.; Suzuki, T.; Ozawa, K. CD25 as an adverse prognostic factor in elderly patients with acute myeloid leukemia. Hematology 2017, 22, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilley, J.W.; Chen, L.; Fry, D.C.; Emerson, S.D.; Powers, G.D.; Biondi, D.; Varnell, T.; Trilles, R.; Guthrie, R.; Mennona, F.; et al. Identification of a small moleucle inhibitor of the IL-2/IL-2Rα receptor interaction which binds to IL-2. J. Am. Chem. Soc. 1997, 119, 7589–7590. [Google Scholar] [CrossRef]
- Braisted, A.C.; Oslob, J.D.; Delano, W.L.; Hyde, J.; McDowell, R.S.; Waal, N.; Yu, C.; Arkin, M.R.; Raimundo, B.C. Discovery of a potent small molecule IL-2 inhibitor through fragment assembly. J. Am. Chem. Soc. 2003, 125, 3714–3715. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.M.; Arkin, M.R. Small-molecule inhibitors of IL-2/IL-2R: Lessons learned and applied. Curr. Top. Microbiol. Immunol. 2011, 348, 25–59. [Google Scholar] [CrossRef] [Green Version]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Du, J.; Zhong, C.; Zhang, D.; Guo, H.; Guo, Y.; Ding, J. Structural basis of immunosuppression by the therapeutic antibody daclizumab. Cell Res. 2010, 20, 1361–1371. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet 2003, 52, 602–623. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 923. [Google Scholar] [CrossRef] [Green Version]
- Heitz, F.; Van Mau, N. Protein structural changes induced by their uptake at interfaces. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2002, 1597, 1–11. [Google Scholar] [CrossRef]
- Salentin, S.; Haupt, V.J.; Daminelli, S.; Schroeder, M. Polypharmacology rescored: Protein-ligand interaction profiles for remote binding site similarity assessment. Prog. Biophys. Mol. Biol. 2014, 116, 174–186. [Google Scholar] [CrossRef]
- Sawada, T.; Fedorov, D.G.; Kitaura, K. Role of the key mutation in the selective binding of avian and human influenza hemagglutinin to sialosides revealed by quantum-mechanical calculations. J. Am. Chem. Soc. 2010, 132, 16862–16872. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, 1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajander, T.; Kahn, P.C.; Passila, S.H.; Cohen, D.C.; Lehtiö, L.; Adolfsen, W.; Warwicker, J.; Schell, U.; Goldman, A. Buried charged surface in proteins. Structure 2000, 8, 1203–1214. [Google Scholar] [CrossRef]
- Rickert, M.; Wang, X.; Boulanger, M.J.; Goriatcheva, N.; Garcia, K.C. The structure of interleukin-2 complexed with its alpha receptor. Science 2005, 308, 1477–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lise, S.; Buchan, D.; Pontil, M.; Jones, D.T. Predictions of Hot Spot Residues at Protein-Protein Interfaces Using Support Vector Machines. PLoS ONE 2011, 6, e16774. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Bhati, A.P.; Zasada, S.J.; Coveney, P.V. Rapid, accurate, precise and reproducible ligand–protein binding free energy prediction. Interface Focus 2020, 10, 20200007. [Google Scholar] [CrossRef]
- Domínguez-Villa, F.X.; Durán-Iturbide, N.A.; Ávila-Zárraga, J.G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl)indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorg. Chem. 2021, 106, 104497. [Google Scholar] [CrossRef]
- Raschi, E.; Ceccarini, L.; De Ponti, F.; Recanatini, M. hERG-related drug toxicity and models for predicting hERG liability and QT prolongation. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1005–1021. [Google Scholar] [CrossRef]
- Franc, I.; Lipinski, A.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y. Enhanced pharmacokinetics and anti-tumor efficacy of PEGylated liposomal rhaponticin and plasma protein binding ability of rhaponticin. J. Nanosci. Nanotechnol. 2012, 12, 7677–7684. [Google Scholar] [CrossRef]
- Liang, X.; Sun, Y.; Zeng, W.; Liu, L.; Ma, X.; Zhao, Y.; Fan, J. Synthesis and biological evaluation of a folate-targeted rhaponticin conjugate. Bioorg. Med. Chem. 2013, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Huang, Y.W.; Oshima, K.; Yearsley, M.; Zhang, J.; Arnold, M.; Yu, J.; Wang, L.S. The immunomodulatory potential of natural compounds in tumor-bearing mice and humans. Crit. Rev. Food Sci. Nutr. 2019, 59, 992. [Google Scholar] [CrossRef] [PubMed]
- Bojadzic, D.; Buchwald, P. Toward Small-Molecule Inhibition of Protein–Protein Interactions: General Aspects and Recent Progress in Targeting Costimulatory and Coinhibitory (Immune Checkpoint) Interactions. Curr. Top. Med. Chem. 2018, 18, 674. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Hydrogen Bonds | Avg. Distance (nm) | π-Interactions | van der Waals Interactions |
|---|---|---|---|---|
| Desoxyrhaponticin | Asp6:OD2-H39 | 0.37 | Asp5, Pro7, Phe15, Ile118 | Leu2, Cys3, Glu116, Arg117, Tyr119 |
| His120:ND1-H40 | 0.52 | |||
| Phe121:HN-O7 | 0.28 | |||
| Rhaponticin | Asp6:OD1-H40 | 0.31 | Pro7, Phe15, Ile118 | Leu2, Cys3, Asp4, Asp5, Glu116, Tyr119, Trp156, |
| Arg117:O-H51 | 0.51 | |||
| His120:ND1-H41 | 0.42 | |||
| Phe121:HN-O7 | 0.24 | |||
| Rhapontigenin | Asp6:OD1-H33 | 0.44 | Phe15, Ile118, Tyr119 | Leu26, Arg117, His120, Phe121 |
| Ala17:HN-O2 | 0.35 |
| Parameters | Standard | Desoxyrhaponticin | Rhaponticin | Rhapontigenin | |
|---|---|---|---|---|---|
| Properties | Molecular Weight (Da) | ≤500 | 404.41 | 420.41 | 258.27 |
| LogP | ≤5 | 0.74 | 0.45 | 2.98 | |
| Rotatable bonds | ≤10 | 6 | 6 | 3 | |
| H-bond acceptors | ≤10 | 8 | 9 | 4 | |
| H-bond donors | ≤5 | 5 | 6 | 3 | |
| TPSA (Å2) | ≤140 | 128.84 | 149.07 | 69.92 | |
| Absorption | Human intestinal absorption (%) | >30%: perfectly absorbed | 58.29 | 50.00 | 91.20 |
| Permeability Caco-2 cell (Log Papp in 10−6 cm/s) | >0.90: well absorbed | −0.15 | −0.07 | 0.86 | |
| Distribution | BBB permeability | <−1: poorly distribute to the brain | −1.10 | −1.25 | −0.82 |
| CNS permeability | <−3: unable to penetrate CNS | −3.63 | −3.80 | −2.22 | |
| Metabolism | Cytochrome P450 inhibitors | - | No | No | Yes (CYP1A2, CYP2C9) |
| Excretion | Total clearance | - | 0.13 | 0.06 | 0.08 |
| Toxicity | AMES | - | No | No | No |
| hERG I/II inhibitor | - | No | No | No | |
| Hepatotoxicity | - | No | No | No | |
| Skin Sensitization | - | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkarni, A.M.; Parate, S.; Lee, G.; Kim, Y.; Jung, T.S.; Lee, K.W.; Ha, M.W. Computational Simulations Highlight the IL2Rα Binding Potential of Polyphenol Stilbenes from Fenugreek. Molecules 2022, 27, 1215. https://doi.org/10.3390/molecules27041215
Kulkarni AM, Parate S, Lee G, Kim Y, Jung TS, Lee KW, Ha MW. Computational Simulations Highlight the IL2Rα Binding Potential of Polyphenol Stilbenes from Fenugreek. Molecules. 2022; 27(4):1215. https://doi.org/10.3390/molecules27041215
Chicago/Turabian StyleKulkarni, Apoorva M., Shraddha Parate, Gihwan Lee, Yongseong Kim, Tae Sung Jung, Keun Woo Lee, and Min Woo Ha. 2022. "Computational Simulations Highlight the IL2Rα Binding Potential of Polyphenol Stilbenes from Fenugreek" Molecules 27, no. 4: 1215. https://doi.org/10.3390/molecules27041215
APA StyleKulkarni, A. M., Parate, S., Lee, G., Kim, Y., Jung, T. S., Lee, K. W., & Ha, M. W. (2022). Computational Simulations Highlight the IL2Rα Binding Potential of Polyphenol Stilbenes from Fenugreek. Molecules, 27(4), 1215. https://doi.org/10.3390/molecules27041215