
Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization


Abstract
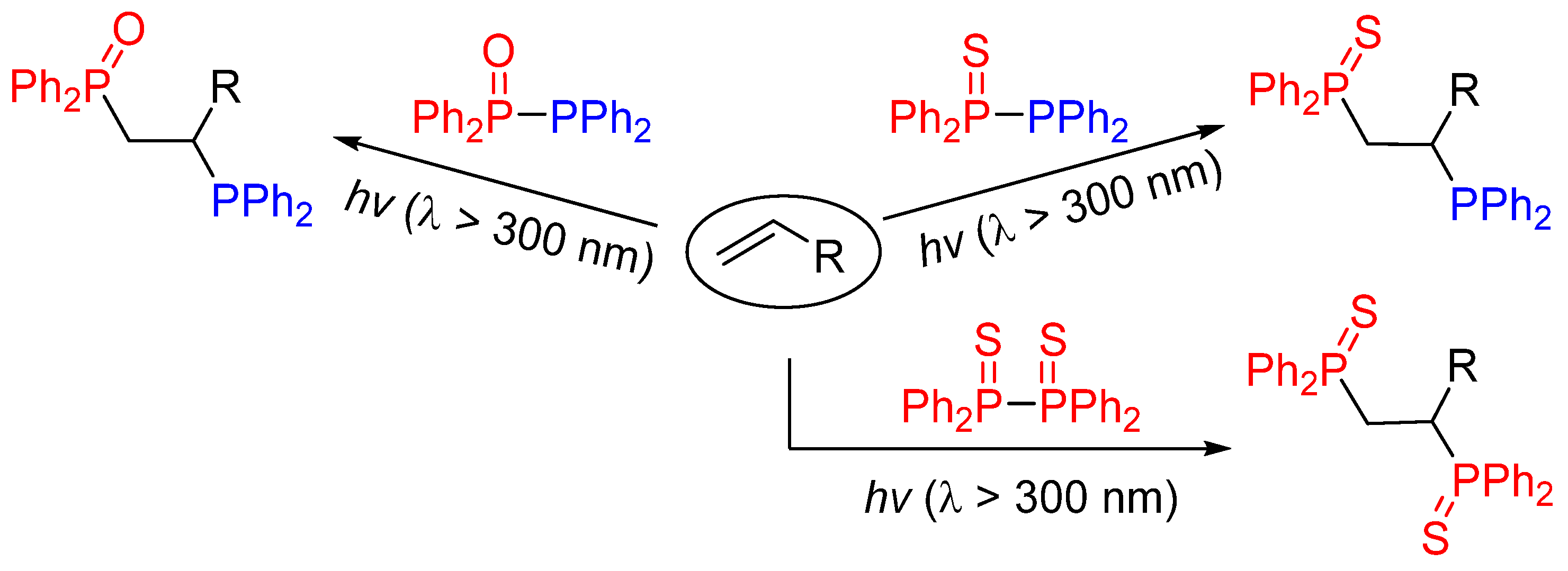
:1. Introduction
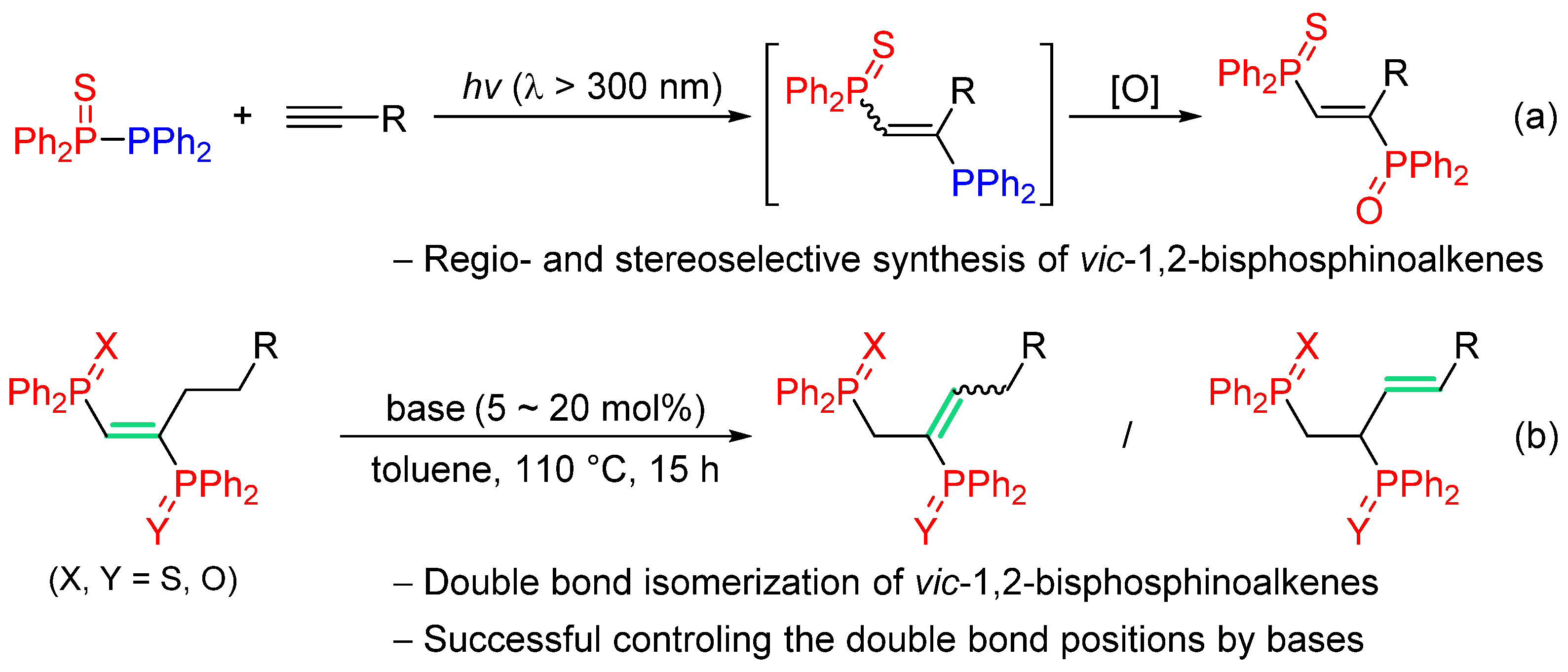
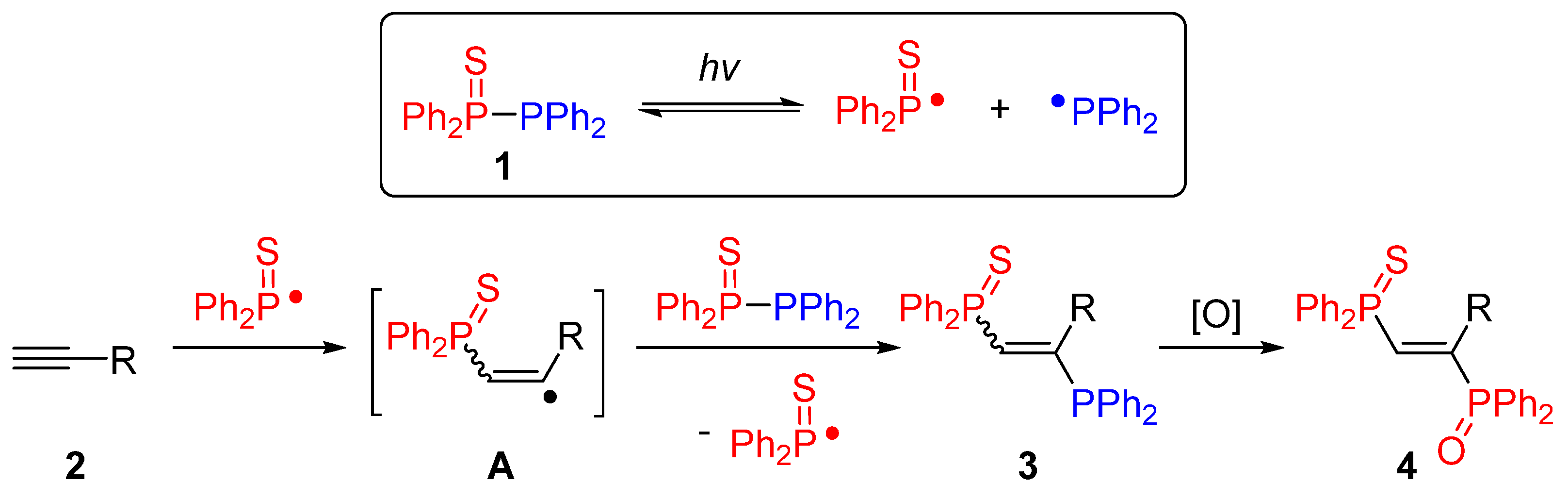
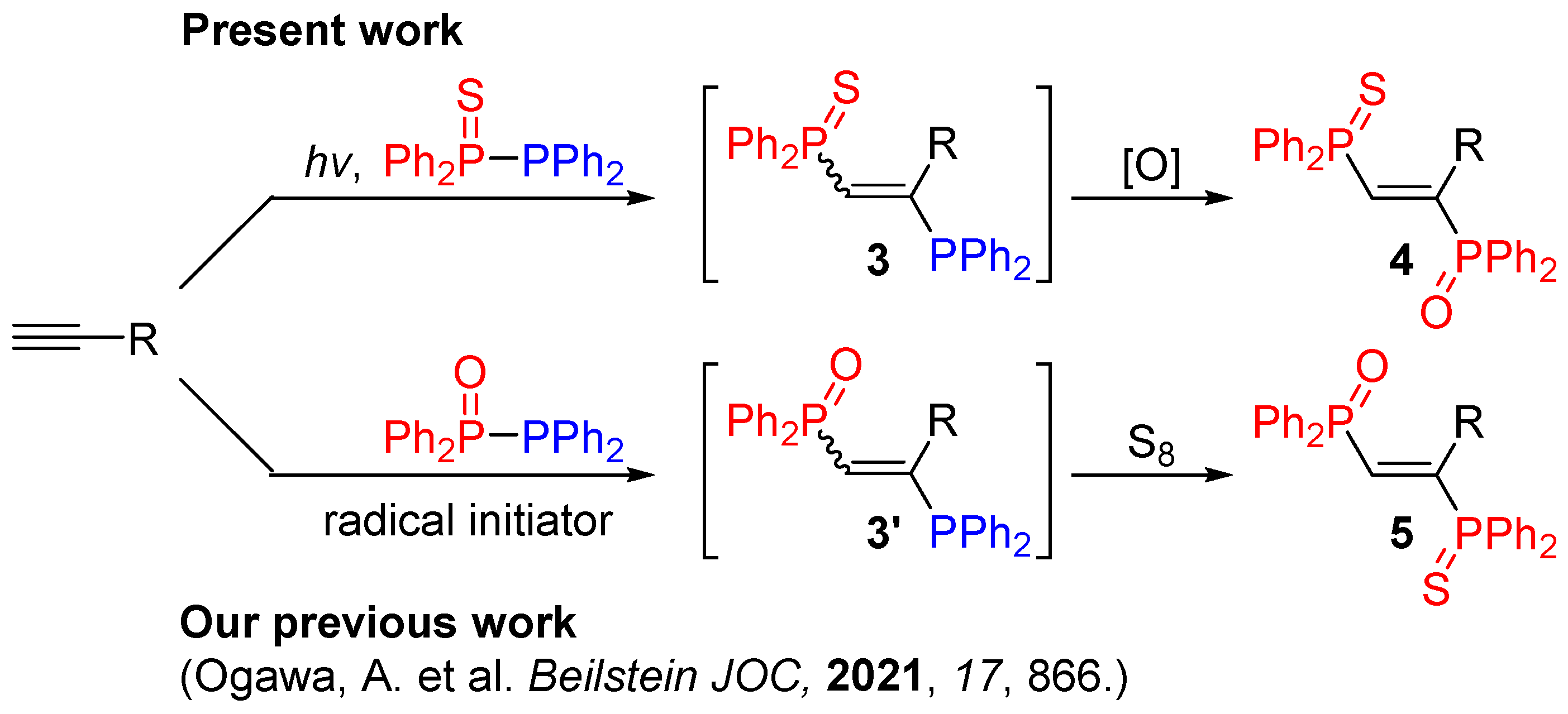
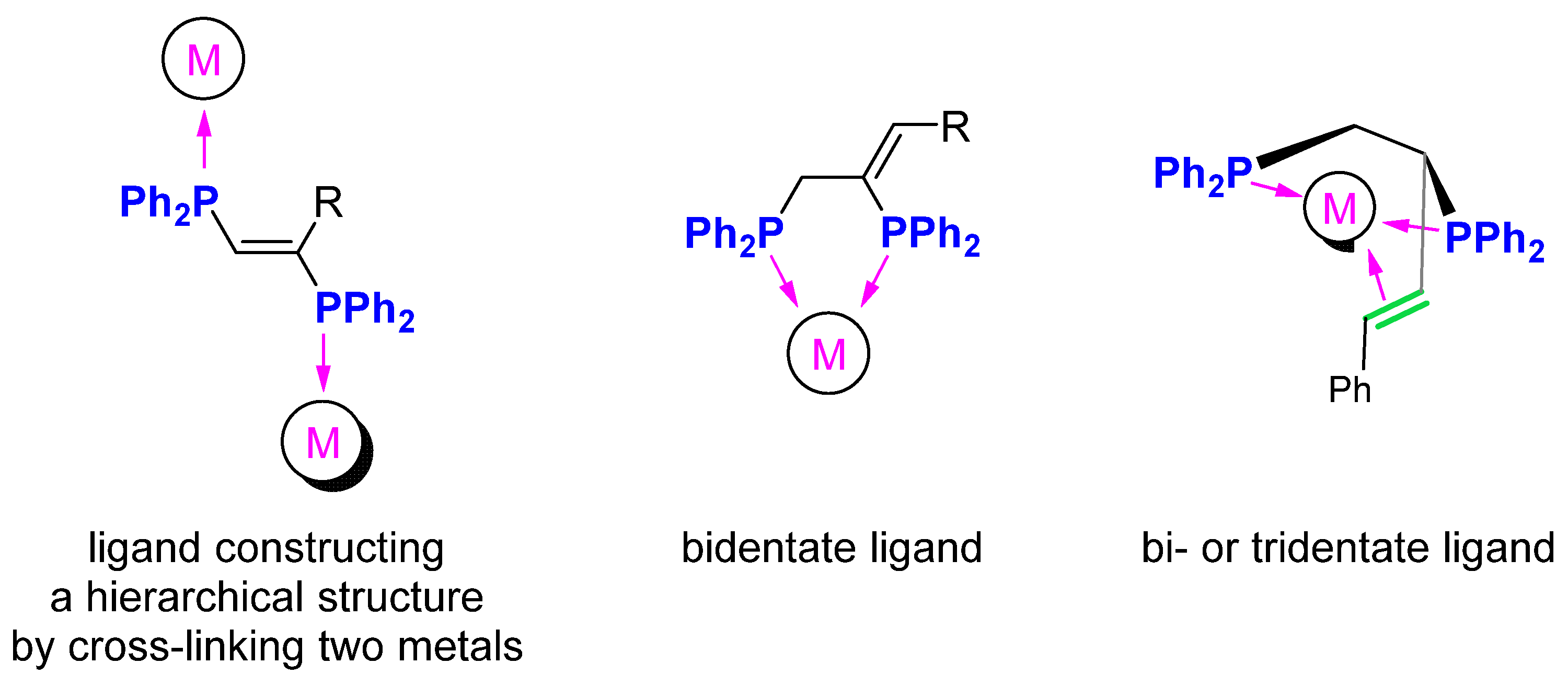
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Photoinduced Bisphosphination of Alkynes with Tetraphenyldiphosphine Monosulfide
3.3. Base-Catalyzed Double Bond Isomerization of vic-1,2-Bisphosphinoalkenes 5a
3.4. Base-Catalyzed Double Bond Isomerization of vic-1,2-Bisphosphinoalkenes 5b
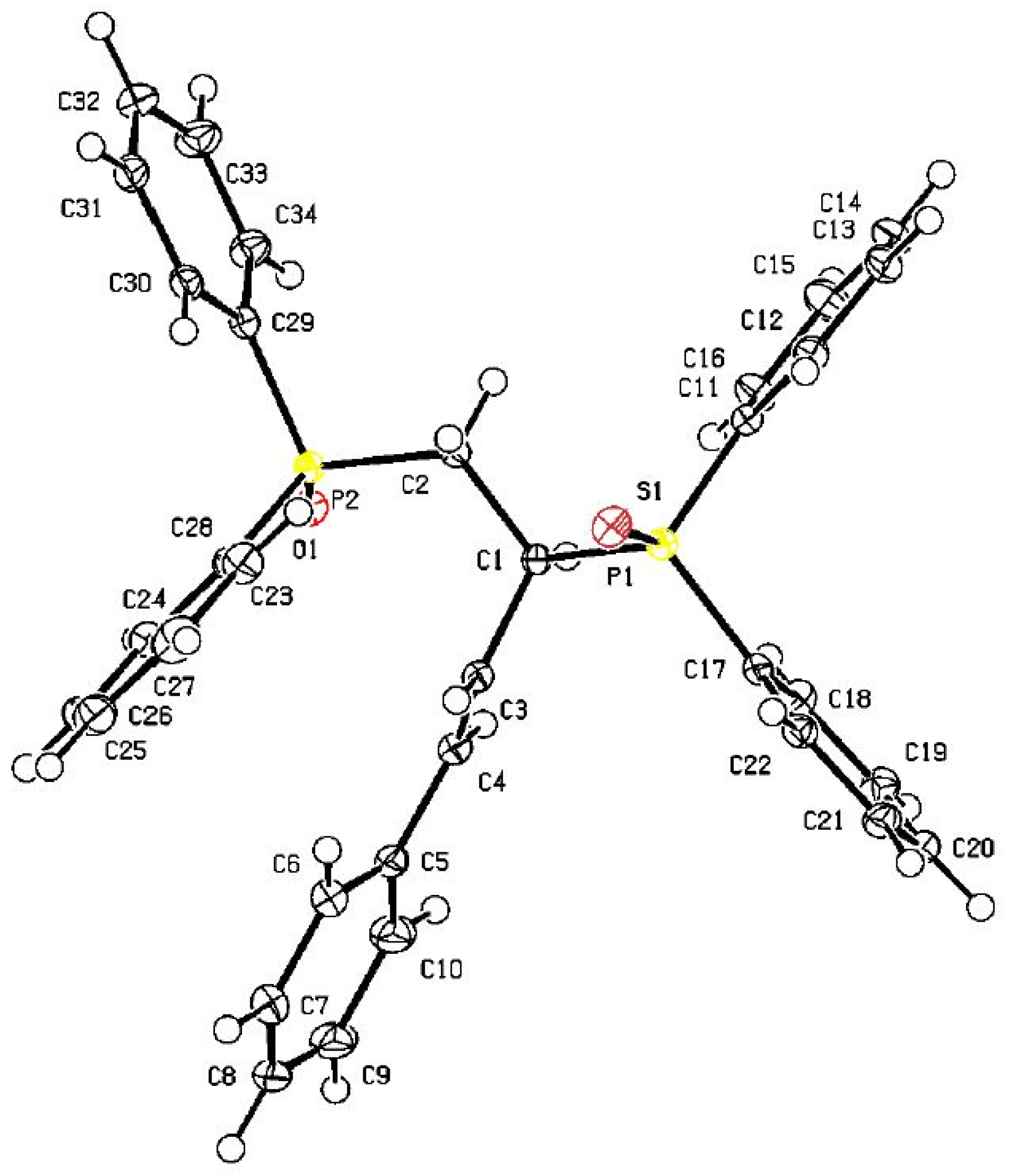
3.5. X-ray Diffraction Studies of 6b
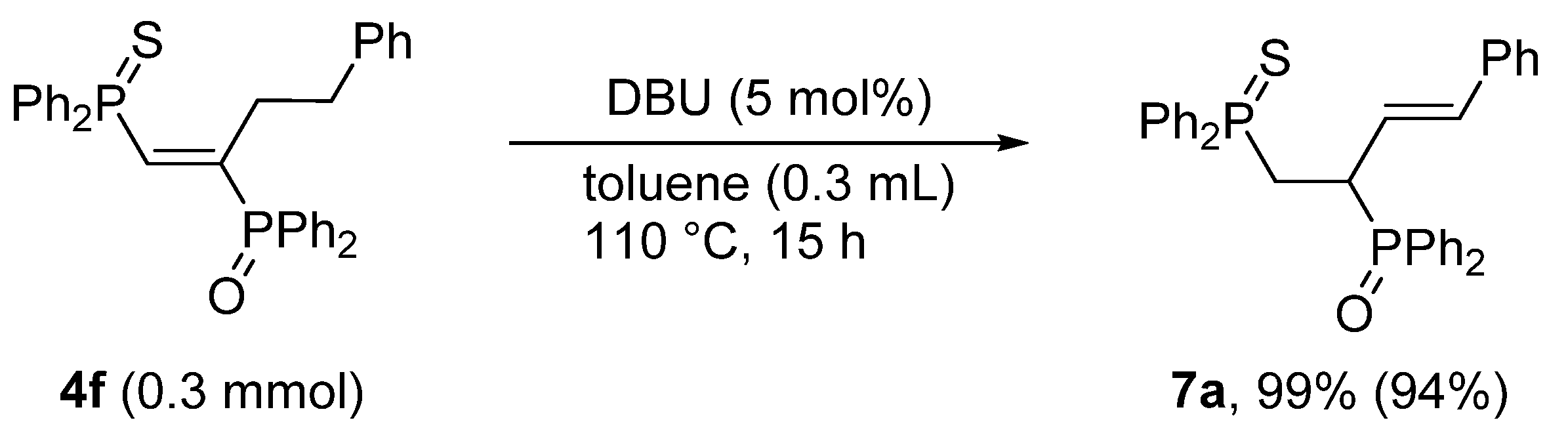
3.6. Base-Catalyzed Double Bond Isomerization of vic-1,2-Bisphosphinoalkenes 4f
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tamao, K.; Yamaguchi, S. Introduction to the chemistry of interelement linkage. J. Organomet. Chem. 2000, 611, 3–4. [Google Scholar] [CrossRef]
- Tokitoh, N. New aspects in the chemistry of low-coordinated inter-element compounds of heavier Group 15 elements. J. Organomet. Chem. 2000, 611, 217–227. [Google Scholar] [CrossRef]
- Hata, T.; Kitagawa, H.; Masai, H.; Kurahashi, T.; Shimizu, M.; Hiyama, T. Geminal difunctionalization of alkenylidene-type carbenoids by using interelement compounds. Angew. Chem. Int. Ed. 2001, 40, 790–792. [Google Scholar] [CrossRef]
- Beletskaya, I.; Moberg, C. Element–element additions to unsaturated carbon-carbon bonds catalyzed by transition metal complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Ogawa, A. Addition of X−Y reagents to alkenes, alkynes, and allenes. Compr. Organic Synth. II 2014, 4, 392–411. [Google Scholar]
- Iwasaki, M.; Nishihara, Y. Synthesis of multisubstituted olefins through regio- and stereoselective addition of interelement compounds having B–Si, B–B, and Cl–S bonds to alkynes, and subsequent cross-couplings. Chem. Rec. 2016, 16, 2031–2045. [Google Scholar] [CrossRef]
- Han, L.-B.; Choi, N.; Tanaka, M. Facile oxidative addition of the phosphorous–selenium bond to Pd(0) and Pt(0) complexes and development of Pd-catalyzed regio- and stereoselective selenophosphorylation of alkynes. J. Am. Chem. Soc. 1996, 118, 7000–7001. [Google Scholar] [CrossRef]
- Han, L.-B.; Tanaka, M. The first platinum(0)-catalyzed regio- and stereoselective thiosilylation of alkynes using disulfides and disilanes: A new strategy for introducing two different heteroatoms into carbon-carbon unsaturated bonds. J. Am. Chem. Soc. 1998, 120, 8249–8250. [Google Scholar] [CrossRef]
- Kondo, T.; Uenoyama, S.-Y.; Fujita, K.-I.; Mitsudo, T.-A. First transition-metal complex catalyzed addition of organic disulfides to alkenes enables the rapid synthesis of vicinal-Dithioethers. J. Am. Chem. Soc. 1999, 121, 482–483. [Google Scholar] [CrossRef]
- Han, L.-B.; Tanaka, M. Transition metal-catalysed addition reactions of H–heteroatom and inter-heteroatom bonds to carbon-carbon unsaturated linkages via oxidative additions. Chem. Commun. 1999, 5, 395–402. [Google Scholar] [CrossRef]
- Kondo, T.; Mitsudo, T.-A. Metal-catalyzed carbon–sulfur bond formation. Chem. Rev. 2000, 100, 3205–3220. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Oshima, K. (Eds.) Main Group Metals in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Sato, A.; Yorimitsu, H.; Oshima, K. Synthesis of (E)-1,2-diphosphanylethene derivatives from alkynes by radical addition of tetraorganodiphosphane generated in situ. Angew. Chem. Int. Ed. 2005, 44, 1694–1696. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yorimitsu, H.; Oshima, K. Radical phosphination of organic halides and alkyl imidazole-1-carbothioates. J. Am. Chem. Soc. 2006, 128, 4240–4241. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.P.; Ananikov, V.P. Addition reactions of E-E and E-H bonds to triple bond of alkynes catalyzed by Pd, Pt, and Ni complexes (E = S, Se). Pure Appl. Chem. 2007, 79, 1041–1056. [Google Scholar] [CrossRef]
- Wada, T.; Kondoh, A.; Yorimitsu, H.; Oshima, K. Intermolecular radical addition of alkylthio-and arylthiodiphenylphosphines to terminal alkynes. Org. Lett. 2008, 10, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yorimistu, H.; Oshima, K. Regio- and stereoselective synthesis of 1-aryl-1-thio-2-thiophosphinylethene derivatives via a radical process. Tetrahedron 2009, 65, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Beletskaya, I.P.; Ananikov, V.P. Transition-metal-catalyzed C–S, C–Se, and C–Te bond formation via cross-coupling and atom-economic addition reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef]
- Yorimitsu, H. Homolytic substitution at phosphorus for C-P bond formation in organic synthesis. Beilstein J. Org. Chem. 2013, 9, 1269–1277. [Google Scholar] [CrossRef]
- Wille, U. Radical cascades initiated by intermolecular radical addition to alkynes and related triple bond systems. Chem. Rev. 2013, 113, 813–853. [Google Scholar] [CrossRef]
- Okugawa, Y.; Hirano, K.; Miura, M. Copper-catalyzed vicinal diphosphination of styrenes: Access to 1,2-bis(diphenylphosphino)ethane-type bidentate ligands from olefins. Angew. Chem. Int. Ed. 2016, 55, 13558–13561. [Google Scholar] [CrossRef]
- Okugawa, Y.; Hirano, K.; Miura, M. Brønsted base mediated stereoselective diphosphination of terminal alkynes with diphosphanes. Org. Lett. 2017, 19, 2973–2976. [Google Scholar] [CrossRef] [PubMed]
- Otomura, N.; Okugawa, Y.; Hirano, K.; Miura, M. vic-Diphosphination of alkenes with silylphosphine under visible-light-promoted photoredox catalysis. Org. Lett. 2017, 19, 4802–4805. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Miura, M. Recent advances in diphosphination of alkynes and alkenes. Tetrahedron Lett. 2017, 58, 4317–4322. [Google Scholar] [CrossRef]
- Otomura, N.; Hirano, K.; Miura, M. Diphosphination of 1,3-dienes with diphosphines under visible-light-promoted photoredox catalysis. Org. Lett. 2018, 20, 7965–7968. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, S.-I.; Yamamoto, Y.; Ogawa, A. Catalytic synthesis of sulfur and phosphorus compounds via atom-economic reactions. Mendeleev Commun. 2020, 30, 129–138. [Google Scholar] [CrossRef]
- Escobedo, R.; Miranda, R.; Martínez, J. Infrared irradiation: Toward green chemistry, a review. Int. J. Mol. Sci. 2016, 17, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlowski, R.; Stanek, F.; Stodulski, M. Recent advances on metal-free, visible-light-induced catalysis for assembling nitrogen- and oxygen-based heterocyclic scaffolds. Molecules 2019, 24, 1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albano, G.; Decandia, G.; Capozzi, M.A.M.; Zappimbulso, N.; Punzi, A.; Farinola, G.M. Infrared irradiation-assisted solvent-free Pd-catalyzed (hetero)aryl-aryl coupling via C−H bond activation. ChemSusChem 2021, 14, 3391–3401. [Google Scholar] [CrossRef]
- Nomoto, A.; Higuchi, Y.; Kobiki, Y.; Ogawa, A. Synthesis of selenium compounds by free radical addition based on visible-light-activated Se-Se bond cleavage. Mini-Rev. Med. Chem. 2013, 13, 814–823. [Google Scholar] [CrossRef]
- Kawaguchi, S.-I.; Ogawa, A. Future trends in organophosphorus chemistry. Organophosphorus Chemistry: From Molecules to Applications; Iaroshenko, V., Ed.; Wiley-VCH: Weinheim, Germany, 2019; pp. 545–556. [Google Scholar]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Sekiguchi, M.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl diselenide to allenes. Tetrahedron Lett. 1990, 31, 5931–5934. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, H.; Yokoyama, K.; Masawaki, T.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl diselenide to acetylenes. J. Org. Chem. 1991, 56, 5721–5723. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Obayashi, R.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl ditelluride to acetylenes. J. Chem. Soc. Chem. Commun. 1991, 24, 1748–1750. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Obayashi, R.; Han, L.-B.; Kambe, N.; Sonoda, N. Photo-induced ditelluration of acetylenes with diphenyl ditelluride. Tetrahedron 1993, 49, 1177–1188. [Google Scholar] [CrossRef]
- Tran, C.C.; Kawaguchi, S.-I.; Sato, F.; Nomoto, A.; Ogawa, A. Photoinduced cyclizations of o-diisocyanoarenes with organic diselenides and thiols that afford chalcogenated quinoxalines. J. Org. Chem. 2020, 85, 7258–7266. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, S.-I.; Nagata, S.; Shirai, T.; Tsuchii, K.; Nomoto, A.; Ogawa, A. Photochemical behaviors of tetraphenyldiphosphine in the presence of alkynes. Tetrahedron Lett. 2006, 47, 3919–3922. [Google Scholar] [CrossRef]
- Sato, Y.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Highly selective phosphinylphosphination of alkenes with tetraphenyldiphosphine monoxide. Angew. Chem. Int. Ed. 2016, 55, 9700–9703. [Google Scholar] [CrossRef]
- Sato, Y.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Synthesis of bis(phosphanyl)alkane monosulfides by the addition of diphosphane monosulfides to alkenes under light. Chem. Eur. J. 2019, 25, 2295–2302. [Google Scholar] [CrossRef]
- Sato, Y.; Nishimura, M.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Reductive rearrangement of tetraphenyldiphosphine disulfide to trigger the bisthiophosphinylation of alkenes and alkynes. Chem. Eur. J. 2019, 25, 6797–6806. [Google Scholar] [CrossRef]
- Kawaguchi, S.-I.; Ogawa, A. Applications of diphosphines in radical reactions. Asian J. Org. Chem. 2019, 8, 1164–1173. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tanaka, R.; Ota, M.; Nishimura, M.; Tran, C.C.; Kawaguchi, S.-I.; Kodama, S.; Nomoto, A.; Ogawa, A. Photoinduced syntheses and reactivities of phosphorus-containing interelement compounds. J. Org. Chem. 2020, 85, 14708–14719. [Google Scholar] [CrossRef]
- Yoshimura, A.; Takamachi, Y.; Han, L.-B.; Ogawa, A. Organosulfide-catalyzed diboration of terminal alkynes under light. Chem. Eur. J. 2015, 21, 13930–13933. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Takamachi, Y.; Mihara, K.; Saeki, T.; Kawaguchi, S.-I.; Han, L.-B.; Nomoto, A.; Ogawa, A. Photoinduced metal-free diboration of alkynes in the presence of organophosphine catalysts. Tetrahedron 2016, 72, 7832–7838. [Google Scholar] [CrossRef]
- Ogawa, A.; Tanaka, H.; Yokoyama, H.; Obayashi, R.; Yokoyama, K.; Sonoda, N. A highly selective thioselenation of olefins using disulfide-diselenide mixed system. J. Org. Chem. 1992, 57, 111–115. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Doi, M.; Sonoda, N.; Hirao, T. A novel photoinduced thioselenation of allenes by use of a disulfide-diselenide binary system. J. Org. Chem. 1998, 63, 4277–4281. [Google Scholar] [CrossRef]
- Ogawa, A.; Ogawa, I.; Obayashi, R.; Umezu, K.; Doi, M.; Hirao, T. Highly selective thioselenation of vinylcyclopropanes with a (PhS)2-(PhSe)2 binary system and its application to thiotelluration. J. Org. Chem. 1999, 64, 86–92. [Google Scholar] [CrossRef]
- Tsuchii, K.; Ogawa, A. A highly selective photoinduced selenoperfluoroalkylation of terminal acetylenes by using a novel binary system of perfluoroalkyl iodide and diphenyl diselenide. Tetrahedron Lett. 2003, 44, 8777–8780. [Google Scholar] [CrossRef]
- Tsuchii, K.; Tsuboi, Y.; Kawaguchi, S.-I.; Takahashi, J.; Sonoda, N.; Nomoto, A.; Ogawa, A. Highly selective double chalcogenation of isocyanides with disulfide−diselenide mixed systems. J. Org. Chem. 2007, 72, 415–423. [Google Scholar] [CrossRef]
- Mitamura, T.; Tsuboi, Y.; Iwata, K.; Tsuchii, K.; Nomoto, A.; Sonoda, M.; Ogawa, A. Photoinduced thiotelluration of isocyanides by using a (PhS)2–(PhTe)2 mixed system, and its application to bisthiolation via radical cyclization. Tetrahedron Lett. 2007, 48, 5953–5957. [Google Scholar] [CrossRef]
- Shirai, T.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Photoinduced highly selective thiophosphination of alkynes using a (PhS)2/(Ph2P)2 binary system. Tetrahedron Lett. 2008, 49, 4043–4046. [Google Scholar] [CrossRef]
- Mitamura, T.; Iwata, K.; Ogawa, A. Photoinduced intramolecular cyclization of o-ethenylaryl isocyanides with organic disulfides mediated by diphenyl ditelluride. J. Org. Chem. 2011, 76, 3880–3887. [Google Scholar] [CrossRef]
- Tamai, T.; Nomoto, A.; Tsuchii, K.; Minamida, Y.; Mitamura, T.; Sonoda, M.; Ogawa, A. Highly selective perfluoroalkylchalcogenation of alkynes by the combination of iodoperfluoroalkanes and organic dichalcogenides upon photoirradiation. Tetrahedron 2012, 68, 10516–10522. [Google Scholar] [CrossRef]
- Littke, A.F.; Fu, G.C. Palladium-catalyzed coupling reactions of aryl chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Marsden, J.A.; Miller, J.J.; Shirtcliff, L.D.; Haley, M.M. Structure−property relationships of donor/acceptor-functionalized tetrakis(phenylethynyl)benzenes and bis(dehydrobenzoannuleno)benzenes. J. Am. Chem. Soc. 2005, 127, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.B.; Jones, A.S.; Thompson, A.L.; Paton, R.S.; Anderson, E.A. Ligand bite angle-dependent palladium-catalyzed cyclization of propargylic carbonates to 2-alkynyl azacycles or cyclic dienamides. Angew. Chem. Int. Ed. 2014, 53, 1915–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locascio, T.M.; Tunge, J.A. Palladium-catalyzed regiodivergent substitution of propargylic carbonates. Chem. Eur. J. 2016, 22, 18140–18146. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ishimaru, T.; Shiota, K.; Yamaguchi, Y. Bottleable NiCl2(dppe) as a catalyst for the Markovnikov-selective hydroboration of styrenes with bis(pinacolato)diboron. Chem. Commun. 2020, 56, 11701–11704. [Google Scholar] [CrossRef]
- Tran, D.P.; Sato, Y.; Yamamoto, Y.; Kawaguchi, S.-I.; Kodama, S.; Nomoto, A.; Ogawa, A. Highly regio-and stereoselective phosphinylphosphination of terminal alkynes with tetraphenyldiphosphine monoxide under radical conditions. Beilstein J. Org. Chem. 2021, 17, 866–872. [Google Scholar] [CrossRef]
- Lozano, E.; Nieuwenhuyzen, M.; James, S.L. Ring-opening polymerisation of silver–diphosphine [M2L3] coordination cages to give [M2L3]∞ coordination polymers. Chem. Eur. J. 2001, 7, 2644–2651. [Google Scholar] [CrossRef]
- DelNegro, A.S.; Woessner, S.M.; Sullivan, B.P.; Dattelbaum, D.M.; Schoonover, J.R. Stereospecific, unsymmetrical photosubstitution in a ligand-bridged dimer. Inorg. Chem. 2001, 40, 5056–5057. [Google Scholar] [CrossRef]
- Brandys, M.-C.; Puddephatt, R.J. Polymeric complexes of silver(I) with diphosphine ligands: Self-assembly of a puckered sheet network structure. J. Am. Chem. Soc. 2002, 124, 3946–3950. [Google Scholar] [CrossRef]
- Ghosh, S.; Mukherjee, P.S. Self-assembly of metallamacrocycles via a rigid phosphorus donor linker. Organometallics 2007, 26, 3362–3367. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
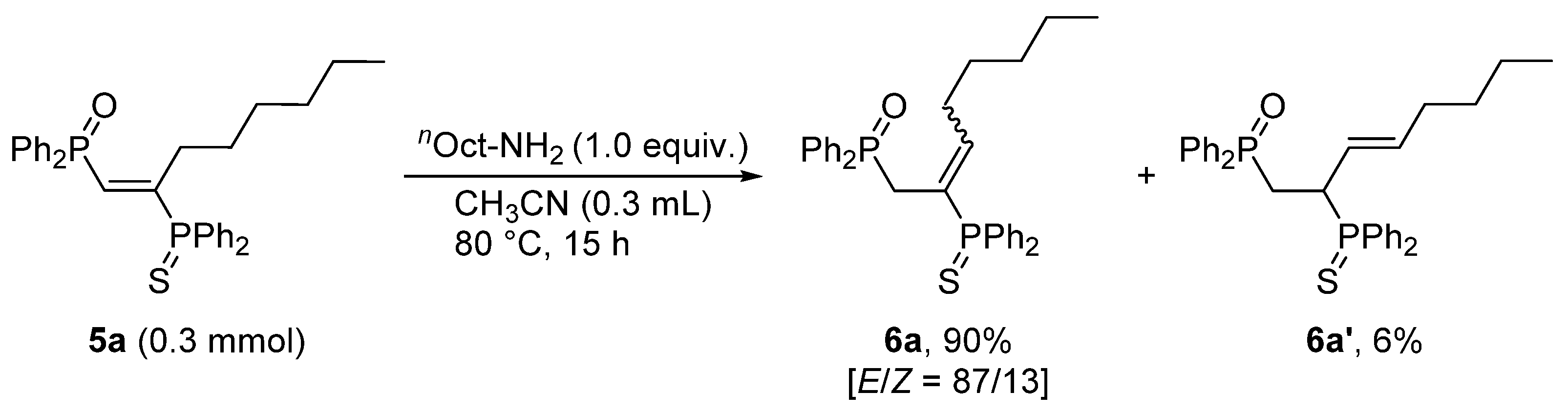{kind=link}
{kind=link}
{kind=link}
 | ||||
| Time (h) | Yield 3a (%) a | E-Selectivity (%) |  | |
| E | Z | |||
| 0.5 | 19 | 0.2 | 99 | |
| 1.0 | 28 | 1 | 97 | |
| 1.5 | 34 | 1 | 97 | |
| 2.0 | 39 | 1 | 98 | |
| 3.5 | 47 | 2 | 96 | |
| 6.0 | 52 | 3 | 95 | |
| 9.0 | 55 | 5 | 92 | |
| 13 | 56 | 7 | 89 | |
 | ||||
| Time (h) | Yield 3b (%) a | E-Selectivity (%) |  | |
| E | Z | |||
| 0.5 | 41 | 2 | 95 | |
| 1.0 | 51 | 3 | 94 | |
| 2.0 | 65 | 8 | 89 | |
| 4.5 | 64 | 14 | 82 | |
| 6.0 | 61 | 19 | 76 | |
| 12 | 52 | 32 | 62 | |
 | |||||
| Entry | Alkyne 2 | 3 | Yield (%) a [E/Z] | Product 4 | Yield (%) b [E/Z] |
| 1 c |  |  | 57 [91/9] |  | 63 [90/10] |
| 2 d |  |  | 67 [90/10] |  | 62 [90/10] |
| 3 c |  |  | 58 [91/9] |  | 48 [100/0] |
| 4 c |  |  | 41 [90/10] |  | 42 [89/11] |
| 5 c |  |  | 50 [90/10] |  | 57 [90/10] |
| 6 c |  |  | 56 [89/11] |  | 50 [98/2] |
| 7 c |  | complex mixture | - | - | - |
| 8 c |  |  | overlapped |  | 54 [91/9] |
| 9 d |  |  | 62 e |  | 65 [99/1] |
| 10 d |  |  | 67 [90/10] |  | 64 [100/0] |
 | |||||
| Entry | Base (mol%) | Solvent | Temp. (°C) | Yield (%) a | |
| 6a [E/Z] | 6a’ | ||||
| 1 | nOct–NH2 (100) | CH3CN | 80 | 90 [87/13] | 6 |
| 2 | nOct–NH2 (20) | Toluene | 110 | 90 [87/13] (42) | 6 |
| 3 b | nOct–NH2 (5) | Toluene | 110 | 29 [83/17] | N. D. |
| 4 b | DBU (5) | Toluene | 110 | 59 [88/12] | 39 |
| 5 | DBU (40) | Toluene | 110 | 15 [93/7] | 83 (56) |
| 6 | - | Toluene | 110 | N. D. | N. D. |
 | ||||
| Entry | Base (mol%) | Solvent | Temp. (°C) | Yield 6b (%) a |
| 1 | nOct-NH2 (100) | CH3CN | 80 | 74 |
| 2 | nBu-NH2 (100) | CH3CN | 80 | 62 |
| 3 | iPr2NH (100) | CH3CN | 80 | 4 |
| 4 | iPr2NEt (100) | CH3CN | 80 | trace |
| 5 | Et3N (100) | CH3CN | 80 | 2 |
| 6 | DBU (100) | CH3CN | 80 | 94 |
| 7 | DMAP (100) | CH3CN | 80 | 23 |
| 8 | Cs2CO3 (100) | CH3CN | 80 | trace |
| 9 | nOct-NH2 (20) | Toluene | 110 | 94 |
| 10 | DBU (20) | Toluene | 110 | 98 |
| 11 b | DBU (5) | Toluene | 110 | 99 (95) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, Y.; Tanaka, R.; Kodama, S.; Nomoto, A.; Ogawa, A. Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization. Molecules 2022, 27, 1284. https://doi.org/10.3390/molecules27041284
Yamamoto Y, Tanaka R, Kodama S, Nomoto A, Ogawa A. Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization. Molecules. 2022; 27(4):1284. https://doi.org/10.3390/molecules27041284
Chicago/Turabian StyleYamamoto, Yuki, Ryo Tanaka, Shintaro Kodama, Akihiro Nomoto, and Akiya Ogawa. 2022. "Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization" Molecules 27, no. 4: 1284. https://doi.org/10.3390/molecules27041284