
The Pathological G51D Mutation in Alpha-Synuclein Oligomers Confers Distinct Structural Attributes and Cellular Toxicity

 , , ,
, , ,
Abstract
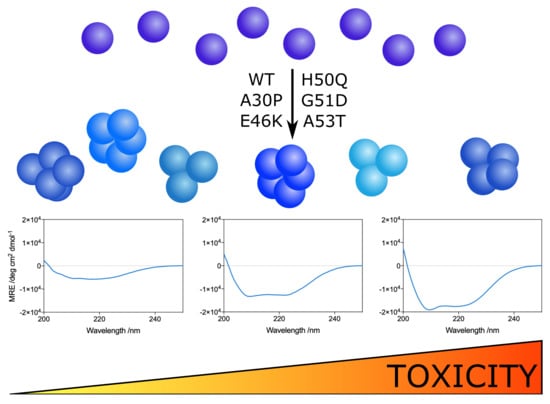
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
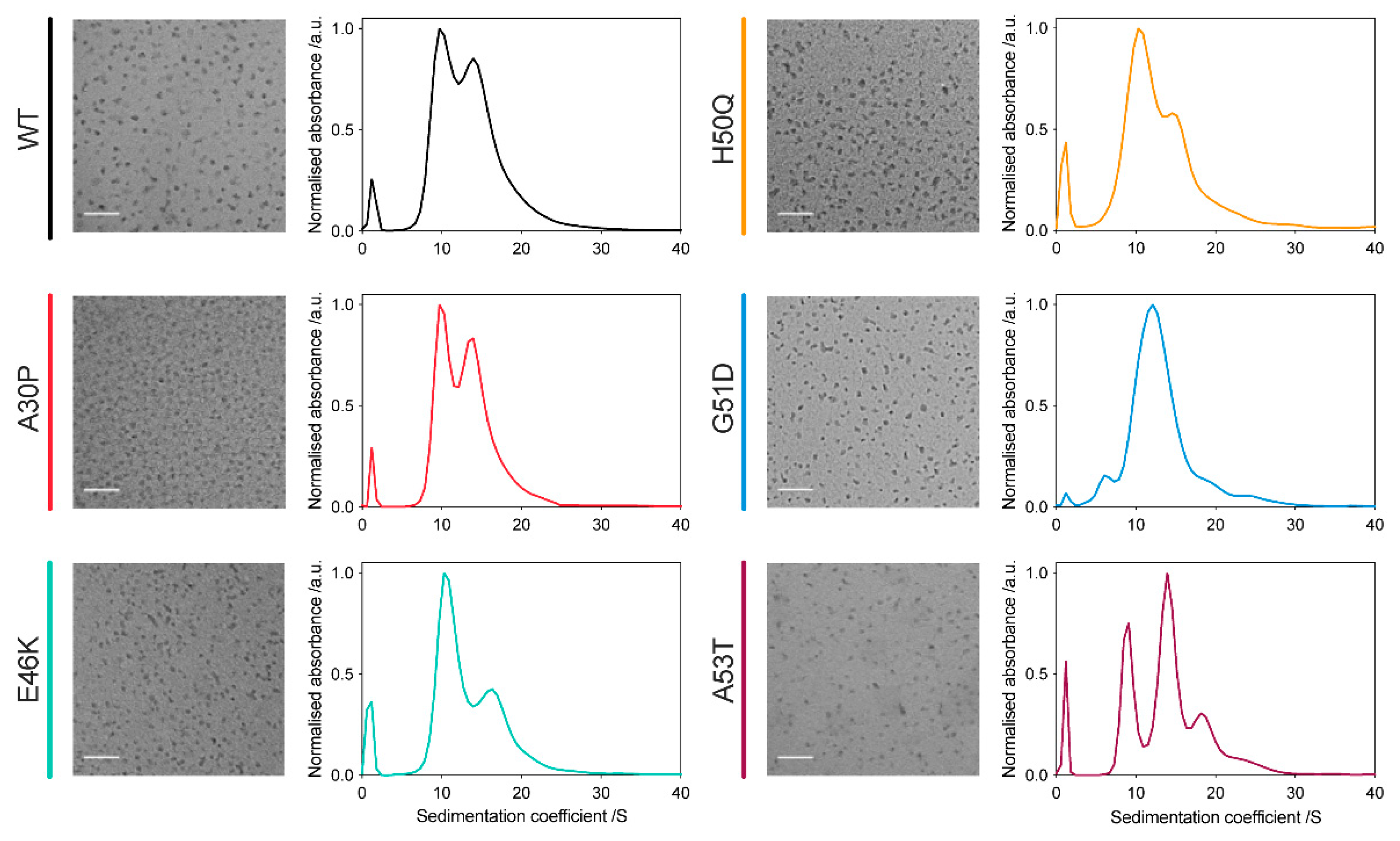
2.1. All α-Synuclein Variants Form Oligomers with Similar Size and Morphology
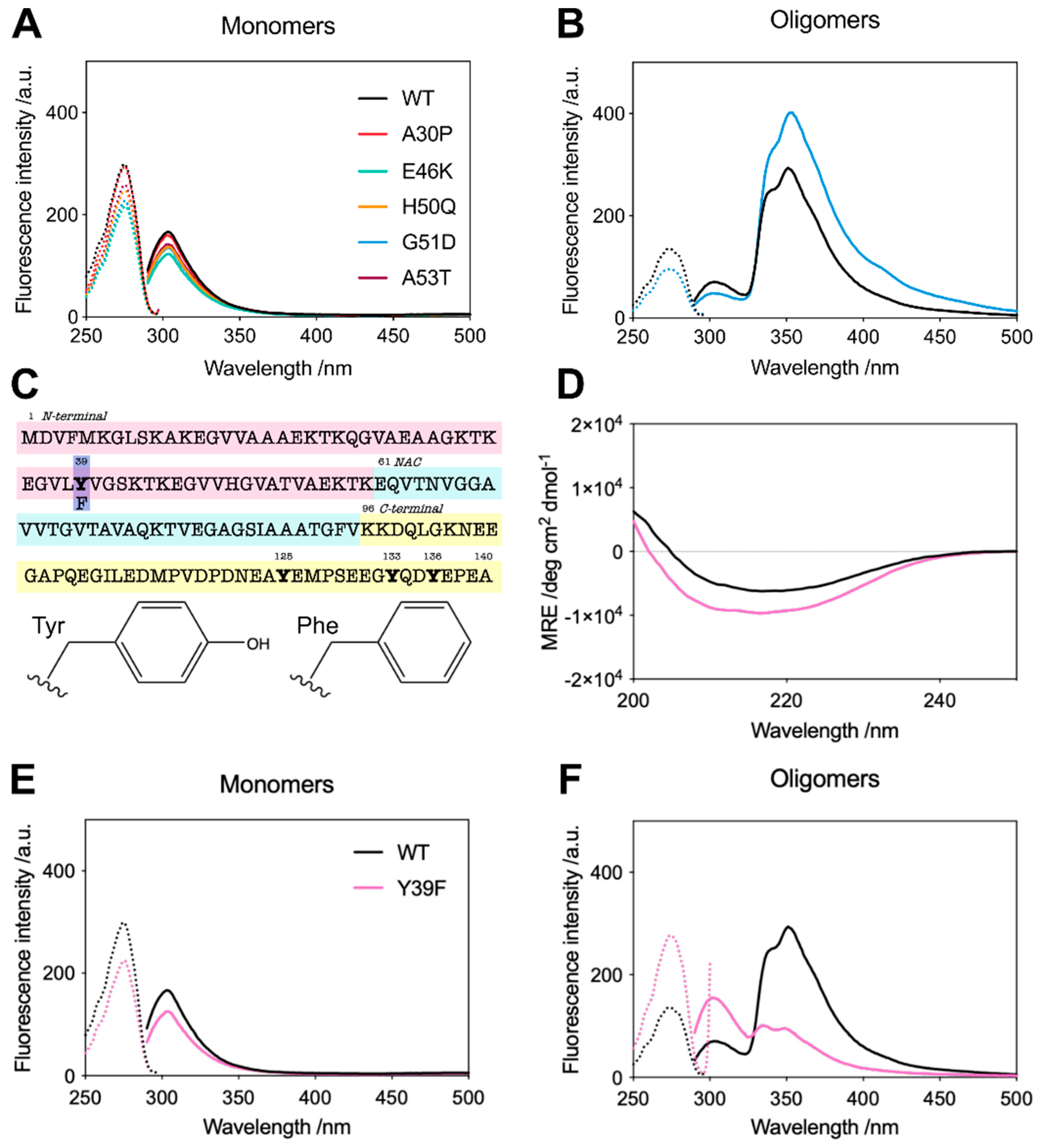
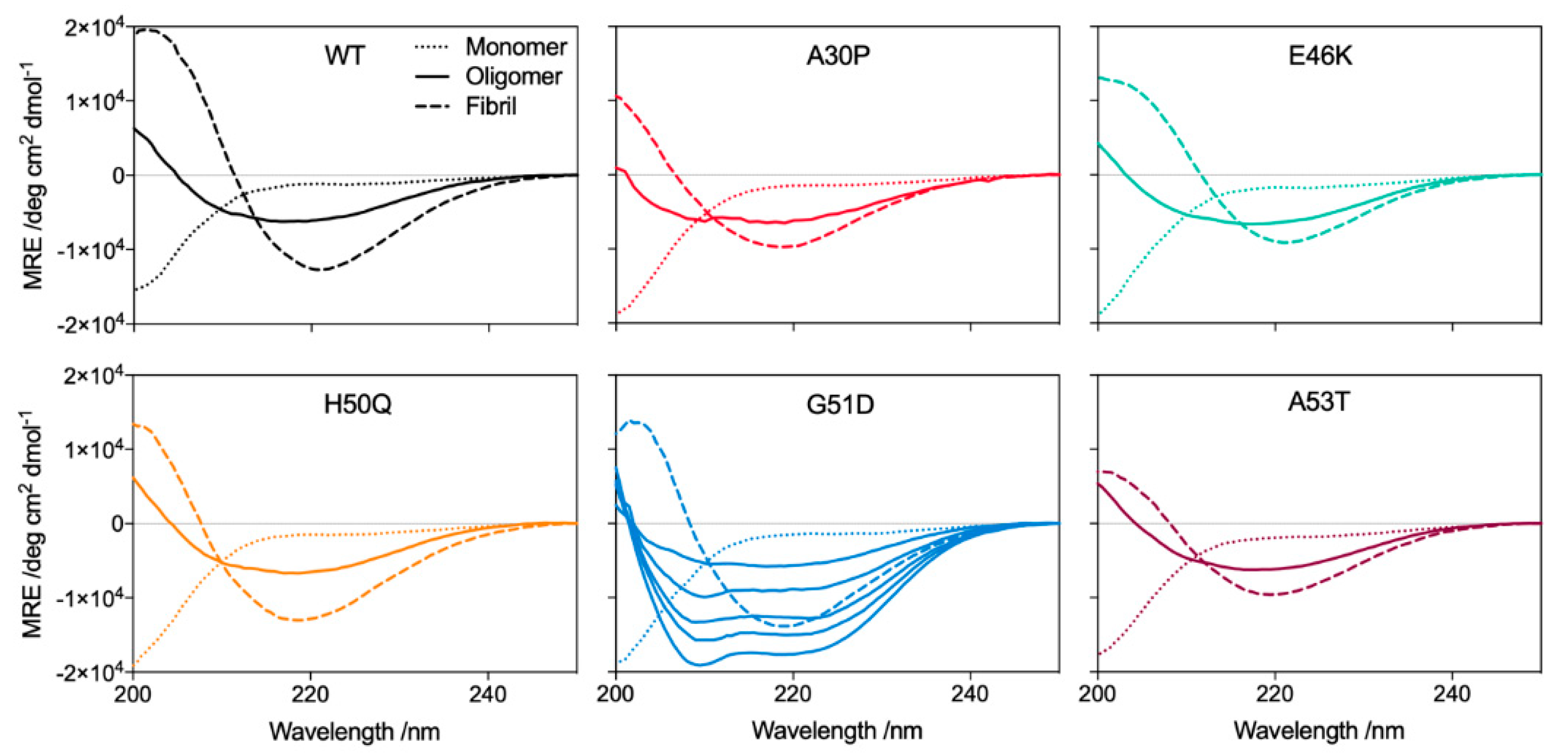
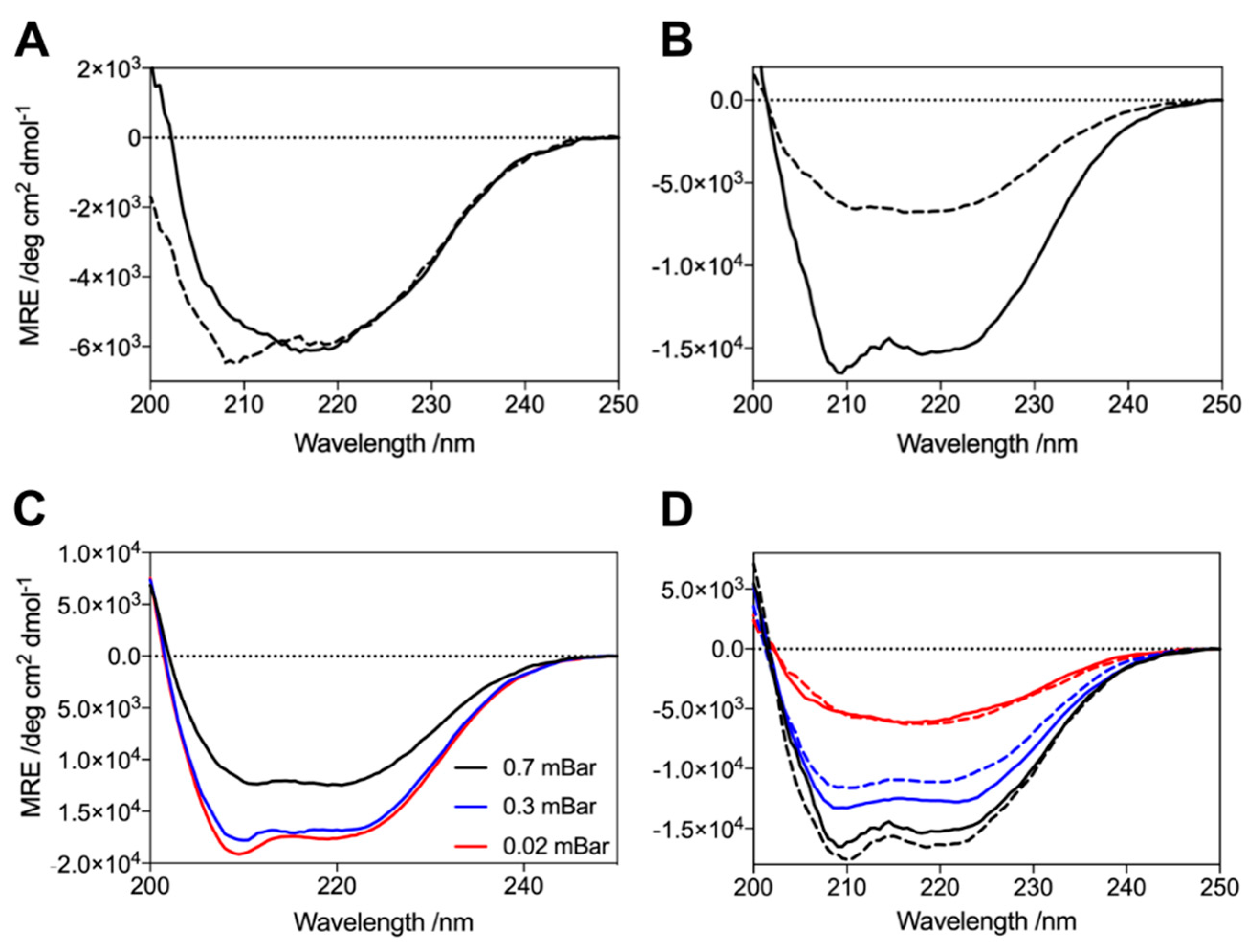
2.2. G51D Oligomers Display Marked Structural Differences, including Increased α-Helical Content and Decreased Surface Hydrophobicity
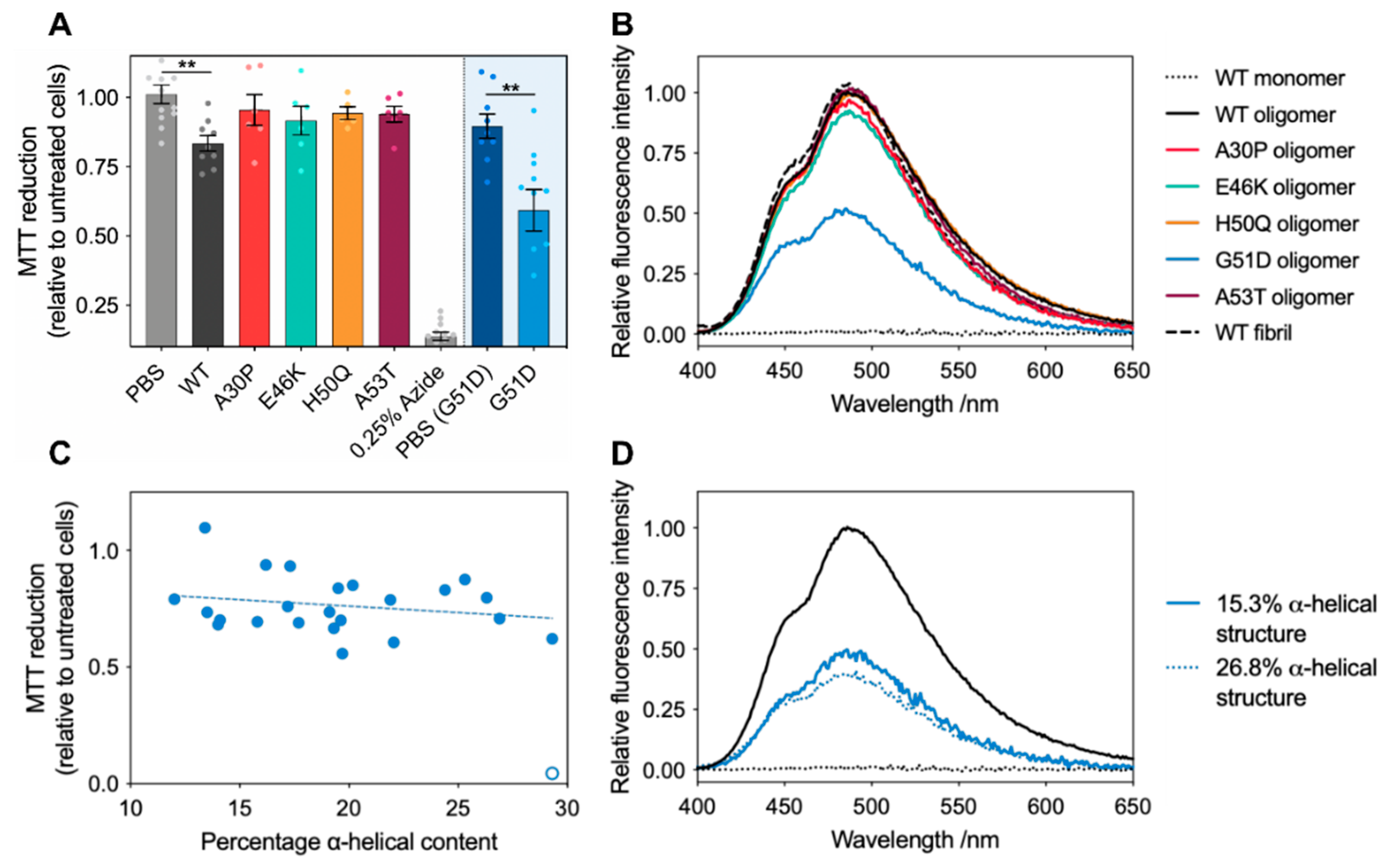
2.3. G51D Oligomer Polymorphs with High Helical Content Exhibit the Highest Cytotoxicity
3. Materials and Methods
3.1. Preparation of Oligomers
3.2. Bicinchoninic Acid Assay
3.3. ANS Binding
3.4. Circular Dichroism Spectroscopy
3.5. Dot Blot Analysis
3.6. FTIR Spectroscopy
3.7. Intrinsic Fluorescence Spectroscopy
3.8. MTT Cell Viability Assay
3.9. Native Polyacrylamide Gel Electrophoresis
3.10. Transmission Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat. Comm. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. Alpha-synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.T.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Bemporad, F.; Chiti, F. Protein misfolded oligomers: Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 2012, 19, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar] [CrossRef]
- Hartl, F.U. Protein misfolding diseases. Annu Rev. Biochem 2017, 86, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Giampà, M.; Amundarain, M.J.; Herrera, M.G.; Tonali, N.; Dodero, V.I. Implementing complementary approaches to shape the mechanism of α-synuclein oligomerization as a model of amyloid aggregation. Molecules 2021, 27, 88. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, M.; Lorenzen, N.; Otzen, D. Interactions between misfolded protein oligomers and membranes: A central topic in neurodegenerative diseases? Biochim. Biophys. Acta 2015, 1848, 1897–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musteikyte, G.; Jayaram, A.K.; Xu, C.K.; Vendruscolo, M.; Krainer, G.; Knowles, T.P.J. Interactions of alpha-synuclein oligomers with lipid membranes. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183536. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T. The impact of the E46K mutation on the properties of α-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.-L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.-J.; et al. The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Lemkau, L.R.; Comellas, G.; Kloepper, K.D.; Woods, W.S.; George, J.M.; Rienstra, C.M. Mutant protein A30P α-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J. Biol. Chem. 2012, 287, 11526–11532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Bodner, C.R.; Maltsev, A.S.; Dobson, C.M.; Bax, A. Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 2010, 49, 862–871. [Google Scholar] [CrossRef]
- Bussell, R.; Eliezer, D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry 2004, 43, 4810–4818. [Google Scholar] [CrossRef]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Anthony Crowther, R.; Goedert, M. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; De Simone, A.; Arosio, P.; Vendruscolo, M.; Veglia, G.; Dobson, C.M. Structural ensembles of membrane-bound α-synuclein reveal the molecular determinants of synaptic vesicle affinity. Sci. Rep. 2016, 6, 27125. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. α-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human α-synuclein and Parkinson’s disease variants with phospholipids: Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, A.N.D.; Lindhoud, S.; Semerdzhiev, S.A.; Claessens, M.M.A.E.; Subramaniam, V. Oligomers of Parkinson’s disease-related α-synuclein mutants have similar structures but distinctive membrane permeabilization properties. Biochemistry 2015, 54, 3142–3150. [Google Scholar] [CrossRef]
- Ysselstein, D.; Joshi, M.; Mishra, V.; Griggs, A.M.; Asiago, J.M.; McCabe, G.P.; Stanciu, L.A.; Post, C.B.; Rochet, J.-C. Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol. Dis. 2015, 79, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, E.R.; Rhoades, E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, M.-B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovicic, A.; Kiely, A.; Holton, J.L.; Lee, S.-J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Mondal, M.; Mohite, G.M.; Singh, P.K.; Ranjan, P.; Anoop, A.; Ghosh, S.; Jha, N.N.; Kumar, A.; Maji, S.K. The Parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry 2013, 52, 6925–6927. [Google Scholar] [CrossRef]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.Y. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Uversky, V.N.; Fink, A.L. Conformational behavior of human α-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology 2002, 23, 553–567. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Paslawski, W.; Mysling, S.; Thomsen, K.; Jørgensen, T.J.D.; Otzen, D.E. Co-existence of two different α-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angew. Chem. Int. Ed. 2014, 53, 7560–7563. [Google Scholar] [CrossRef]
- Tosatto, L.; Horrocks, M.H.; Dear, A.J.; Knowles, T.P.J.; Dalla Serra, M.; Cremades, N.; Dobson, C.M.; Klenerman, D. Single-molecule FRET studies on alpha-synuclein oligomerization of Parkinson’s disease genetically related mutants. Sci. Rep. 2015, 5, 16696. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, F.G.; Hampton, P.D.; Jones, B. Characteristics of tyrosinate fluorescence emission in α and β-purothionins. Biochemistry 1984, 23, 6690–6697. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.G.; Lynn, K.R.; Krajcarski, D.T.; Rayner, D.M. Tyrosinate fluorescence maxima at 345 nm in proteins lacking tryptophan at pH 7. FEBS Lett. 1978, 94, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Camino, J.D.; Gracia, P.; Chen, S.W.; Sot, J.; de la Arada, I.; Sebastian, V.; Arrondo, J.L.R.; Goni, F.M.; Dobson, C.M.; Cremades, N. The extent of protein hydration dictates the preference for heterogeneous or homogeneous nucleation generating either parallel or antiparallel beta-sheet alpha-synuclein aggregates. Chem. Sci. 2020, 11, 11902–11914. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, B.; Kumita, J.R.; Barros, T.P.; Esbjörner, E.K.; Luheshi, L.M.; Crowther, D.C.; Wilson, M.R.; Dobson, C.M.; Favrin, G.; Yerbury, J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010, 5, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Oma, Y.; Kino, Y.; Toriumi, K.; Sasagawa, N.; Ishiura, S. Interactions between homopolymeric amino acids (HPAAs). Protein Sci. 2007, 16, 2195–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Stefanovic, A.N.D.; Stöckl, M.T.; Claessens, M.M.A.E.; Subramaniam, V. Alpha-synuclein oligomers distinctively permeabilize complex model membranes. FEBS J. 2014, 281, 2838–2850. [Google Scholar] [CrossRef] [Green Version]
- Apetri, M.M.; Maiti, N.C.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Secondary structure of α-synuclein oligomers: Characterization by Raman and Atomic Force Microscopy. J. Mol. Biol. 2006, 355, 63–71. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Salinas, N.; Colletier, J.-P.; Moshe, A.; Landau, M. Extreme amyloid polymorphism in Staphylococcus aureus virulent PSMα peptides. Nat. Commun. 2018, 9, 3512. [Google Scholar] [CrossRef] [Green Version]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.-P.; Landau, M. The cytotoxic Staphylococcus aureus PSMα reveals a cross-a amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.K.; Castellana-Cruz, M.; Chen, S.W.; Du, Z.; Meisl, G.; Levin, A.; Mannini, B.; Itzhaki, L.S.; Knowles, T.P.J.; Dobson, C.M.; et al. The Pathological G51D Mutation in Alpha-Synuclein Oligomers Confers Distinct Structural Attributes and Cellular Toxicity. Molecules 2022, 27, 1293. https://doi.org/10.3390/molecules27041293
Xu CK, Castellana-Cruz M, Chen SW, Du Z, Meisl G, Levin A, Mannini B, Itzhaki LS, Knowles TPJ, Dobson CM, et al. The Pathological G51D Mutation in Alpha-Synuclein Oligomers Confers Distinct Structural Attributes and Cellular Toxicity. Molecules. 2022; 27(4):1293. https://doi.org/10.3390/molecules27041293
Chicago/Turabian StyleXu, Catherine K., Marta Castellana-Cruz, Serene W. Chen, Zhen Du, Georg Meisl, Aviad Levin, Benedetta Mannini, Laura S. Itzhaki, Tuomas P. J. Knowles, Christopher M. Dobson, and et al. 2022. "The Pathological G51D Mutation in Alpha-Synuclein Oligomers Confers Distinct Structural Attributes and Cellular Toxicity" Molecules 27, no. 4: 1293. https://doi.org/10.3390/molecules27041293