
Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine

 , ,
, ,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
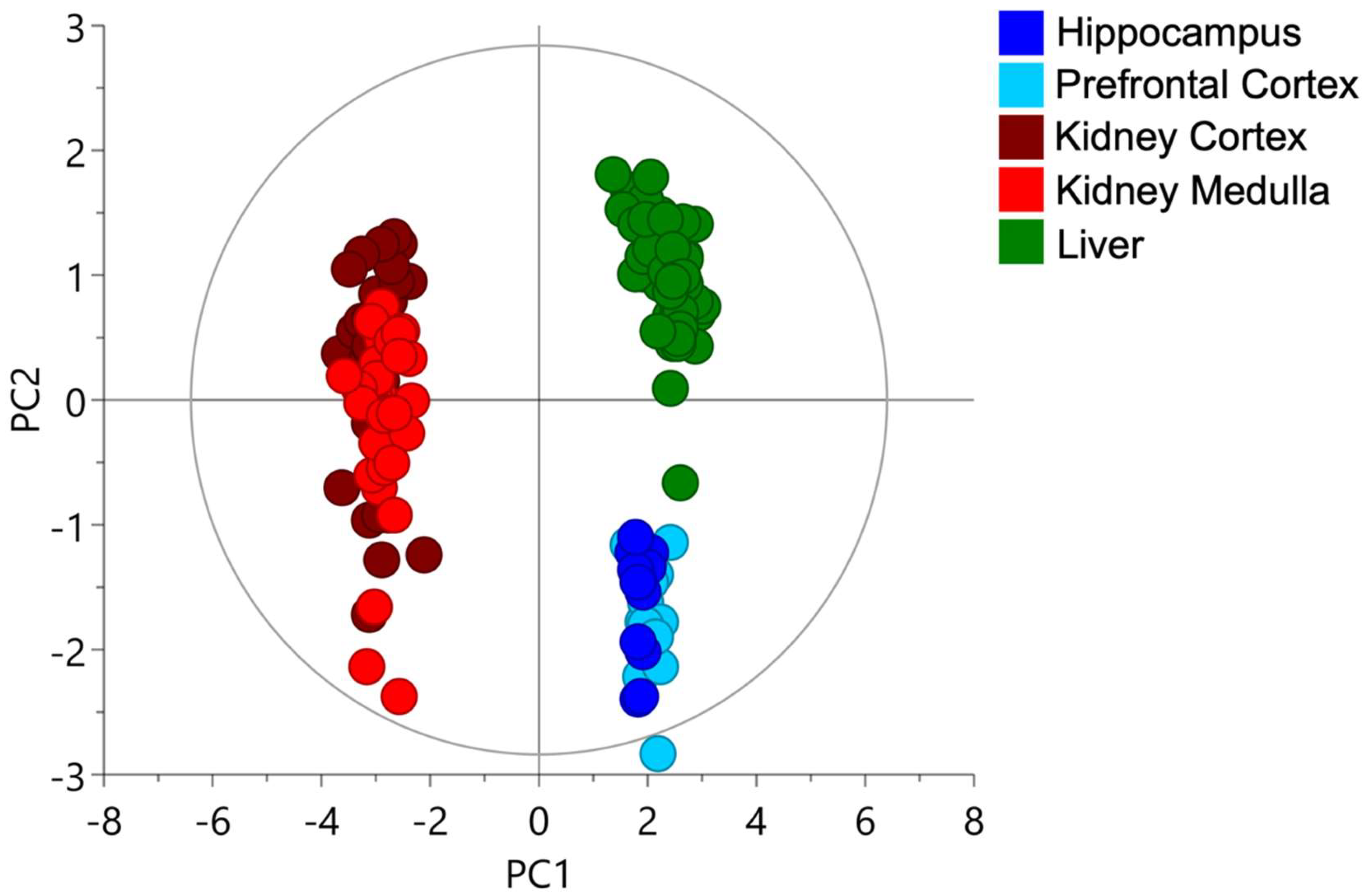
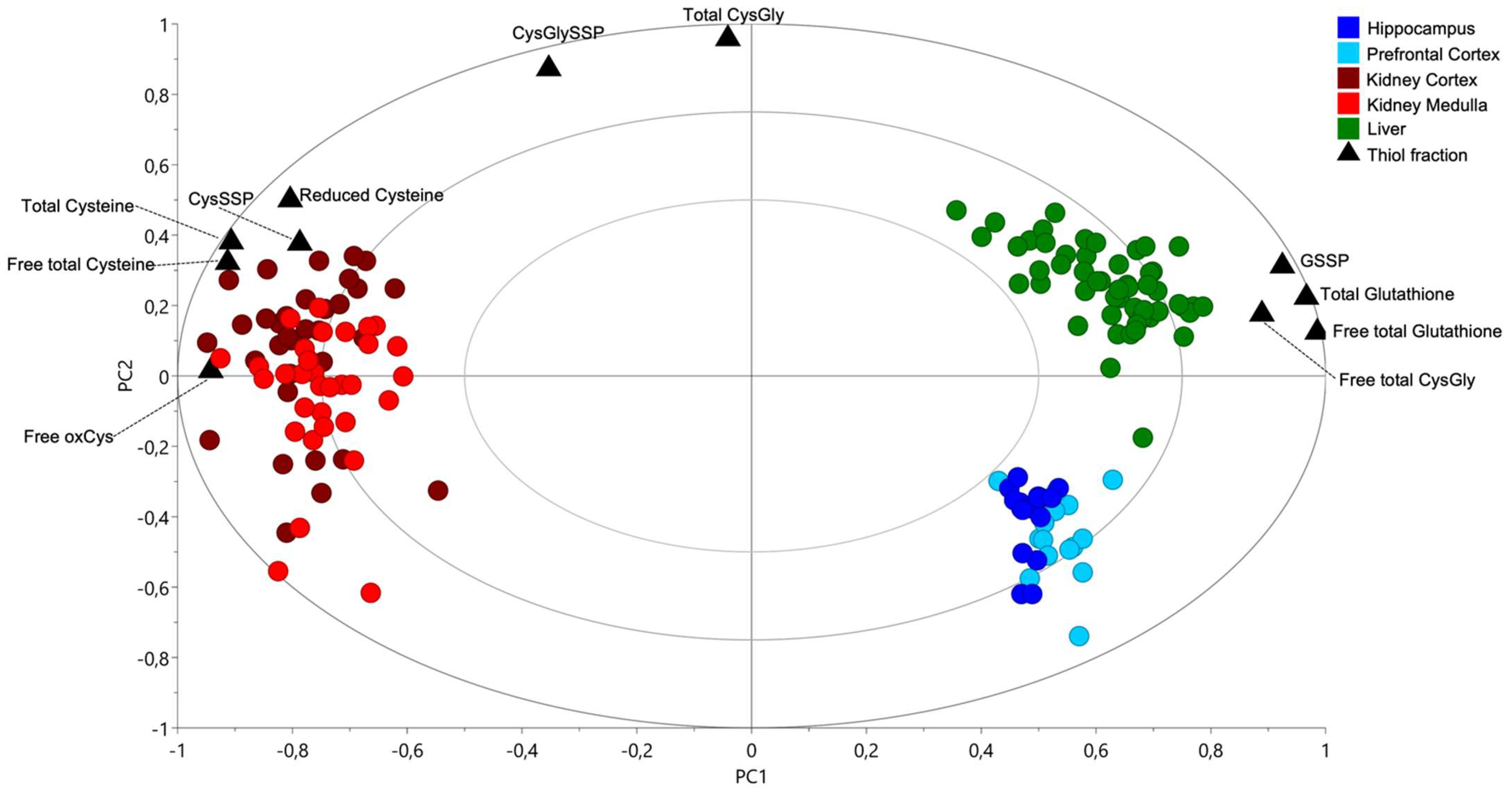
2.1. Cysteine-Related Thiolome in Different Organs
2.2. The Kidney Is a Cysteine-Rich Organ
3. Evidence Supporting That Cysteine and Its Renal Metabolism Justify the Kidney’s Reliance on Cysteine: A Literature Review
3.1. Cysteine De Novo Synthesis
3.2. Cysteine Catabolism in the Control of Cysteine Excess and a Source of Relevant Metabolites
3.3. Cysteine Is a Pivotal Molecule for Kidney Biomass and Bioenergetics
3.3.1. β-Oxidation and Oxidative Phosphorylation
3.3.2. Gluconeogenesis
3.3.3. Pentose Phosphate Pathway
3.4. Cysteine as an Alternative Source of Energy through H2S Production
3.5. Cysteine Is Preventive of Ferroptosis and Promoter of Cell Survival
3.6. Cysteine Contribution for Kidney Adaptation to Hypoxia
3.7. Implications for Precision Medicine in Arterial Hypertension and Kidney Disease
4. Materials and Methods
4.1. Animals
4.2. Quantification of Cysteine-Related Thiolome
4.3. Data Analysis
4.3.1. Multivariate Analysis
4.3.2. Univariate Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bowes, A.D.P.; Church, H.N.; Pennington, J.A.T. Supplementary tables—amino acids. In Bowes and Church’s Food Values of Portions Commonly Used; Allen, A., Ed.; J.B. Lippincott: Philadelphia, CA, USA, 1994; pp. 325–377. [Google Scholar]
- Reeds, P.J. Dispensable and indispensable amino acids for humans. J. Nutr. 2000, 130, 1835S–1840S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Miguelsanz, J.; Vallecillo, N.; Garrido, F.; Reytor, E.; Pérez-Sala, D.; Pajares, M.A. Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1165–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stipanuk, M.H. Metabolism of Sulfur-Containing Amino Acids: How the Body Copes with Excess Methionine, Cysteine, and Sulfide. J. Nutr. 2020, 150, 2494S–2505S. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, D.A.; Baker, D.H. Sulfur amino acid nutrition of the growing puppy: Determination of dietary requirements for methionine and cystine. Nutr. Res. 1985, 5, 631–642. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Martin, J.J.; Harris, B.J. Effect of Dietary Cystine on Methionine Metabolism in Rat Liver. J. Nutr. 1986, 116, 985–990. [Google Scholar] [CrossRef]
- Serpa, J. Cysteine as a Carbon Source, a Hot Spot in Cancer Cells Survival. Front. Oncol. 2020, 10, 947. [Google Scholar] [CrossRef]
- Van Goor, H.; Van Den Born, J.C.; Hillebrands, J.L.; Joles, J.A. Hydrogen sulfide in hypertension. Curr. Opin. Nephrol. Hypertens. 2016, 25, 107–113. [Google Scholar] [CrossRef]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Abraham, J.R.; Apostolopoulos, V.; Zulli, A. The Anti-Inflammatory Effect of Taurine on Cardiovascular Disease. Nutrients 2020, 12, 2847. [Google Scholar] [CrossRef]
- Coelho, N.R.; Dias, C.G.; João Correia, M.; Grácio, P.; Serpa, J.; Monteiro, E.C.; Diogo, L.N.; Pereira, S.A. Cysteine Oxidative Dynamics Underlies Hypertension and Kidney Dysfunction Induced by Chronic Intermittent Hypoxia. Adv. Exp. Med. Biol. 2018, 1071, 83–88. [Google Scholar]
- Correia, M.J.; Pimpão, A.B.; Lopes-Coelho, F.; Sequeira, C.O.; Coelho, N.R.; Gonçalves-Dias, C.; Barouki, R.; Coumoul, X.; Serpa, J.; Morello, J.; et al. Aryl Hydrocarbon Receptor and Cysteine Redox Dynamics Underlie (Mal)adaptive Mechanisms to Chronic Intermittent Hypoxia in Kidney Cortex. Antioxidants 2021, 10, 1484. [Google Scholar] [CrossRef]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Giustarini, D.; Milzani, A.; Dalle-Donne, I. Cysteinylation and homocysteinylation of plasma protein thiols during ageing of healthy human beings. J. Cell. Mol. Med. 2009, 13, 3131–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, P.V.S.; Laurindo, F.R.M. Implications of plasma thiol redox in disease. Clin. Sci. 2018, 132, 1257–1280. [Google Scholar] [CrossRef] [PubMed]
- Sumayao, R.; Newsholme, P.; McMorrow, T. The Role of Cystinosin in the Intermediary Thiol Metabolism and Redox Homeostasis in Kidney Proximal Tubular Cells. Antioxidants 2018, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Rebbeor, J.F.; Wang, W.; Clifton, D.; Ballatori, N. Glutathione S-conjugate formation and metabolism in HepG2 cells: A cell model of mercapturic acid biosynthesis. J. Toxicol. Environ. Health 1998, 53, 651–663. [Google Scholar]
- Goncalves-Dias, C.; Morello, J.; Correia, M.J.; Coelho, N.R.; Antunes, A.M.M.; MacEdo, M.P.; Monteiro, E.C.; Soto, K.; Pereira, S.A. Mercapturate Pathway in the Tubulocentric Perspective of Diabetic Kidney Disease. Nephron 2019, 143, 17–23. [Google Scholar] [CrossRef]
- Park, S.; Imlay, J.A. High levels of intracellular cysteine promote oxidative DNA damage by driving the fenton reaction. J. Bacteriol. 2003, 185, 1942–1950. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Vina, J.; Saez, G.T.; Wiggins, D.; Roberts, A.F.; Hems, R.; Krebs, H.A. The effect of cysteine oxidation on isolated hepatocytes. Biochem. J. 1983, 212, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Munday, R. Toxicity of thiols and disulphides: Involvement of free-radical species. Free Radic. Biol. Med. 1989, 7, 659–673. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Galván, I.; Ghanem, G.; Møller, A.P. Has removal of excess cysteine led to the evolution of pheomelanin? Pheomelanogenesis as an excretory mechanism for cysteine. Bioessays 2012, 34, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Yabunaka, N.; Fujisawa, H.; Watanabe, I.; Agishi, Y. Effect of thermal stress on glutathione metabolism in human erythrocytes. Eur. J. Appl. Physiol. Occup. Physiol. 1994, 68, 87–91. [Google Scholar] [CrossRef] [PubMed]
- García-Borrón, J.C.; Olivares Sánchez, M.C. Biosynthesis of Melanins. In Melanins and Melanosomes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 87–116. [Google Scholar]
- Guégan, J.F.; Thomas, F.; Hochberg, M.E.; De Meeus, T.; Renaud, F. Disease diversity and human fertility. Evolution 2001, 55, 1308–1314. [Google Scholar] [CrossRef]
- Camasmie Abe, K.; de Campos Brandão, L.; Aguilar Calegare, B.F.; Tufik, S.; do Nascimento Saldiva, P.H.; D’Almeida, V. Homocysteine and cysteine concentrations are modified by recent exposure to environmental air pollution in São Paulo, Brazil. Environ. Res. 2009, 109, 887–890. [Google Scholar] [CrossRef]
- Gonzalez-Jaramillo, V.; Portilla-Fernandez, E.; Glisic, M.; Voortman, T.; Bramer, W.; Chowdhury, R.; Roks, A.J.M.; Jan Danser, A.H.; Muka, T.; Nano, J.; et al. The role of DNA methylation and histone modifications in blood pressure: A systematic review. J. Hum. Hypertens. 2019, 33, 703–715. [Google Scholar] [CrossRef]
- Schulman, J.D.; Bradley, K.H.; Seegmiller, J.E. Cystine: Compartmentalization within lysosomes in cystinotic leukocytes. Science 1969, 166, 1152–1154. [Google Scholar] [CrossRef]
- Thoene, J.G.; Lemons, R. Modulation of the intracellular cystine content of cystinotic fibroblasts by extracellular albumin. Pediatr. Res. 1980, 14, 785–787. [Google Scholar] [CrossRef] [Green Version]
- Danpure, C.J.; Jennings, P.R.; Fyfe, D.A. Further studies on the effect of chloroquine on the uptake, metabolism and intracellular translocation of [35S]cystine in cystinotic fibroblasts. Biochim. Biophys. Acta 1986, 885, 256–265. [Google Scholar] [CrossRef]
- Sumayao, R.; McEvoy, B.; Newsholme, P.; McMorrow, T. Lysosomal cystine accumulation promotes mitochondrial depolarization and induction of redox-sensitive genes in human kidney proximal tubular cells. J. Physiol. 2016, 594, 3353–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliubadaló, L.; Font, M.; Purroy, J.; Rousaud, F.; Estivill, X.; Nunes, V.; Golomb, E.; Centola, M.; Aksentijevich, I.; Kreiss, Y.; et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat. Genet. 1999, 23, 52–57. [Google Scholar] [CrossRef]
- Fernández, E.; Carrascal, M.; Rousaud, F.; Abián, J.; Zorzano, A.; Palacín, M.; Chillarón, J. rBAT-b(0,+)AT heterodimer is the main apical reabsorption system for cystine in the kidney. Am. J. Physiol. Renal Physiol. 2002, 283, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Aliaga, I.; Frey, I.; Boll, M.; Groneberg, D.A.; Eichinger, H.M.; Balling, R.; Daniel, H. Targeted Disruption of the Peptide Transporter Pept2 Gene in Mice Defines Its Physiological Role in the Kidney. Mol. Cell. Biol. 2003, 23, 3247–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, E.; Torrents, D.; Chillarón, J.; Martín del Rio, R.; Zorzano, A.; Palacín, M. Basolateral LAT-2 Has a Major Role in the Transepithelial Flux of L-Cystine in the Renal Proximal Tubule Cell Line OK. J. Am. Soc. Nephrol. 2003, 14, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves-Dias, C.; Morello, J.; Semedo, V.; Correia, M.J.; Coelho, N.R.; Monteiro, E.C.; Antunes, A.M.M.; Pereira, S.A. The mercapturomic profile of health and non-communicable diseases. High-Throughput 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves-Dias, C.; Sequeira, C.O.; Vicente, J.B.; Correia, M.J.; Coelho, N.R.; Morello, J.; Antunes, A.M.M.; Soto, K.; Monteiro, E.C.; Pereira, S.A. A Mechanistic-Based and Non-invasive Approach to Quantify the Capability of Kidney to Detoxify Cysteine-Disulfides. Adv. Exp. Med. Biol. 2021, 1306, 109–120. [Google Scholar] [PubMed]
- Dröge, W.; Eck, H.P.; Gmünder, H.; Mihm, S. Modulation of lymphocyte functions and immune responses by cysteine and cysteine derivatives. Am. J. Med. 1991, 91, 140–144. [Google Scholar] [CrossRef]
- Pompella, A.; Corti, A.; Paolicchi, A.; Giommarelli, C.; Zunino, F. Gamma-glutamyltransferase, redox regulation and cancer drug resistance. Curr. Opin. Pharmacol. 2007, 7, 360–366. [Google Scholar] [CrossRef]
- Lieberman, M.W.; Wiseman, A.L.; Shi, Z.Z.; Carter, B.Z.; Barrios, R.; Ou, C.N.; Chévez-Barrios, P.; Wang, Y.; Habib, G.M.; Goodman, J.C.; et al. Growth retardation and cysteine deficiency in gamma-glutamyl transpeptidase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 7923–7926. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Forman, H.J.; Choi, J. Gamma-glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005, 401, 468–483. [Google Scholar] [PubMed]
- De Carvalho, J.A.M.; Piva, S.J.; Hausen, B.S.; Bochi, G.V.; Kaefer, M.; Coelho, A.C.; Duarte, M.M.M.F.; Moresco, R.N. Assessment of urinary γ-glutamyltransferase and alkaline phosphatase for diagnosis of diabetic nephropathy. Clin. Chim. Acta 2011, 412, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Zhang, W.; Lord, G.M.; Van Der Harst, P.; Lawlor, D.A.; Sehmi, J.S.; Gale, D.P.; Wass, M.N.; Ahmadi, K.R.; Bakker, S.J.L.; et al. Genetic loci influencing kidney function and chronic kidney disease. Nat. Genet. 2010, 42, 373–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga-da-Cunha, M.; Tyteca, D.; Stroobant, V.; Courtoy, P.J.; Opperdoes, F.R.; Van Schaftingen, E. Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J. Biol. Chem. 2010, 285, 18888–18898. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Grunwell, J.R.; Gillespie, S.E.; Ward, J.M.; Fitzpatrick, A.M.; Brown, L.A.; Gauthier, T.W.; Hebbar, K.B. Comparison of Glutathione, Cysteine, and Their Redox Potentials in the Plasma of Critically Ill and Healthy Children. Front. Pediatr. 2015, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Szwergold, B.S. Alpha-thiolamines such as cysteine and cysteamine act as effective transglycating agents due to formation of irreversible thiazolidine derivatives. Med. Hypotheses 2006, 66, 698–707. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl leukotrienes and their receptors; emerging concepts. Allergy. Asthma Immunol. Res. 2014, 6, 288–295. [Google Scholar] [CrossRef]
- Jian, W.; Yao, M.; Zhang, D.; Zhu, M. Rapid detection and characterization of in vitro and urinary N-acetyl-L-cysteine conjugates using quadrupole-linear ion trap mass spectrometry and polarity switching. Chem. Res. Toxicol. 2009, 22, 1246–1255. [Google Scholar] [CrossRef]
- Juhanson, P.; Kepp, K.; Org, E.; Veldre, G.; Kelgo, P.; Rosenberg, M.; Viigimaa, M.; Laan, M. N-acetyltransferase 8, a positional candidate for blood pressure and renal regulation: Resequencing, association and in silico study. BMC Med. Genet. 2008, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Grácio, P.C.; Gonçalves-Dias, C.; Lopes-Coelho, F.; Monteiro, E.C.; Serpa, J.; Da Silva, C.L.; Pereira, S.A. Changes in N-acetyltransferase 8 in kidney tubular cell: Injury, recovery and mesenchymal stromal cell-based therapy. In Proceedings of the 6th IEEE Portuguese Meeting on Bioengineering (ENBENG); Institute of Electrical and Electronics Engineers Inc., Lisbon, Portugal, 22–23 February 2019. [Google Scholar]
- Nagamori, S.; Wiriyasermkul, P.; Guarch, M.E.; Okuyama, H.; Nakagomi, S.; Tadagaki, K.; Nishinaka, Y.; Bodoy, S.; Takafuji, K.; Okuda, S.; et al. Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1. Proc. Natl. Acad. Sci. USA 2016, 113, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifton, R.P.; Somlo, S.; Giebisch, G.; Seldin, D. Genetic Diseases of the Kidney; Elsevier Inc.: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Barbosa, M.; Lopes, A.; Mota, C.; Martins, E.; Oliveira, J.; Alves, S.; de Bonis, P.; do Céu Mota, M.; Dias, C.; Rodrigues-Santos, P.; et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin. Genet. 2012, 81, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Gallen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Singh, V.K.; Rahman, M.N.; Munro, K.; Uversky, V.N.; Smith, S.P.; Jia, Z. Free cysteine modulates the conformation of human C/EBP homologous protein. PLoS ONE 2012, 7, e34680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Wu, Z.; Dai, Z.; Sun, K.; Zhang, Q.; Wu, G. Excessive L-cysteine induces vacuole-like cell death by activating endoplasmic reticulum stress and mitogen-activated protein kinase signaling in intestinal porcine epithelial cells. Amino Acids 2016, 48, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Shibui, Y.; Sakai, R.; Manabe, Y.; Masuyama, T. Comparisons of l-cysteine and d-cysteine toxicity in 4-week repeated-dose toxicity studies of rats receiving daily oral administration. J. Toxicol. Pathol. 2017, 30, 217–229. [Google Scholar] [CrossRef] [Green Version]
- States, B.; Foreman, J.W.; Segal, S. Cysteine and glutathione levels in developing rat kidney and liver. Pediatr. Res. 1987, 22, 605–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regazzoni, L.; Del Vecchio, L.; Altomare, A.; Yeum, K.J.; Cusi, D.; Locatelli, F.; Carini, M.; Aldini, G. Human serum albumin cysteinylation is increased in end stage renal disease patients and reduced by hemodialysis: Mass spectrometry studies. Free Radic. Res. 2013, 47, 172–180. [Google Scholar] [CrossRef]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.G.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS ONE 2014, 9, e85216. [Google Scholar] [CrossRef]
- Banjac, A.; Perisic, T.; Sato, H.; Seiler, A.; Bannai, S.; Weiss, N.; Kölle, P.; Tschoep, K.; Issels, R.D.; Daniel, P.T.; et al. The cystine/cysteine cycle: A redox cycle regulating susceptibility versus resistance to cell death. Oncogene 2008, 27, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 2005, 111, 2973–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, P. Monocytes as immune targets in arterial hypertension. Br. J. Pharmacol. 2019, 176, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.L.; Escobar, J.; Izquierdo-Álvarez, A.; Gil, A.; Pérez, S.; Pereda, J.; Zapico, I.; Vento, M.; Sabater, L.; Marina, A.; et al. Disulfide stress: A novel type of oxidative stress in acute pancreatitis. Free Radic. Biol. Med. 2014, 70, 265–277. [Google Scholar] [CrossRef]
- Parakh, S.; Atkin, J.D. Novel roles for protein disulphide isomerase in disease states: A double edged sword? Front. Cell Dev. Biol. 2015, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.; Godinho-Pereira, J.; Oudot, C.; Sequeira, C.O.; Macià, A.; Carvalho, F.; Motilva, M.J.; Pereira, S.A.; Matzapetakis, M.; Brenner, C.; et al. Berry fruits modulate kidney dysfunction and urine metabolome in Dahl salt-sensitive rats. Free Radic. Biol. Med. 2020, 154, 119–131. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Dominy, J.E.; Lee, J.I.; Coloso, R.M. Mammalian cysteine metabolism: New insights into regulation of cysteine metabolism. J. Nutr. 2006, 136, 1652–1659. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Ueki, I.; Dominy, J.E.; Simmons, C.R.; Hirschberger, L.L. Cysteine Dioxygenase: A Robust System for Regulation of Cellular Cysteine Levels. Amino Acids 2009, 37, 63. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.D. The metabolism of homocysteine: Pathways and regulation. Eur. J. Pediatr. 1998, 157, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Roorda, M.; Miljkovic, J.L.; van Goor, H.; Henning, R.H.; Bouma, H.R. Spatiotemporal regulation of hydrogen sulfide signaling in the kidney. Redox Biol. 2021, 43, 101961. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Isaak, C.K.; Siow, Y.L.; Karmin, O. Downregulation of cystathionine β-synthase and cystathionine γ-lyase expression stimulates inflammation in kidney ischemia-reperfusion injury. Physiol. Rep. 2014, 2, e12251. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Song, H.; Pan, X.; Xue, H.; Wan, Y.; Wang, T.; Tian, Z.; Hou, E.; Lanza, I.R.; Liu, P.; et al. Urinary metabolites associated with blood pressure on a low- or high-sodium diet. Theranostics 2018, 8, 1468–1480. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Londono, M.; Lee, J.I.; Hu, M.; Yu, A.F. Enzymes and metabolites of cysteine metabolism in nonhepatic tissues of rats show little response to changes in dietary protein or sulfur amino acid levels. J. Nutr. 2002, 132, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Bella, D.L.; Hahn, C.; Stipanuk, M.H. Effects of nonsulfur and sulfur amino acids on the regulation of hepatic enzymes of cysteine metabolism. Am. J. Physiol. 1999, 277, 144–153. [Google Scholar] [CrossRef]
- Laidlaw, S.A.; Grosvenor, M.; Kopple, J.D. The taurine content of common foodstuffs. J. Parenter. Enter. Nutr. 1990, 14, 183–188. [Google Scholar] [CrossRef]
- Lourenço, R.; Camilo, M.E. Taurine: A conditionally essential amino acid in humans? An overview in health and disease. Nutr. Hosp. 2002, 17, 262–270. [Google Scholar]
- Bouckenooghe, T.; Remacle, C.; Reusens, B. Is taurine a functional nutrient? Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E.; Simmons, C.R.; Hirschberger, L.L.; Hwang, J.; Coloso, R.M.; Stipanuk, M.H. Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J. Biol. Chem. 2007, 282, 25189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Takahashi, H.; Lee, L.C.; Sasaki, S.; Kohno, Y.; Takeda, K.; Yoshimura, M.; Nakagawa, M. Retardation of the development of hypertension in DOCA salt rats by taurine supplement. Cardiovasc. Res. 1988, 22, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Ideishi, M.; Miura, S.; Sakai, T.; Sasaguri, M.; Misumi, Y.; Arakawa, K. Taurine amplifies renal kallikrein and prevents salt-induced hypertension in Dahl rats. J. Hypertens. 1994, 12, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Hagar, H.H.; El Etter, E.; Arafa, M. Taurine attenuates hypertension and renal dysfunction induced by cyclosporine A in rats. Clin. Exp. Pharmacol. Physiol. 2006, 33, 189–196. [Google Scholar] [CrossRef]
- Mozaffari, M.S.; Patel, C.; Abdelsayed, R.; Schaffer, S.W. Accelerated NaCl-induced hypertension in taurine-deficient rat: Role of renal function. Kidney Int. 2006, 70, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Xu, X.; Yang, J.; Wu, G.; Sun, C.; Lv, Q. Antihypertensive effect of taurine in rat. Adv. Exp. Med. Biol. 2009, 643, 75–84. [Google Scholar]
- Brodin-Sartorius, A.; Tête, M.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Wang, B.; Li, Y.; Sun, F.; Li, P.; Xia, W.; Zhou, X.; Li, Q.; Wang, X.; Chen, J.; et al. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension 2016, 67, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Mitchell, R.D.; Houben, T.; Palo, A.; Yadati, T.; Parnell, A.J.; Patel, K.; Shiri-Sverdlov, R.; Leake, D.S. Cysteamine Decreases Low-Density Lipoprotein Oxidation, Causes Regression of Atherosclerosis, and Improves Liver and Muscle Function in Low-Density Lipoprotein Receptor-Deficient Mice. J. Am. Heart Assoc. 2021, 10, e017524. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The Quantitative Significance of the Transsulfuration Enzymes for H2S Production in Murine Tissues. Antioxid. Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Prathapasinghe, G.; Wu, N.; Hwang, S.Y.; Siow, Y.L.; O, K. Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am. J. Physiol. Renal Physiol. 2009, 297, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Banerjee, R. Regulation of the redox metabolome and thiol proteome by hydrogen sulfide. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1170. [Google Scholar] [CrossRef] [PubMed]
- Bithi, N.; Link, C.; Henderson, Y.O.; Kim, S.; Yang, J.; Li, L.; Wang, R.; Willard, B.; Hine, C. Dietary restriction transforms the mammalian protein persulfidome in a tissue-specific and cystathionine γ-lyase-dependent manner. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, A.; Vicente, J.B. Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- Perna, A.F.; Zacchia, M.; Trepiccione, F.; Ingrosso, D. The Sulfur Metabolite Lanthionine: Evidence for a Role as a Novel Uremic Toxin. Toxins 2017, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Russo, L.; D’esposito, V.; Formisano, P.; Bruzzese, D.; Vigorito, C.; Coppola, A.; Lombari, P.; Russo, D.; Ingrosso, D. Lanthionine, a Novel Uremic Toxin, in the Vascular Calcification of Chronic Kidney Disease: The Role of Proinflammatory Cytokines. Int. J. Mol. Sci. 2021, 22, 6875. [Google Scholar] [CrossRef]
- Hipólito, A.; Nunes, S.C.; Vicente, J.B.; Serpa, J. Cysteine Aminotransferase (CAT): A Pivotal Sponsor in Metabolic Remodeling and an Ally of 3-Mercaptopyruvate Sulfurtransferase (MST) in Cancer. Molecules 2020, 25, 3984. [Google Scholar] [CrossRef]
- Akbari, M.; Sogutdelen, E.; Juriasingani, S.; Sener, A. Hydrogen Sulfide: Emerging Role in Bladder, Kidney, and Prostate Malignancies. Oxid. Med. Cell. Longev. 2019, 2019, 2360945. [Google Scholar] [CrossRef] [PubMed]
- Randi, E.B.; Casili, G.; Jacquemai, S.; Szabo, C. Selenium-Binding Protein 1 (SELENBP1) Supports Hydrogen Sulfide Biosynthesis and Adipogenesis. Antioxidants 2021, 10, 361. [Google Scholar] [CrossRef]
- Lee, E.K.; Shin, Y.J.; Park, E.Y.; Kim, N.D.; Moon, A.; Kwack, S.J.; Son, J.Y.; Kacew, S.; Lee, B.M.; Bae, O.N.; et al. Selenium-binding protein 1: A sensitive urinary biomarker to detect heavy metal-induced nephrotoxicity. Arch. Toxicol. 2017, 91, 1635–1648. [Google Scholar] [CrossRef]
- Bełtowski, J. Hypoxia in the renal medulla: Implications for hydrogen sulfide signaling. J. Pharmacol. Exp. Ther. 2010, 334, 358–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.J.; Jang, H.S.; Kim, J.I.; Han, S.J.; Park, J.W.; Park, K.M. Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim. Biophys. Acta 2013, 1832, 1989–1997. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ 10 supplementation rescues nephrotic syndrome through normalization of H 2 S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Carter, R.N.; Gibbins, M.T.G.; Barrios-Llerena, M.E.; Wilkie, S.E.; Freddolino, P.L.; Libiad, M.; Vitvitsky, V.; Emerson, B.; Le Bihan, T.; Brice, M.; et al. The hepatic compensatory response to elevated systemic sulfide promotes diabetes. Cell Rep. 2021, 37, 109958. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Huang, C.W.; Moore, P.K. H2S Synthesizing Enzymes: Biochemistry and Molecular Aspects. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide; Moore, P.K., Whiteman, M., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–25. [Google Scholar]
- Adeva-Andany, M.M.; López-Maside, L.; Donapetry-García, C.; Fernández-Fernández, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 2017, 49, 1005–1028. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Price, L.B.; Gappmaier, E.; Cantor, N.L.; Ernst, S.L.; Bailey, C.; Pasquali, M. Anaplerotic therapy in propionic acidemia. Mol. Genet. Metab. 2017, 122, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Shimizu, T.; Shimizu, S.; Higashi, Y.; Nakamura, K.; Ono, H.; Aratake, T.; Saito, M. Possible role of hydrogen sulfide as an endogenous relaxation factor in the rat bladder and prostate. Neurourol. Urodyn. 2018, 37, 2519–2526. [Google Scholar] [CrossRef]
- Pascale, R.M.; Peitta, G.; Simile, M.M.; Feo, F. Alterations of Methionine Metabolism as Potential Targets for the Prevention and Therapy of Hepatocellular Carcinoma. Medicina 2019, 55, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, S.; Guo, J. Lipotoxic Proximal Tubular Injury: A Primary Event in Diabetic Kidney Disease. Front. Med. 2021, 8, 751529. [Google Scholar] [CrossRef]
- Thongnak, L.; Pongchaidecha, A.; Lungkaphin, A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am. J. Med. Sci. 2020, 359, 84–99. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Lewy, P.R.; Quintanilla, A.; Levin, N.W.; Kessler, R.H. Renal energy metabolism and sodium reabsorption. Annu. Rev. Med. 1973, 24, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guan, Q.; Liu, Y.; Zhang, Y.; Chen, Y.; Chen, J.; Liu, Y.; Su, Z. Regulation of hepatic gluconeogenesis by nuclear factor Y transcription factor in mice. J. Biol. Chem. 2018, 293, 7894–7904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Guder, W.G.; Ross, B.D. Enzyme distribution along the nephron. Kidney Int. 1984, 26, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, L.J. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu. Rev. Physiol. 1985, 47, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. Suppl. 2011, 79, S1–S6. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Thompson, S.C.; McDonough, A.A. Metabolic Basis of Solute Transport. In Brenner & Rector’s The Kidney; Elsevier: Amsterdam, The Netherlands, 2019; pp. 133–155. [Google Scholar]
- Pfaller, W.; Rittinger, M. Quantitative morphology of the rat kidney. Int. J. Biochem. 1980, 12, 17–22. [Google Scholar] [CrossRef]
- Irokawa, H.; Tachibana, T.; Watanabe, T.; Matsuyama, Y.; Motohashi, H.; Ogasawara, A.; Iwai, K.; Naganuma, A.; Kuge, S. Redox-dependent Regulation of Gluconeogenesis by a Novel Mechanism Mediated by a Peroxidatic Cysteine of Peroxiredoxin. Sci. Rep. 2016, 6, 33536. [Google Scholar] [CrossRef]
- Stumvoll, M.; Meyer, C.; Perriello, G.; Kreider, M.; Welle, S.; Gerich, J. Human kidney and liver gluconeogenesis: Evidence for organ substrate selectivity. Am. J. Physiol. 1998, 274, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, M. Cysteine. In Nutrient Metabolism; Elsevier: Amsterdam, The Netherlands, 2003; pp. 348–356. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Zeng, X.; Cowan, A.J.; Wang, L.; Chen, L.; Morscher, R.J.; Reyes, J.; Frezza, C.; Hwang, H.Y.; et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 2019, 30, 594–606.e3. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Bodin, S.; Ferrier, B.; Delage, H.; Boghossian, M.; Martin, M.; Baverel, G.; Conjard, A. Intrinsic gluconeogenesis is enhanced in renal proximal tubules of Zucker diabetic fatty rats. J. Am. Soc. Nephrol. 2006, 17, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Legouis, D.; Faivre, A.; Cippà, P.E.; de Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant 2020, 1–9. [Google Scholar] [CrossRef]
- Akhtar, S.; Culver, S.A.; Siragy, H.M. Novel regulation of renal gluconeogenesis by Atp6ap2 in response to high fat diet via PGC1-α/AKT-1 pathway. Sci. Rep. 2021, 11, 11367. [Google Scholar] [CrossRef]
- Sharma, R.; Tiwari, S. Renal gluconeogenesis in insulin resistance: A culprit for hyperglycemia in diabetes. World J. Diabetes 2021, 12, 556–568. [Google Scholar] [CrossRef]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Yamagishi, S.I. Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol. Metab. Syndr. 2015, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Martinez, J.I. The regulation of the pentose phosphate pathway: Remember Krebs. Arch. Biochem. Biophys. 2017, 614, 50–52. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef]
- Smith, J.A.; Jay Stallons, L.; Schnellmann, R.G. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2014, 307, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Guo, R.; Wang, Y.; Cunningham, P.N. The role of ICAM-1 in endotoxin-induced acute renal failure. Am. J. Physiol. Renal Physiol. 2007, 293, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Schley, G.; Klanke, B.; Schödel, J.; Forstreuter, F.; Shukla, D.; Kurtz, A.; Amann, K.; Wiesener, M.S.; Rosen, S.; Eckardt, K.U.; et al. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 2004–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, J.A.; Venkatachalam, M.A.; Weinberg, J.M. Proximal Tubular Oxidative Metabolism in Acute Kidney Injury and the Transition to CKD. Kidney360 2021, 2, 355–364. [Google Scholar] [CrossRef]
- Shaik, Z.P.; Fifer, E.K.; Nowak, G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am. J. Physiol. Renal Physiol. 2008, 294, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuhra, K.; Tomé, C.S.; Forte, E.; Vicente, J.B.; Giuffrè, A. The multifaceted roles of sulfane sulfur species in cancer-associated processes. Biochim. Biophys. Acta-Bioenerg. 2021, 1862, 148338. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, P.; Marshall, D.C.; Cooper, C.E.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Landry, A.P.; Guha, A.; Vitvitsky, V.; Lee, H.J.; Seike, K.; Reddy, P.; Lyssiotis, C.A.; Banerjee, R. A redox cycle with complex II prioritizes sulfide quinone oxidoreductase-dependent H2S oxidation. J. Biol. Chem. 2021, 298, 101435. [Google Scholar] [CrossRef]
- Módis, K.; Ju, Y.J.; Ahmad, A.; Untereiner, A.A.; Altaany, Z.; Wu, L.; Szabo, C.; Wang, R. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 2016, 113, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [Green Version]
- Untereiner, A.A.; Oláh, G.; Módis, K.; Hellmich, M.R.; Szabo, C. H 2 S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem. Pharmacol. 2017, 136, 86–98. [Google Scholar] [CrossRef]
- Poltorack, C.D.; Dixon, S.J. Understanding the role of cysteine in ferroptosis: Progress & paradoxes. FEBS J. 2021, 289, 374–385. [Google Scholar] [CrossRef]
- Shi, Z.; Naowarojna, N.; Pan, Z.; Zou, Y. Multifaceted mechanisms mediating cystine starvation-induced ferroptosis. Nat. Commun. 2021, 12, 1–4. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Carlo Maurer, H.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1181. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, N.; Zhou, Y.; Wang, K.; Sun, Y.; Yan, H.; Han, W.; Wang, X.; Wei, B.; Ke, Y.; et al. Oridonin induces ferroptosis by inhibiting gamma-glutamyl cycle in TE1 cells. Phyther. Res. 2021, 35, 494–503. [Google Scholar] [CrossRef]
- Zhu, J.; Berisa, M.; Schwörer, S.; Qin, W.; Cross, J.R.; Thompson, C.B. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019, 30, 876. [Google Scholar] [CrossRef]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Leu, J.I.J.; Murphy, M.E.; George, D.L. Functional interplay among thiol-based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc. Natl. Acad. Sci. USA 2020, 117, 26804–26811. [Google Scholar] [CrossRef] [PubMed]
- Borini Etichetti, C.M.; Arel Zalazar, E.; Cocordano, N.; Girardini, J. Beyond the Mevalonate Pathway: Control of Post-Prenylation Processing by Mutant p53. Front. Oncol. 2020, 10, 595034. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Rosenfeldt, F.L.; Haas, S.J.; Krum, H.; Hadj, A.; Ng, K.; Leong, J.Y.; Watts, G.F. Coenzyme Q10 in the treatment of hypertension: A meta-analysis of the clinical trials. J. Hum. Hypertens. 2007, 21, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell. Signal. 2021, 78, 109870. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef]
- Tanaka, T.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 2004, 165, 1979–1992. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.C.; Ramos, C.; Santos, I.; Mendes, C.; Silva, F.; Vicente, J.B.; Pereira, S.A.; Félix, A.; Gonçalves, L.G.; Serpa, J. Cysteine Boosts Fitness Under Hypoxia-Mimicked Conditions in Ovarian Cancer by Metabolic Reprogramming. Front. Cell Dev. Biol. 2021, 9, 722412. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.G.; Smith, D.W.; Lee, C.J.; Ngo, J.P.; Gardiner, B.S. What Makes the Kidney Susceptible to Hypoxia? Anat. Rec. 2020, 303, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Wirthensohn, G.; Guder, W.G. Renal substrate metabolism. Physiol. Rev. 1986, 66, 469–497. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Mandel, L.J. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am. J. Physiol. 1988, 254, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants 2021, 10, 1650. [Google Scholar] [CrossRef] [PubMed]
- Aukland, K.; Krog, J. Renal oxygen tension. Nature 1960, 188, 671. [Google Scholar] [CrossRef]
- Epstein, F.H.; Agmon, Y.; Brezis, M. Physiology of renal hypoxia. Ann. N. Y. Acad. Sci. 1994, 718, 72–82. [Google Scholar] [CrossRef]
- Evans, R.G.; Gardiner, B.S.; Smith, D.W.; O’Connor, P.M. Intrarenal oxygenation: Unique challenges and the biophysical basis of homeostasis. Am. J. Physiol. Renal Physiol. 2008, 295, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J. Exp. Biol. 2006, 209, 4011–4023. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, N.L.; Kreimier, E.L.; Verdial, F.C.; Skovgaard, N.; Olson, K.R. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, 1930–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, T.A.; Gustafsson, H.; Palm, F. Kidney outer medulla mitochondria are more efficient compared with cortex mitochondria as a strategy to sustain ATP production in a suboptimal environment. Am. J. Physiol. Renal Physiol. 2018, 315, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malagrinò, F.; Zuhra, K.; Mascolo, L.; Mastronicola, D.; Vicente, J.B.; Forte, E.; Giuffrè, A. Hydrogen sulfide oxidation: Adaptive changes in mitochondria of SW480 colorectal cancer cells upon exposure to hypoxia. Oxid. Med. Cell. Longev. 2019, 2019, 8102936. [Google Scholar] [CrossRef]
- Marutani, E.; Morita, M.; Hirai, S.; Kai, S.; Grange, R.M.H.; Miyazaki, Y.; Nagashima, F.; Traeger, L.; Magliocca, A.; Ida, T.; et al. Sulfide catabolism ameliorates hypoxic brain injury. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Perna, A.F.; Luciano, M.G.; Ingrosso, D.; Pulzella, P.; Sepe, I.; Lanza, D.; Violetti, E.; Capasso, R.; Lombardi, C.; De Santo, N.G. Hydrogen sulphide-generating pathways in haemodialysis patients: A study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrol. Dial. Transplant. 2009, 24, 3756–3763. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Zeng, P.; Friedrich, J.L.; Leonard, E.C.; Yoder, M.C. Low proliferative potential and impaired angiogenesis of cultured rat kidney endothelial cells. Microcirculation 2012, 19, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Smith, D.W.; Gardiner, B.S.; Evans, R.G. Stimulation of erythropoietin release by hypoxia and hypoxemia: Similar but different. Kidney Int. 2019, 95, 23–25. [Google Scholar] [CrossRef]
- Montero, D.; Lundby, C. Arterial oxygen content regulates plasma erythropoietin independent of arterial oxygen tension: A blinded crossover study. Kidney Int. 2019, 95, 173–177. [Google Scholar] [CrossRef]
- Donnelly, S. Why is erythropoietin made in the kidney? The kidney functions as a “critmeter” to regulate the hematocrit. Adv. Exp. Med. Biol. 2003, 543, 73–87. [Google Scholar]
- Goldfarb-Rumyantzev, A.S.; Alper, S.L. Short-term responses of the kidney to high altitude in mountain climbers. Nephrol. Dial. Transplant. 2014, 29, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Coelho, F.; Martins, F.; Hipólito, A.; Mendes, C.; Sequeira, C.O.; Pires, R.F.; Almeida, A.M.; Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J. The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy. Front. Oncol. 2021, 11, 656229. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kumar, G.; Chhabra, A.; Sethy, N.K.; Jain, N.; Meena, R.N.; Tulsawani, R.; Prasad, D.N.; Kumar, B.; Sharma, M. Cysteine becomes conditionally essential during hypobaric hypoxia and regulates adaptive neuro-physiological responses through CBS/H2S pathway. Biochim. Biophys. Acta Mol. basis Dis. 2020, 1866, 165769. [Google Scholar] [CrossRef] [PubMed]
- Takano, N.; Peng, Y.J.; Kumar, G.K.; Luo, W.; Hu, H.; Shimoda, L.A.; Suematsu, M.; Prabhakar, N.R.; Semenza, G.L. Hypoxia-inducible factors regulate human and rat cystathionine β-synthase gene expression. Biochem. J. 2014, 458, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Pan, L.L.; Zhuo, Y.; Gong, Q.H.; Rose, P.; Zhu, Y.Z. Hypoxia-inducible factor-1α is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol. Pharm. Bull. 2010, 33, 1550–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, W.J. Intrarenal oxygen and hypertension. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Nordquist, L. Renal oxidative stress, oxygenation, and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef] [Green Version]
- O’neill, J.; Jasionek, G.; Drummond, S.E.; Brett, O.; Lucking, E.F.; Abdulla, M.A.; O’halloran, K.D. Renal cortical oxygen tension is decreased following exposure to long-term but not short-term intermittent hypoxia in the rat. Am. J. Physiol. Renal Physiol. 2019, 316, 635–645. [Google Scholar] [CrossRef]
- Hasegawa, S.; Tanaka, T.; Saito, T.; Fukui, K.; Wakashima, T.; Susaki, E.A.; Ueda, H.R.; Nangaku, M. The oral hypoxia-inducible factor prolyl hydroxylase inhibitor enarodustat counteracts alterations in renal energy metabolism in the early stages of diabetic kidney disease. Kidney Int. 2020, 97, 934–950. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Shino, M.; Fujikawa, T.; Itoh, Y.; Ueda, E.; Hashimoto, T.; Kuji, T.; Kobayashi, N.; Ohnishi, T.; Hirawa, N.; et al. Plasma Cystine Levels and Cardiovascular and All-Cause Mortality in Hemodialysis Patients. Ther. Apher. Dial. 2018, 22, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Alexander, R.W.; Harrison, D.G.; Quyyumi, A.A. Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension 2008, 52, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Ghasemzadeh, N.; Eapen, D.J.; Sher, S.; Arshad, S.; Ko, Y.A.; Veledar, E.; Samady, H.; Zafari, A.M.; Sperling, L.; et al. Novel Biomarker of Oxidative Stress Is Associated With Risk of Death in Patients With Coronary Artery Disease. Circulation 2016, 133, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Lorenzini, S.; Milzani, A.; Rossi, R. Age-related influence on thiol, disulfide, and protein-mixed disulfide levels in human plasma. J. Gerontol. A. Biol. Sci. Med. Sci. 2006, 61, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Harrison, D.G.; Quyyumi, A.A. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J. Am. Coll. Cardiol. 2006, 47, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Kum, F.; Wong, K.; Game, D.; Bultitude, M.; Thomas, K. Hypertension and renal impairment in patients with cystinuria: Findings from a specialist cystinuria centre. Urolithiasis 2019, 47, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Vasdev, S.; Singal, P.; Gill, V. The antihypertensive effect of cysteine. Int. J. Angiol. 2009, 18, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Vasdev, S.; Stuckless, J. Antihypertensive effects of dietary protein and its mechanism. Int. J. Angiol. 2010, 19, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Yousaf, F.; Spinowitz, B. Hypoxia-Inducible Factor Stabilizers: A New Avenue for Reducing BP While Helping Hemoglobin? Curr. Hypertens. Rep. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Jose, P.A.; Raj, D. Gut microbiota in hypertension. Curr. Opin. Nephrol. Hypertens. 2015, 24, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, N.R.; Matos, C.; Pimpão, A.B.; Correia, M.J.; Sequeira, C.O.; Morello, J.; Pereira, S.A.; Monteiro, E.C. AHR canonical pathway: In vivo findings to support novel antihypertensive strategies. Pharmacol. Res. 2021, 165, 105407. [Google Scholar] [CrossRef] [PubMed]
- Coelho, N.R.; Tomkiewicz, C.; Correia, M.J.; Gonçalves-Dias, C.; Barouki, R.; Pereira, S.A.; Coumoul, X.; Monteiro, E.C. First evidence of aryl hydrocarbon receptor as a druggable target in hypertension induced by chronic intermittent hypoxia. Pharmacol. Res. 2020, 159, 104869. [Google Scholar] [CrossRef]
- Diogo, L.N.; Pereira, S.A.; Nunes, A.R.; Afonso, R.A.; Santos, A.I.; Monteiro, E.C. Efficacy of carvedilol in reversing hypertension induced by chronic intermittent hypoxia in rats. Eur. J. Pharmacol. 2015, 765, 58–67. [Google Scholar] [CrossRef]
- Grilo, N.M.; João Correia, M.; Miranda, J.P.; Cipriano, M.; Serpa, J.; Matilde Marques, M.; Monteiro, E.C.; Antunes, A.M.M.; Diogo, L.N.; Pereira, S.A. Unmasking efavirenz neurotoxicity: Time matters to the underlying mechanisms. Eur. J. Pharm. Sci. 2017, 105, 47–54. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, M.J.; Pimpão, A.B.; Fernandes, D.G.F.; Morello, J.; Sequeira, C.O.; Calado, J.; Antunes, A.M.M.; Almeida, M.S.; Branco, P.; Monteiro, E.C.; et al. Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine. Molecules 2022, 27, 1416. https://doi.org/10.3390/molecules27041416
Correia MJ, Pimpão AB, Fernandes DGF, Morello J, Sequeira CO, Calado J, Antunes AMM, Almeida MS, Branco P, Monteiro EC, et al. Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine. Molecules. 2022; 27(4):1416. https://doi.org/10.3390/molecules27041416
Chicago/Turabian StyleCorreia, Maria João, António B. Pimpão, Dalila G. F. Fernandes, Judit Morello, Catarina O. Sequeira, Joaquim Calado, Alexandra M. M. Antunes, Manuel S. Almeida, Patrícia Branco, Emília C. Monteiro, and et al. 2022. "Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine" Molecules 27, no. 4: 1416. https://doi.org/10.3390/molecules27041416
APA StyleCorreia, M. J., Pimpão, A. B., Fernandes, D. G. F., Morello, J., Sequeira, C. O., Calado, J., Antunes, A. M. M., Almeida, M. S., Branco, P., Monteiro, E. C., Vicente, J. B., Serpa, J., & Pereira, S. A. (2022). Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine. Molecules, 27(4), 1416. https://doi.org/10.3390/molecules27041416