
Intriguing Chloride: Involvement of Chloride Ions in Proton Transfers


Abstract
:1. Introduction
2. Results and Discussion
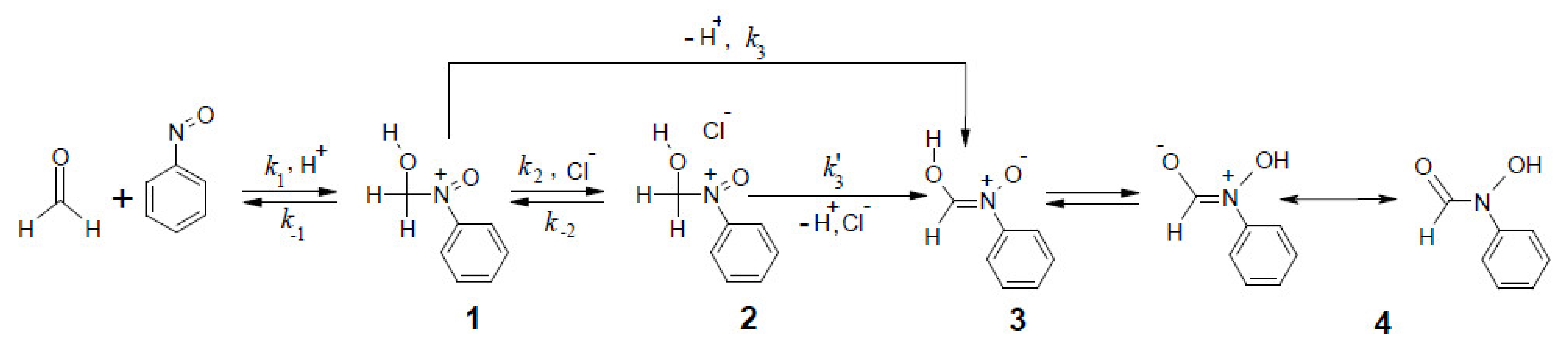
2.1. The Mechanism of the Reaction
2.2. The Effect of the Cosolvent on the Rate of Reaction
2.3. The Case of Chloride Ions and the Ion Pairs
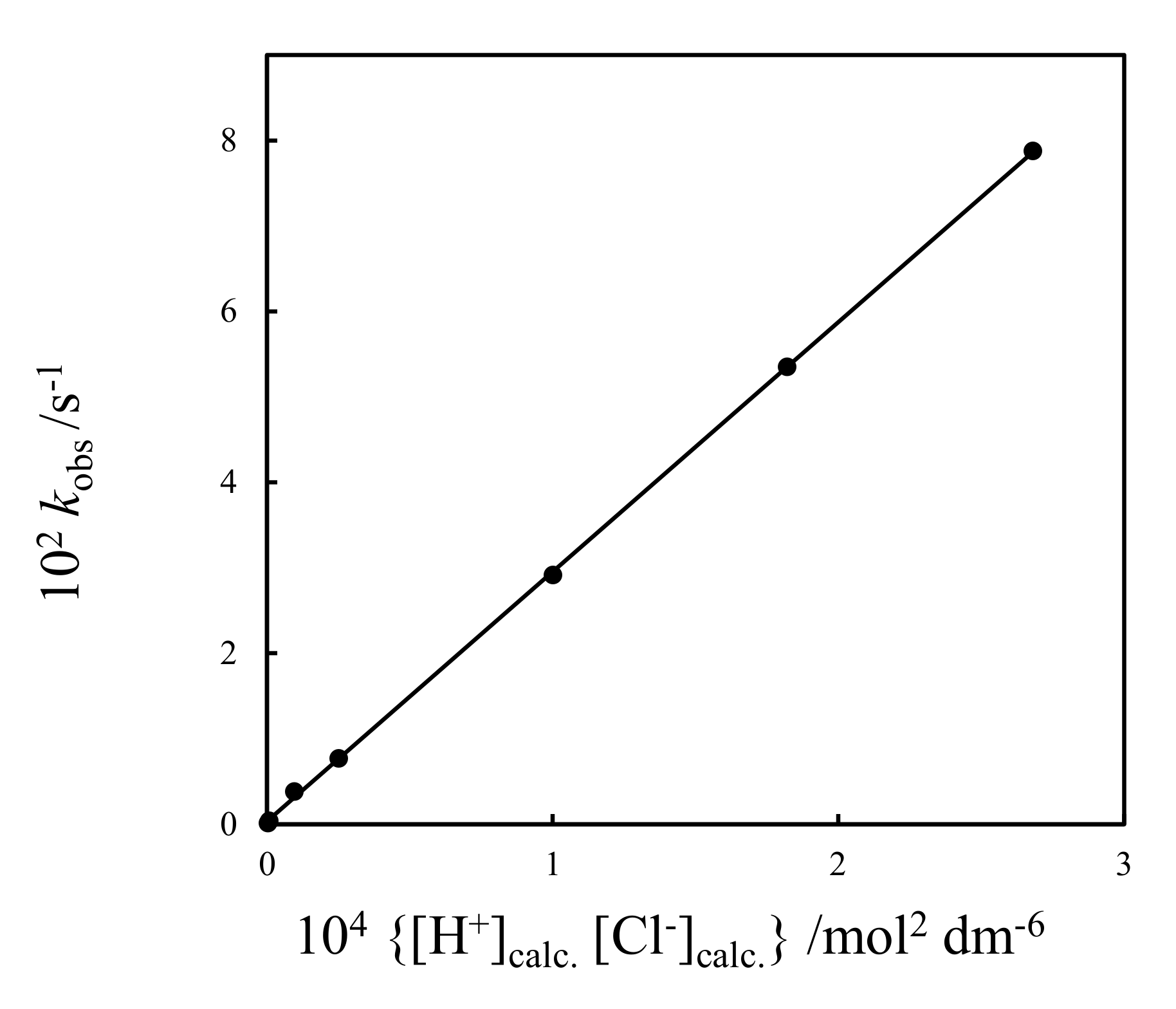
2.3.1. The Involvement of Ion Pairs
2.3.2. Kinetic Isotope Effects. The Involvement of Chloride in the Proton Transfer and the Role of Ion Pairs
2.3.3. Other Kinetic Evidence
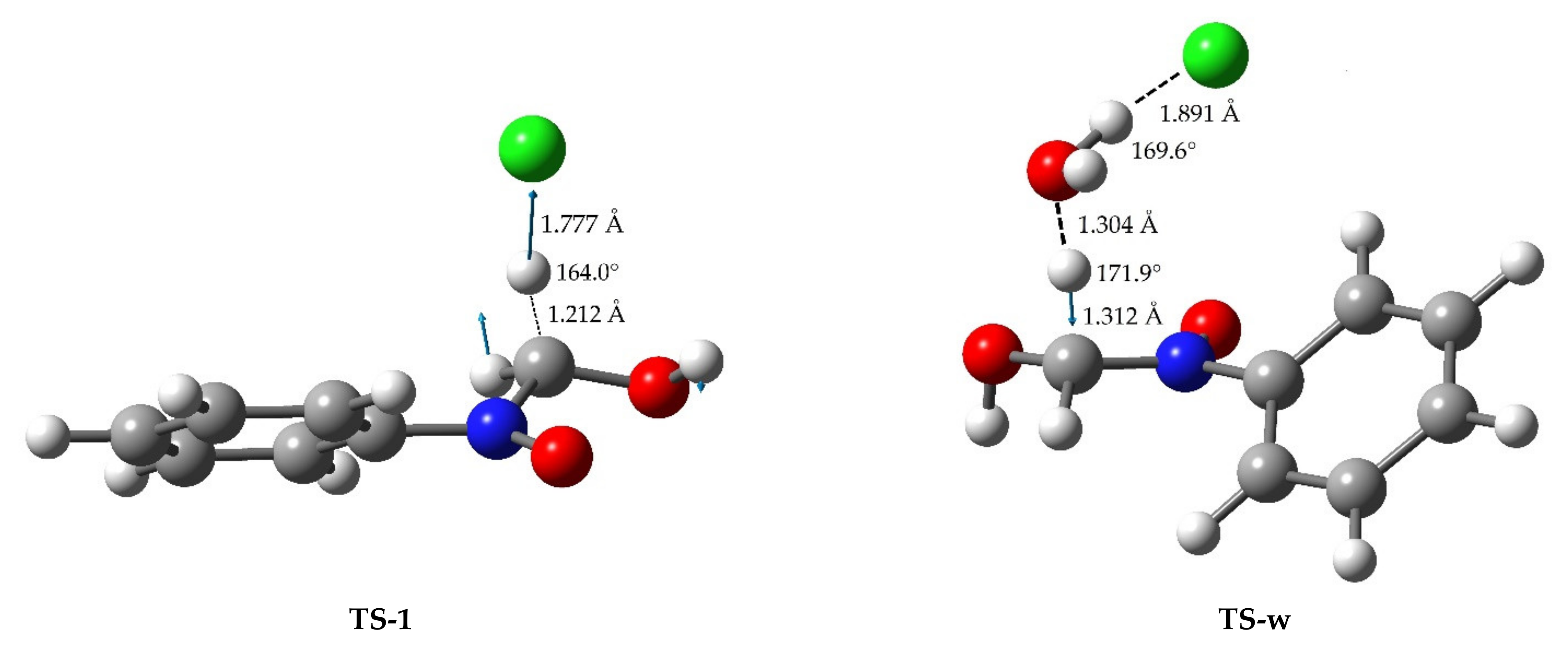
2.4. Theoretical Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brahmachari, U.; Gonthier, J.F.; Sherrill, C.D.; Barry, B.A. Chloride Maintains a Protonated Internal Water Network in the Photosynthetic Oxygen Evolving Complex. J. Phys. Chem. B 2017, 121, 10327–10337. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Mandal, M.; Ishikita, H. Energetics of Ionized Water Molecules in the H-Bond Network near the Ca2+ and Cl− Binding Sites in Photosystem II. Biochemistry 2020, 59, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, U.; Guo, Z.; Konecny, S.E.; Obi, E.N.C.; Barry, B.A. Engineering Proton Transfer in Photosynthetic Oxygen Evolution: Chloride, Nitrate, and Trehalose Reorganize a Hydrogen-Bonding Network. J. Phys. Chem. B 2018, 122, 6702–6711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobušić Brala, C.; Pilepić, V.; Sajenko, I.; Karković, A.; Uršić, S. Ions Can Move a Proton-Coupled Electron-Transfer Reaction into Tunneling Regime. Helv. Chim. Acta 2011, 94, 1718–1731. [Google Scholar] [CrossRef]
- Karković Marković, A.; Jakobušić Brala, C.; Pilepić, V.; Uršić, S. Kinetic Isotope Effects and Hydrogen Tunnelling in PCET Oxidations of Ascorbate: New Insights into Aqueous Chemistry? Molecules 2020, 25, 1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellmer, M.A.; Sanpitakseree, C.; Demir, B.; Ma, K.; Elliott, W.A.; Bai, P.; Johnson, R.L.; Walker, T.W.; Shanks, B.H.; Rioux, R.M.; et al. Effects of Chloride Ions in Acid-Catalyzed Biomass Dehydration Reactions in Polar Aprotic Solvents. Nat. Commun. 2019, 10, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Thibblin, A. Mechanisms of solvolytic elimination reactions of tertiary substrates: Stereospecific 1,2-elimination reactions. J. Chem. Soc. Perkin Trans. 2 1999, 7, 1397–1404. [Google Scholar] [CrossRef]
- Uršić, S.; Lovrek, M.; Vrček, I.V.; Pilepić, V. Salt effects and kinetic isotope effects interconnected. Evidence for the involvement of chloride ion in the C-H bond breaking in aqueous solution? J. Chem. Soc.-Perkin Trans. 2 1999, 7, 1295–1297. [Google Scholar] [CrossRef]
- Moss, R.A.; Zheng, F.; Sauers, R.R.; Toscano, J.P. The 2-Norbornyl Cation via the Fragmentations of exo- and endo-2-Norbornyloxychlorocarbenes: Distinction without Much Difference. J. Am. Chem. Soc. 2001, 123, 8109–8116. [Google Scholar] [CrossRef]
- Caldin, E.; Gold, V. Proton-Transfer Reactions; Chapman and Hall: London, UK, 1975. [Google Scholar]
- Karas, L.J.; Wu, C.-H.; Ottosson, H.; Wu, J.I. Electron-driven proton transfer relieves excited-state antiaromaticity in photoexcited DNA base pairs. Chem. Sci. 2020, 11, 10071–10077. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T. Proton Transfer Reaction Rates in Phenol–Ammonia Cluster Cation. J. Phys. Chem. A 2020, 124, 7893–7900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Harpe, K.d.L.; Kohl, F.R.; Kohler, B. Isotopic substitution affects excited state branching in a DNA duplex in aqueous solution. Chem. Commun. 2019, 55, 4174–4177. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCaherty, D.G.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S. Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc. 2015, 137, 8860–8871. [Google Scholar] [CrossRef] [Green Version]
- Parada, G.A.; Goldsmith, Z.K.; Kolmar, S.; Rimgard, B.P.; Mercado, B.Q.; Hammarstrom, L.; Hammes-Schiffer, S.; Mayer, J.M. Concerted proton-electron transfer reactions in the Marcus inverted region. Science 2019, 364, 471–475. [Google Scholar] [CrossRef]
- Karković Marković, A.; Jakobušić Brala, C.; Pilepić, V.; Uršić, S. Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry? Molecules 2019, 25, 172. [Google Scholar] [CrossRef] [Green Version]
- Huynh, M.H.V.; Meyer, T.J. Colossal kinetic isotope effects in proton-coupled electron transfer. Proc. Natl. Acad. Sci. USA 2004, 101, 13138–13141. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Dong, H.; Zheng, Y. Elaborating the excited state multiple proton transfer mechanism for 9H-pyrido[3,4-b]indole. J. Lumin. 2018, 195, 228–233. [Google Scholar] [CrossRef]
- Markle, T.F.; Darcy, J.W.; Mayer, J.M. A new strategy to efficiently cleave and form C–H bonds using proton-coupled electron transfer. Sci. Adv. 2018, 4, eaat5776. [Google Scholar] [CrossRef] [Green Version]
- Jarczewski, A.; Hubbard, C.D. A review of proton transfer reactions between various carbon-acids and amine bases in aprotic solvents. J. Mol. Struct. 2003, 649, 287–307. [Google Scholar] [CrossRef]
- Góbi, S.; Nunes, C.M.; Reva, I.; Tarczay, G.; Fausto, R. S–H rotamerization via tunneling in a thiol form of thioacetamide. Phys. Chem. Chem. Phys. 2019, 21, 17063–17071. [Google Scholar] [CrossRef] [PubMed]
- Kronja, O.; Matijević-Sosa, J.; Uršić, S. Reaction of substituted nitrosobenzenes with formaldehyde. J. Chem. Soc. Chem. Commun. 1987, 6, 463–464. [Google Scholar] [CrossRef]
- Uršić, S. Reactions of Carbonyl Group with Nitroso Compounds: Reaction of Formaldehyde with Substituted Nitrosobenzenes. Helv. Chim. Acta 1993, 76, 131–138. [Google Scholar] [CrossRef]
- Uršić, S.; Vrček, V.; Gabričević, M.; Zorc, B. Reaction of pyruvic acid with nitrosobenzenes. J. Chem. Soc. Chem. Commun. 1992, 4, 296–298. [Google Scholar] [CrossRef]
- Uršić, S.; Pilepić, V.; Vrček, V.; Gabričević, M.; Zorc, B. Reactions of the Carbonyl Group with Nitroso-Compounds—The Cases of Pyruvic-Acid and Acetaldehyde. J. Chem. Soc.-Perkin Trans. 2 1993, 7, 509–514. [Google Scholar] [CrossRef]
- Pilepić, V.; Uršić, S. Reaction of 2-Nitroso-2-Methyl Propane with Formaldehyde, Glyoxylate and Glyoxylic-Acid. Tetrahedron Lett. 1994, 35, 7425–7428. [Google Scholar] [CrossRef]
- Pilepić, V.; Uršić, S. Nucleophilic reactivity of the nitroso group. Fukui function DFT calculations for nitrosobenzene and 2-methyl-2-nitrosopropane. J. Mol. Struct. 2001, 538, 41–49. [Google Scholar] [CrossRef]
- Pilepić, V.; Jakobušić, C.; Vikić-Topić, D.; Uršić, S. Evidence for proton transfer from carbon to chloride ion in solution. Tetrahedron Lett. 2006, 47, 371–375. [Google Scholar] [CrossRef]
- Lovrek, M.; Pilepić, V.; Uršić, S. Intriguing Salt Effects in the Formation of Hydroxamic Acids from Aldehydes and Nitroso Compounds. Croat. Chem. Acta 2000, 73, 715–731. [Google Scholar]
- He, J.; Liu, M.; Walker, T.W.; Maravelias, C.T.; Dumesic, J.A.; Huber, G.W. Production of levoglucosenone and 5-hydroxymethylfurfural from cellulose in polar aprotic solvent–water mixtures. Green. Chem. 2017, 19, 3642–3653. [Google Scholar] [CrossRef]
- Barbarić, M.; Uršić, S.; Pilepić, V.; Zorc, B.; Hergold-Brundić, A.; Nagl, A.; Grdiša, M.; Pavelić, K.I.; Snoeck, R.; Andrei, G.; et al. Synthesis, X-ray crystal structure study, and cytostatic and antiviral evaluation of the novel cycloalkyl-N-aryl-hydroxamic acids. J. Med. Chem. 2005, 48, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Saraswati, A.P.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K. Acid-Base Dissociation Constants in Dipolar Aprotic Solvents; Blackwell Scientific Publications: Oxford, UK, 1990. [Google Scholar]
- Szwarc, M. Ion and Ion Pairs; Wiley-Interscience: New York, NY, USA, 1972. [Google Scholar]
- Lowry, T.H.; Richardson, K.S. Addition and elimination reactions. In Mechanism and Theory in Organic Chemistry; Harper-Collins: New York, NY, USA, 1987; pp. 341–349. [Google Scholar]
- Isaacs, S.N. Physical Organic Chemistry; Longman Scientific & Tehnical: Harlow, UK, 1995; Chapter VII; pp. 445–447. [Google Scholar]
- Richard, J.P.; Amyes, T.L.; Toteva, M.M.; Tsui, Y. Dynamics for the reactions of ion pair intermediates of solvolysis. Adv. Phys. Org. Chem. 2004, 39, 1–29. [Google Scholar] [CrossRef]
- Loupy, A.; Tchoubar, B. Salt Effects in Organic and Organometallic Chemistry; VCH: Weinheim, Germany, 1992; pp. 43–47. [Google Scholar]
- Perrin, C.; Pressing, J. Simple Model for Linear Salt Efects in Solvolysis Reactions. J. Am. Chem. Soc. 1971, 93, 5705. [Google Scholar] [CrossRef]
- More O’Ferrall, R.A. Substrate Isotope Effects. In Proton-Transfer Reactions; Caldin, E., Gold, V., Eds.; Springer: New York, NY, USA, 1975; Chapter 8. [Google Scholar]
- Lowry, T.H.; Richardson, K.S. Unimolecular substitutions and related reactions. In Mechanism and Theory in Organic Chemistry; Harper-Collins: New York, NY, USA, 1987; pp. 232–244. [Google Scholar]
- Isaacs, S.N. Physical Organic Chemistry; Longman Scientific & Tehnical: Harlow, UK, 1995; Chapter II; pp. 244–288. [Google Scholar]
- Conway, B.E. Ionic Hydration in Chemistry and Biophysics; Elsevier: Amsterdam, The Netherlands, 1981; Chapter 19; pp. 436–443. [Google Scholar]
- Bunton, C.A.; Carrasco, N.; Cully, N.; Watts, W.E. A kinetic study of concomitant addition and deprotonation reactions of ferrocenyl-stabilised carbocations in aqueous acetonitrile and of the reverse reactions. J. Chem. Soc. Perkin Trans. 2 1980, 12, 1859–1867. [Google Scholar] [CrossRef]
- Zeng, X.F.; Thibblin, A. Competing solvolytic elimination and substitution reactions via very short-lived ion-pair intermediates. J. Chem. Soc.-Perkin Trans. 2 2001, 9, 1600–1607. [Google Scholar] [CrossRef]
- Hunt, P.A.; Gould, I.R. Structural Characterization of the 1-Butyl-3-methylimidazolium Chloride Ion Pair Using ab initio Methods. J. Phys. Chem. A 2006, 110, 2269–2282. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
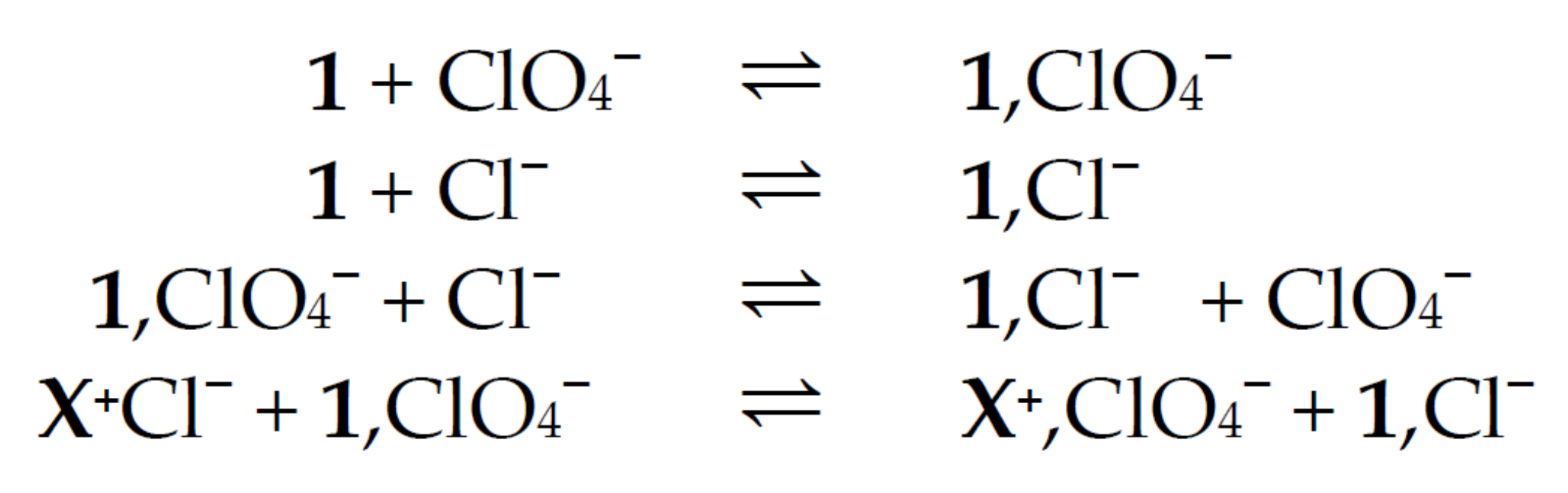

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anion | 104 kH/s−1 | 104 kD/s−1 | PKIE c | |
|---|---|---|---|---|
| 1 | ClO4− (HClO4, 0.1 M) d | 12.36 (0.75) | 1.34 (0.06) | 7.58 (0.32) |
| 2 | ClO4− (HClO4, 0.1 M) e | 1.35 (0.04) | ||
| 3 | ClO4− (HClO4, 0.1 M) d,f | 2.32 (0.06) | 0.30 (0.01) | 6.34 (0.17) |
| 4 | HSO4− (H2SO4, 0.005 M) | 1.22 (0.01) | 0.16 (0.01) | 7.53 (0.38) |
| 5 | Cl− (HCl, 0.005 M) | 39.30 (1.07) | 19.4 (0.75) | 2.02 (0.10) |
| 6 | Cl− (HCl, 0.050 M) | 251.6 (7.16) | 135.0 (5.65) | 1.86 (0.09) |
| 7 | Br− (HBr, 0.005 M) | 50.7 (0.50) | 22.1 (0.40) | 2.29 (0.05) |
| 8 | CCl3COO− (CCl3COOH, 0.050 M) | 1.83 (0.08) | 0.44 (0.01) | 4.11 (0.20) |
| 9 | Cl− (HCl, 0.050 M) g | 5.54 (0.21) | 2.37 (0.20) | 2.34 (0.22) |
| 10 | ClO4− (HClO4, 0.1 M) h | 0.40 (0.04) | ||
| 11 | Cl− (HCl, 0.050 M) h | 3.16 (0.10) |
| Anion | 103 kH/s−1 | 103 kD/s−1 | PKIE c | |
|---|---|---|---|---|
| 1 | Cl−, ClO4− (PhCH2(CH3)3N+ Cl− 0.01 M) d | 1.01 (0.04) | ||
| 2 | Cl−, ClO4− (PhCH2(CH3)3N+ Cl− 0.005 M) e,f | 62.3 (0.08) | 13.07 (0.09) | 3.89 (0.06) |
| 3 | Cl−, ClO4− (PhCH2(CH3)3N+ Cl− 0.001 M) e,f | 13.43 (0.04) | 2.92 (0.06) | 3.76 (0.12) |
| 4 | Cl−, ClO4− (PhCH2(CH3)3N+ Cl− 0.0001 M) | 4.40 (0.13) | 0.98 (0.14) | 4.49 (0.20) |
| 5 | Br−, ClO4− (PhCH2(CH3)3N+ Br− 0.005 M) | 274.0 (5.0) | 119.0 (1.0) | 2.30 (0.05) |
| 6 | Br−, ClO4− (PhCH2(CH3)3N+ Br− 0.001 M) | 32.5 (1.2) | 8.44 (0.05) | 3.85 (0.14) |
| 7 | Br−, ClO4− (PhCH2(CH3)3N+ Br− 0.0001 M) | 6.65 (0.03) | 1.54 (0.01) | 4.32 (0.03) |
| 8 | Cl−, ClO4− ((CH3)4N+ Cl− 0.001 M) | 5.35 (0.02) | 1.15 (0.02) | 4.65 (0.08) |
| 9 | Cl−, ClO4− ((CH3CH2)4N+ Cl− 0.001 M) | 9.24 (0.18) | 3.01 (0.09) | 3.07 (0.11) |
| 10 | Cl−, ClO4− (Betaine hydrochloride 0.001 M) | 25.0 (0.2) | 11.1 (0.2) | 2.25 (0.04) |
| 11 | Cl−, ClO4− (1-Buthyl-3-Methyl Imidazolium Cl− 0.001 M) | 7.92 (0.06) | 1.87 (0.08) | 4.24 (0.18) |
| 12 | BF4−, ClO4− (1-Buthyl-3-Methyl Imidazolium BF4− 0.001 M) | 2.47 (0.03) | 0.444 (0.008) | 5.61 (0.12) |
| 13 | Cl−, ClO4− (LiCl 0.0044 M) g | 9.91 (0.37) | ||
| 14 | Cl−, ClO4− (LiCl 0.0088 M) g | 14.54 (0.55) | 5.44 (0.63) | 2.19 (0.08) |
| 15 | Cl−, ClO4− (MgCl2 0.01 M) h | 0.171 (0.004) | 0.057 (0.002) | 3.01 (0.14) |
| 16 | Cl−, ClO4− (MgCl2 0.01 M) i | 0.586 (0.017) | 0.182 (0.007) | 3.22 (0.16) |
| 17 | Cl−, ClO4− (PhCH2(CH3)3N+ Cl− 0.005 M) j | 0.18 (0.003) |
| H Atom Acceptor | C⋯H/Å b | C⋯H⋯A/° c | v/cm−1 d | ΔG‡/kJ mol−1 e | |
|---|---|---|---|---|---|
| w-TS | water | 1.334 | 169.9 | 1468 i | +74.8 |
| TS-1 | Cl− | 1.212 | 164.0 | 946 i | +24.6 |
| TS-w | water⋯Cl− | 1.312 | 171.9 | 1546 i | +56.0 |
| TS-p | ClO4− | 1.290 | 177.2 | 1601 i | +60.0 |
| TS-wp | water⋯ClO4− | 1.283 | 175.6 | 1400 i | +63.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilepić, V.; Jakobušić Brala, C.; Uršić, S. Intriguing Chloride: Involvement of Chloride Ions in Proton Transfers. Molecules 2022, 27, 1401. https://doi.org/10.3390/molecules27041401
Pilepić V, Jakobušić Brala C, Uršić S. Intriguing Chloride: Involvement of Chloride Ions in Proton Transfers. Molecules. 2022; 27(4):1401. https://doi.org/10.3390/molecules27041401
Chicago/Turabian StylePilepić, Viktor, Cvijeta Jakobušić Brala, and Stanko Uršić. 2022. "Intriguing Chloride: Involvement of Chloride Ions in Proton Transfers" Molecules 27, no. 4: 1401. https://doi.org/10.3390/molecules27041401
APA StylePilepić, V., Jakobušić Brala, C., & Uršić, S. (2022). Intriguing Chloride: Involvement of Chloride Ions in Proton Transfers. Molecules, 27(4), 1401. https://doi.org/10.3390/molecules27041401