
Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations



Abstract
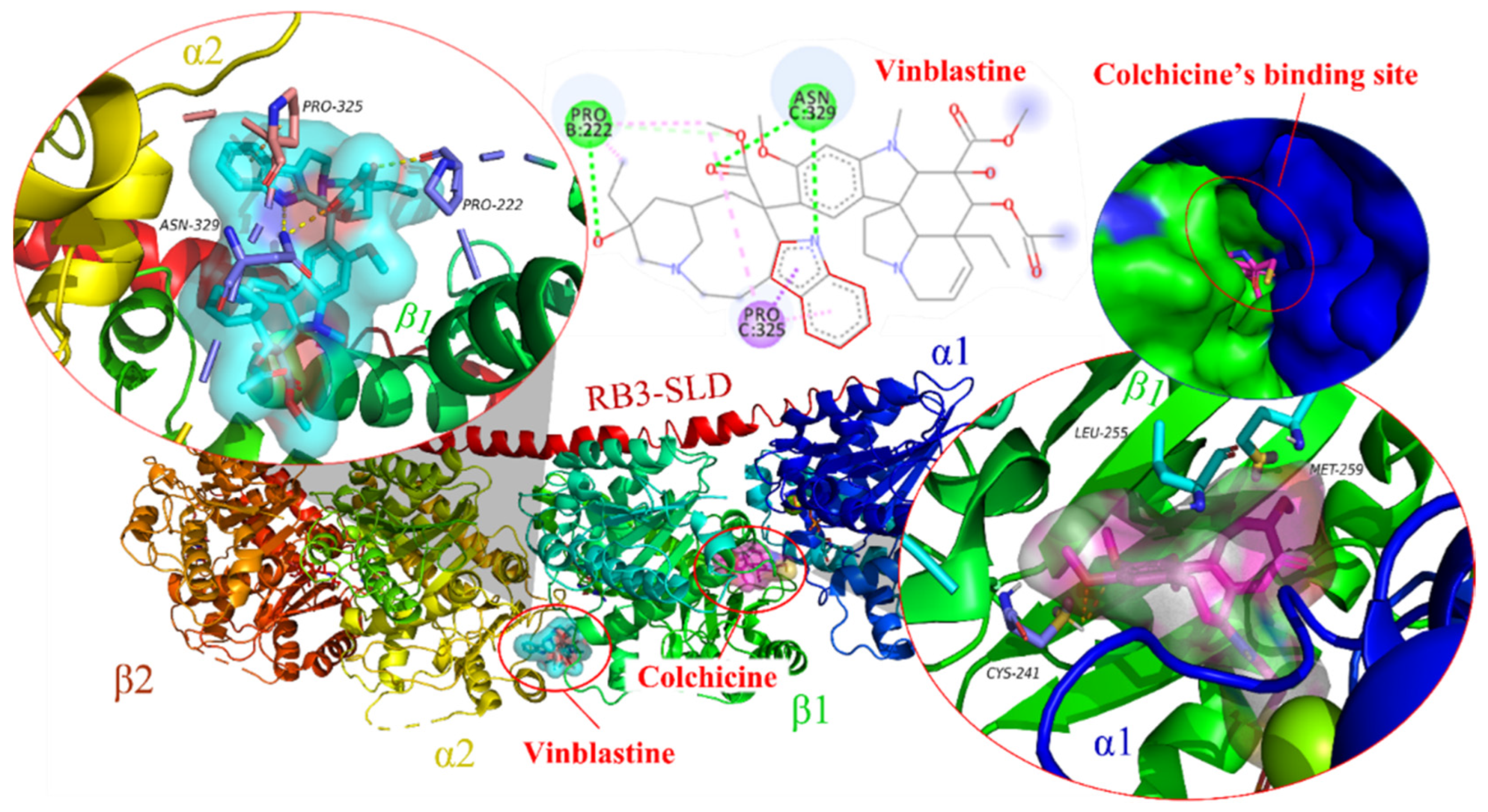
:1. Introduction
2. Indole-Based Tubulin Inhibitors
2.1. TMP Analogues
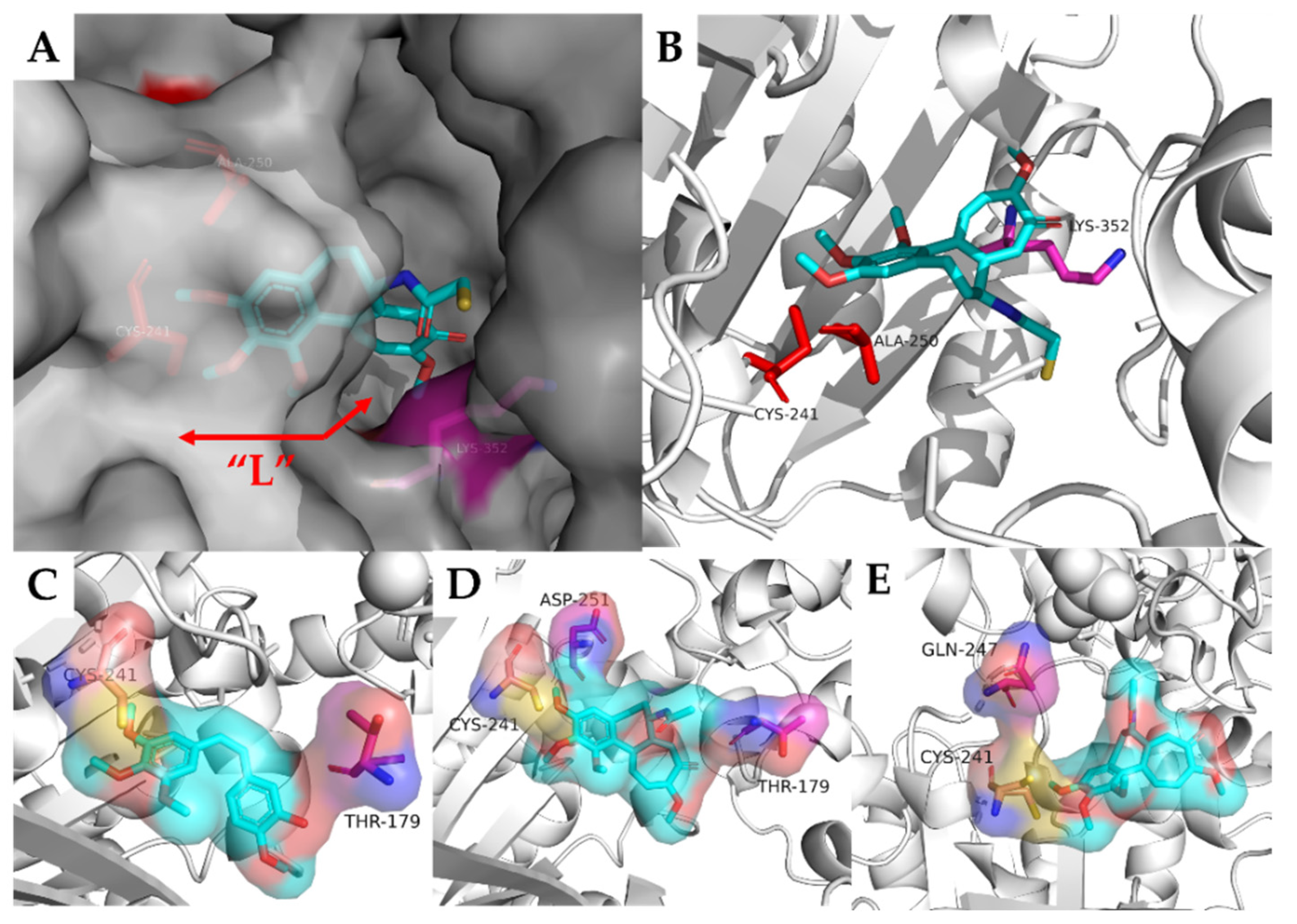
2.2. Arythioindoles
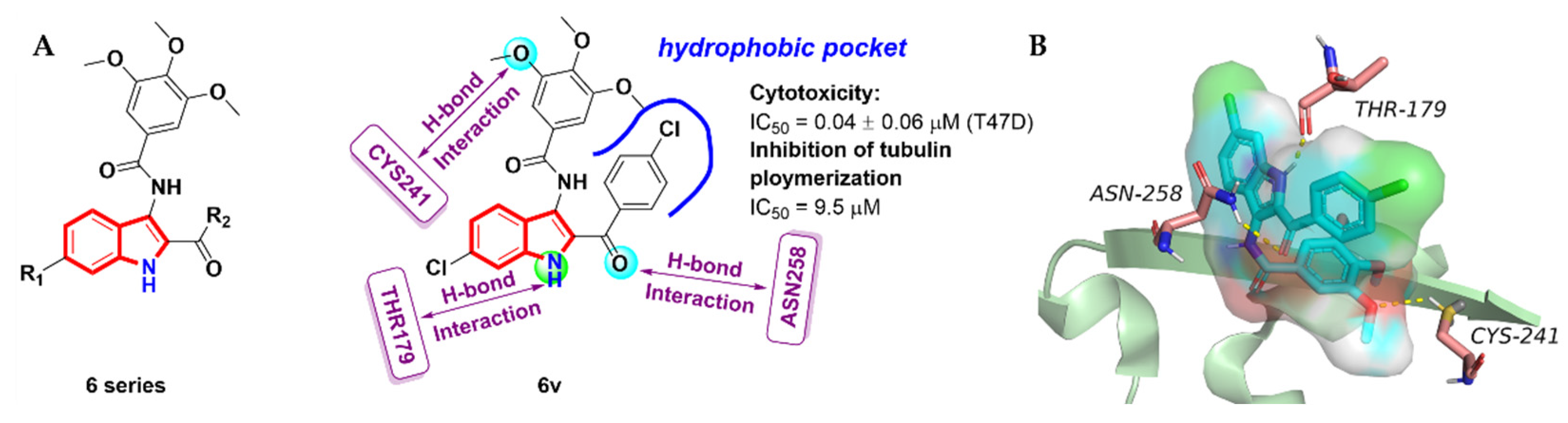
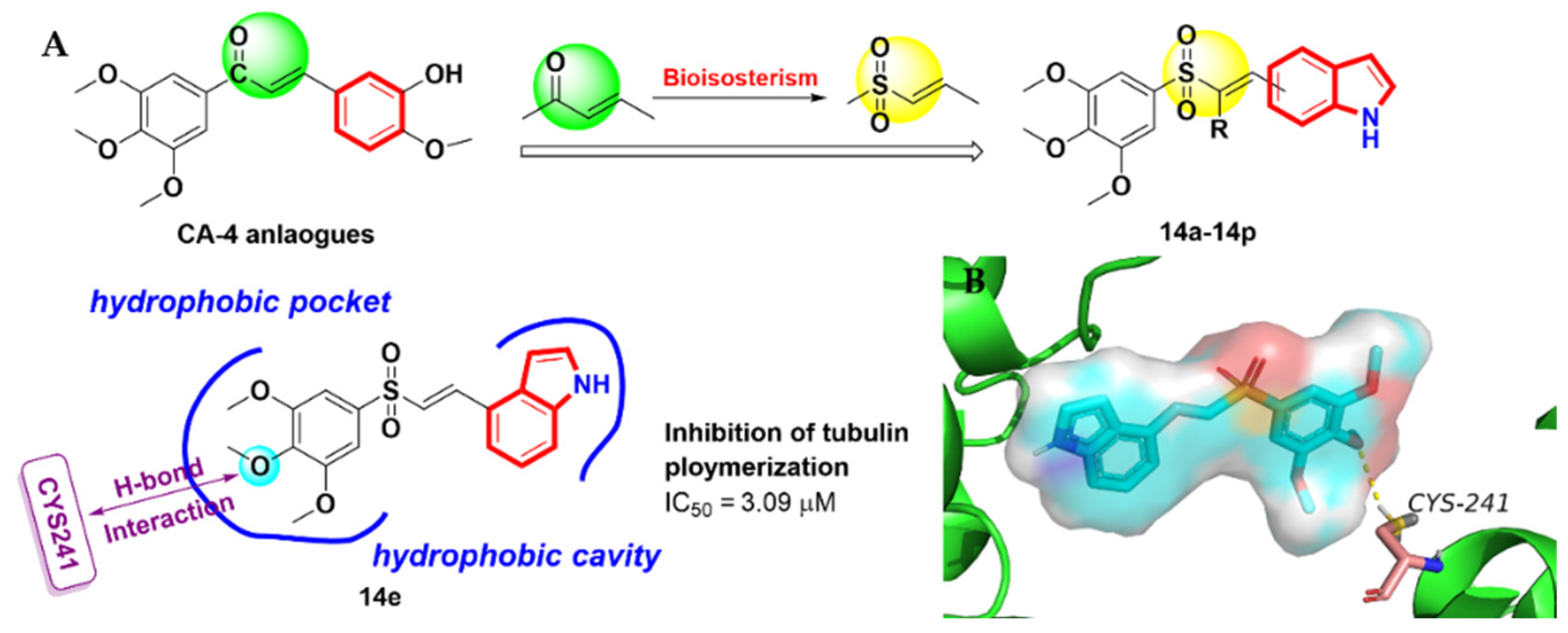
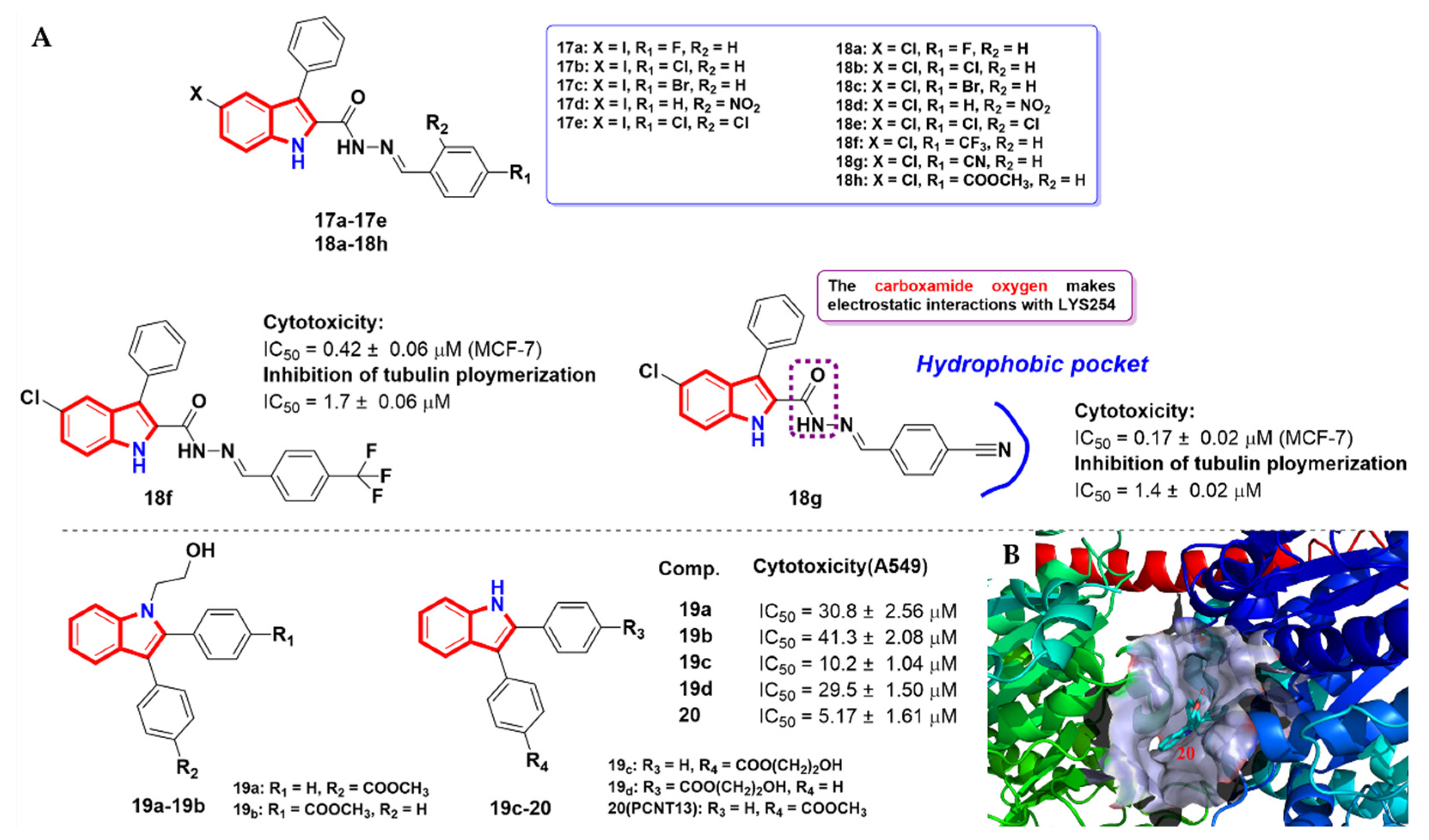
2.3. Aroyindoles
2.4. Fused Indole
2.5. Carbazoles
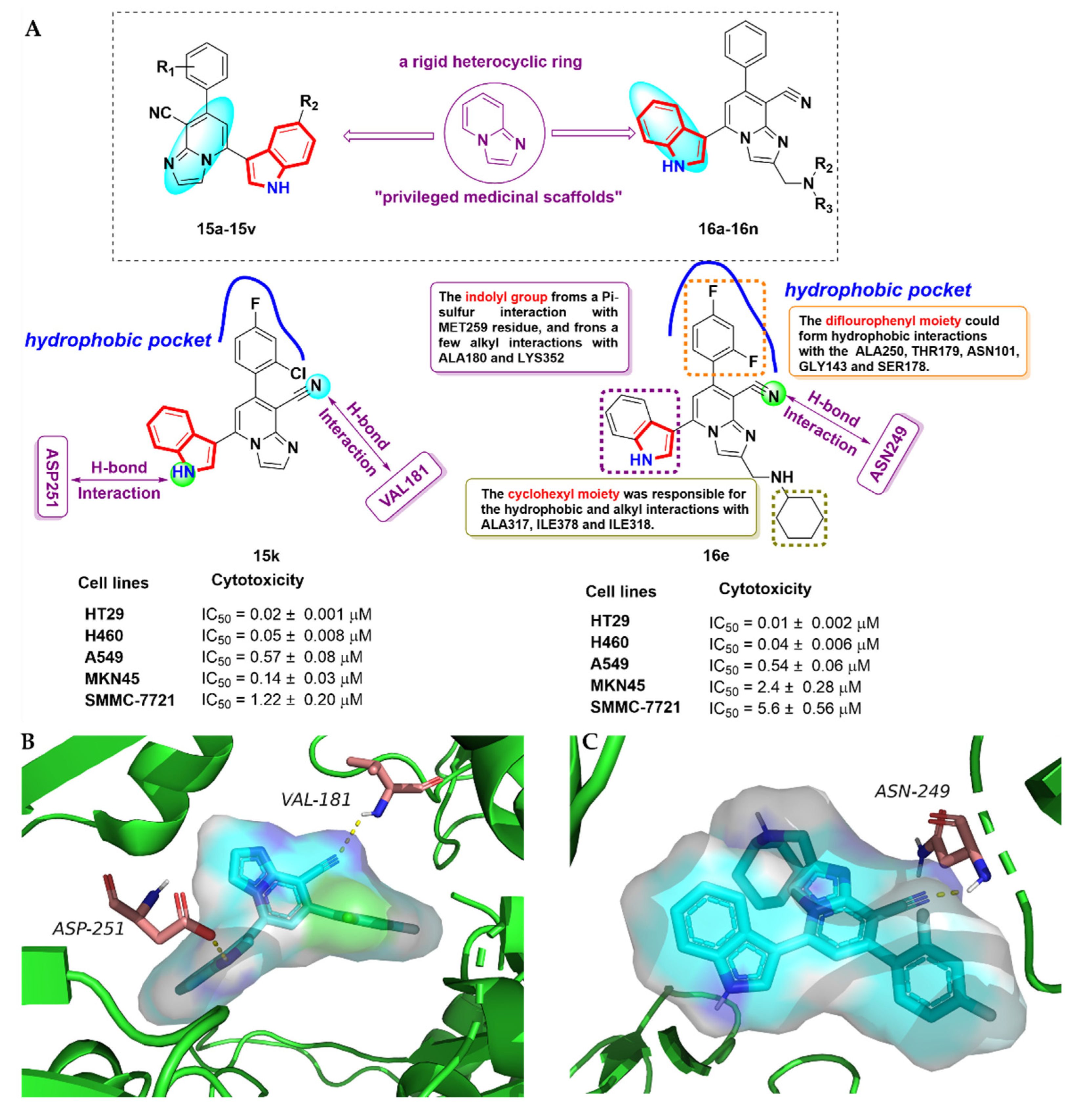
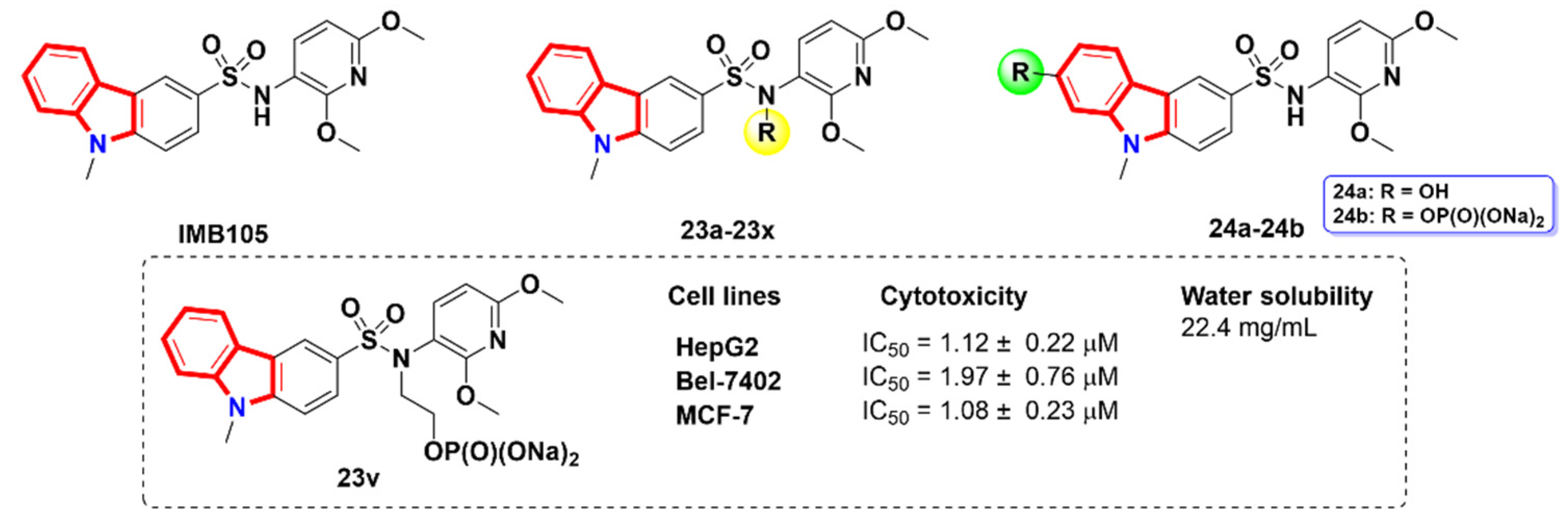
2.6. Azacarbolines
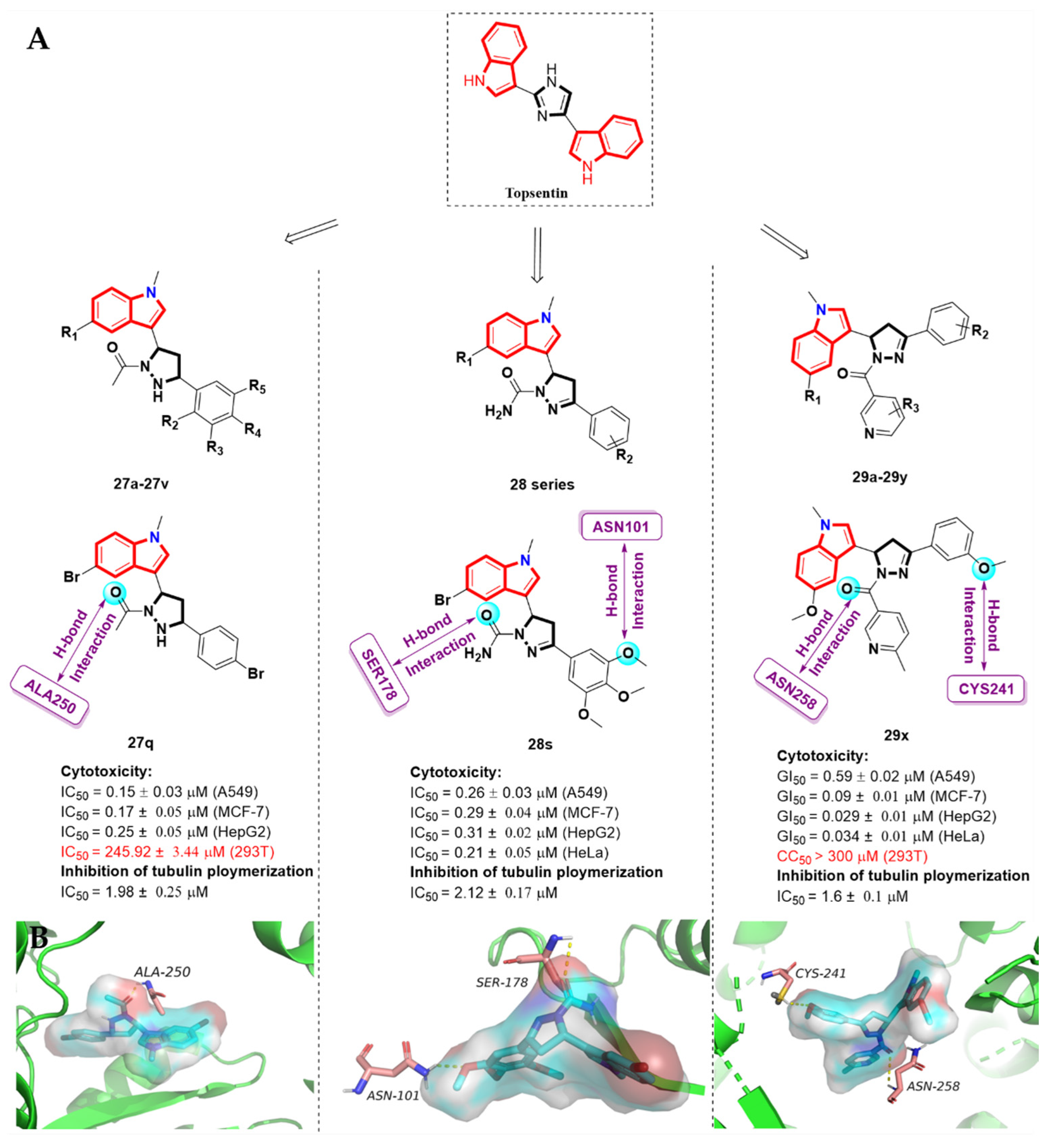
2.7. Alkaloid Nortopsentin Analogues
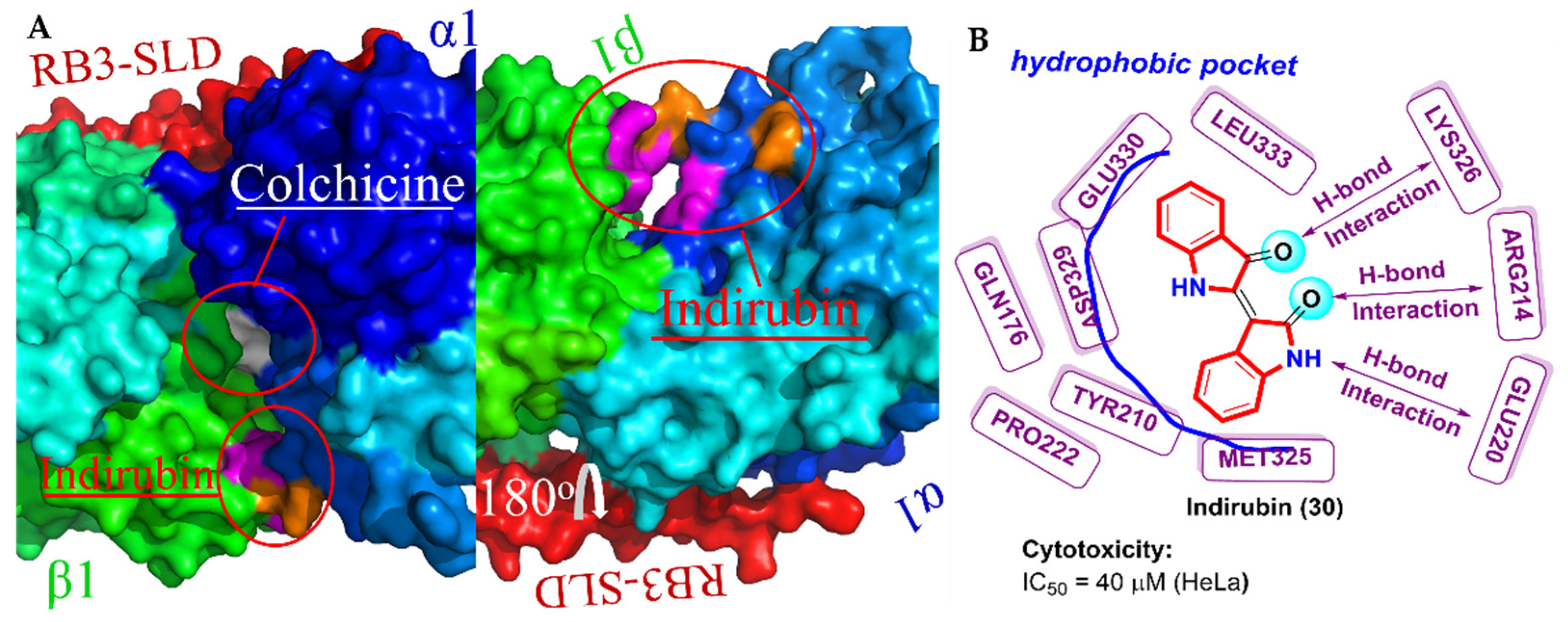
2.8. Bis-Indole Derivatives
2.9. Others
3. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gigant, B.; Wang, C.G.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Dash, C.; Lu, J.C.; Parikh, V.; Wathen, S.; Shah, S.; Chaudhari, R.S.; Adams-Campbell, L. Disparities in colorectal cancer screening among breast and prostate cancer survivors. Cancer Med. 2021, 10, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Koutros, S.; Decker, K.L.; Baris, D.; Pardo, L.A.; Johnson, A.; Hosain, G.M.M.; Rothman, N.; Karagas, M.R.; Schwenn, M.R.; Silverman, D.T. Bladder cancer risk associated with family history of cancer. Int. J. Cancer 2021, 148, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.N.; Zhang, L.X.; Hei, R.X.; Li, X.; Cai, H.N.; Wu, X.; Zheng, Q.P.; Cai, C.G. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Thilakasiri, P.S.; Dmello, R.S.; Nero, T.L.; Parker, M.W.; Ernst, M.; Chand, A.L. Repurposing of drugs as STAT3 inhibitors for cancer therapy. Semin. Cancer Biol. 2021, 68, 31–46. [Google Scholar] [CrossRef]
- Georas, S.N.; Donohue, P.; Connolly, M.; Wechsler, M.E. JAK inhibitors for asthma. J. Allergy Clin. Immunol. 2021, 148, 953–963. [Google Scholar] [CrossRef]
- Van Order, R.B.; Lindwall, H.G. Indole. Chem. Rev. 1942, 30, 69–96. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, F.; Budman, D.R. Review: Tubulin function, action of antitubulin drugs, and new drug development. Cancer Investig. 2005, 23, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Gunning, P.W.; Ghoshdastider, U.; Whitaker, S.; Popp, D.; Robinson, R.C. The evolution of compositionally and functionally distinct actin filaments. J. Cell Sci. 2015, 128, 2009–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A.; Himes, R.H.; Wilson, L. Comparison of the effects of vinblastine, vincristine, vindesine, and vinepidine on microtubule dynamics and cell proliferation in vitro. Cancer Res. 1985, 45, 2741–2747. [Google Scholar]
- Sravanthi, T.V.; Manju, S.L. Indoles—A promising scaffold for drug development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef]
- Singh, A.; Fatima, K.; Srivastava, A.; Khwaja, S.; Priya, D.; Singh, A.; Mahajan, G.; Alam, S.; Saxena, A.K.; Mondhe, D.M.; et al. Anticancer activity of gallic acid template-based benzylidene indanone derivative as microtubule destabilizer. Chem. Biol. Drug Des. 2016, 88, 625–634. [Google Scholar] [CrossRef]
- Rastogi, S.K.; Dunnigan, J.K.; Towne, A.C.; Zhao, Z.Z.; Du, L.Q.; Brittain, W.J. Photopharmacology of Azo-Combretastatin-A4: Utilizing Tubulin Polymerization Inhibitors and Green Chemistry as the Key Steps. Curr. Org. Chem. 2021, 25, 2457–2474. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Bai, R.; Covell, D.G.; Pei, X.F.; Ewell, J.B.; Nguyen, N.Y.; Brossi, A.; Hamel, E. Mapping the binding site of colchicinoids on beta -tubulin. 2-Chloroacetyl-2-demethylthiocolchicine covalently reacts predominantly with cysteine 239 and secondarily with cysteine 354. J. Biol. Chem. 2000, 275, 40443–40452. [Google Scholar] [CrossRef] [Green Version]
- dos Santos Edos, A.; Hamel, E.; Bai, R.; Burnett, J.C.; Tozatti, C.S.; Bogo, D.; Perdomo, R.T.; Antunes, A.M.; Marques, M.M.; Matos Mde, F.; et al. Synthesis and evaluation of diaryl sulfides and diaryl selenide compounds for antitubulin and cytotoxic activity. Bioorg. Med. Chem. Lett. 2013, 23, 4669–4673. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Kajita, D.; Matsumoto, Y.; Hashimoto, Y. Design and synthesis of silicon-containing tubulin polymerization inhibitors: Replacement of the ethylene moiety of combretastatin A-4 with a silicon linker. Bioorg. Med. Chem. 2013, 21, 7381–7391. [Google Scholar] [CrossRef] [PubMed]
- Soussi, M.A.; Provot, O.; Bernadat, G.; Bignon, J.; Wdzieczak-Bakala, J.; Desravines, D.; Dubois, J.; Brion, J.D.; Messaoudi, S.; Alami, M. Discovery of azaisoerianin derivatives as potential antitumors agents. Eur. J. Med. Chem. 2014, 78, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Preti, D.; Tabrizi, M.A.; Balzarini, J.; Bassetto, M.; Brancale, A.; Fu, X.H.; Gao, Y.; et al. Concise Synthesis and Biological Evaluation of 2-Aroyl-5-Amino Benzo b thiophene Derivatives As a Novel Class of Potent Antimitotic Agents. J. Med. Chem. 2013, 56, 9296–9309. [Google Scholar] [CrossRef] [Green Version]
- La Regina, G.; Bai, R.; Coluccia, A.; Naccarato, V.; Famiglini, V.; Nalli, M.; Masci, D.; Verrico, A.; Rovella, P.; Mazzoccoli, C.; et al. New 6-and 7-heterocyclyl-1H-indole derivatives as potent tubulin assembly and cancer cell growth inhibitors. Eur. J. Med. Chem. 2018, 152, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.Y.; Xu, J.W.; Wang, Z.W.; Qi, H.; Xu, Q.L.; Bai, Z.S.; Zhang, Q.; Bao, K.; Wu, Y.L.; Zhang, W.G. 3-(3,4,5-Trimethoxyphenylselenyl)-1H-indoles and their selenoxides as combretastatin A-4 analogs: Microwave-assisted synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 90, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Salvador, M.K.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef]
- Chen, P.; Zhuang, Y.X.; Diao, P.C.; Yang, F.; Wu, S.Y.; Lv, L.; You, W.W.; Zhao, P.L. Synthesis, biological evaluation, and molecular docking investigation of 3-amidoindoles as potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2019, 162, 525–533. [Google Scholar] [CrossRef]
- Liou, J.-P.; Chang, Y.-L.; Kuo, F.-M.; Chang, C.-W.; Tseng, H.-Y.; Wang, C.-C.; Yang, Y.-N.; Chang, J.-Y.; Lee, S.-J.; Hsieh, H.-P. Concise Synthesis and Structure−Activity Relationships of Combretastatin A-4 Analogues, 1-Aroylindoles and 3-Aroylindoles, as Novel Classes of Potent Antitubulin Agents. J. Med. Chem. 2004, 47, 4247–4257. [Google Scholar] [CrossRef]
- Mullagiri, K.; Nayak, V.L.; Sunkari, S.; Mani, G.S.; Guggilapu, S.D.; Nagaraju, B.; Alarifi, A.; Kamal, A. New (3-(1H-benzo[d]imidazol-2-yl))/(3-(3H-imidazo[4,5-b]pyridin-2-yl))-(1H-indol-5-yl)(3,4,5-trimethoxyphenyl)methanone conjugates as tubulin polymerization inhibitors. MedChemComm 2018, 9, 275–281. [Google Scholar] [CrossRef]
- Wang, Y.T.; Qin, Y.J.; Yang, N.; Zhang, Y.L.; Liu, C.H.; Zhu, H.L. Synthesis, biological evaluation, and molecular docking studies of novel 1-benzene acyl-2-(1-methylindol-3-yl)-benzimidazole derivatives as potential tubulin polymerization inhibitors. Eur. J. Med. Chem. 2015, 99, 125–137. [Google Scholar] [CrossRef]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.H.; Huang, L.; Li, X.S. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Ranjith, P.K.; Pakkath, R.; Haridas, K.R.; Kumari, S.N. Synthesis and characterization of new N-(4-(4-chloro-1H-imidazol-1-yl)-3-methoxyphenyl)amide/sulfonamide derivatives as possible antimicrobial and antitubercular agents. Eur. J. Med. Chem. 2014, 71, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Barie, P.S. Non-Operative Management of Appendicitis: Evolution, not Revolution. Surg. Infect. 2021, 22, 991–1003. [Google Scholar] [CrossRef]
- Shah, S.S.A.; Rivera, G.; Ashfaq, M. Recent Advances in Medicinal Chemistry of Sulfonamides. Rational Design as Anti-Tumoral, Anti-Bacterial and Anti-Inflammatory Agents. Mini-Rev. Med. Chem. 2013, 13, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Carta, F.; Supuran, C.T. Secondary and tertiary sulfonamides: A patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.X.; Li, Z.R.; Jiang, J.D.; Boykin, D.W. Novel Diaryl or Heterocyclic Sulfonamides as Antimitotic Agents. Anticancer. Agents Med. Chem. 2008, 8, 739–745. [Google Scholar] [CrossRef]
- Aceves-Luquero, C.; Galiana-Rosello, C.; Ramis, G.; Villalonga-Planells, R.; Garcia-Espana, E.; de Mattos, S.F.; Pelaez, R.; Llinares, J.M.; Gonzalez-Rosende, M.E.; Villalonga, P. N-(2-methyl-indol-1H-5-yl)-1-naphthalenesulfonamide: A novel reversible antimitotic agent inhibiting cancer cell motility. Biochem. Pharmacol. 2016, 115, 28–42. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, J.J.; Chen, J.L.; Ji, P.; Qiao, C.H. N-Arylsulfonylsubstituted-1H indole derivatives as small molecule dual inhibitors of signal transducer and activator of transcription 3 (STAT3) and tubulin. Biorg. Med. Chem. 2018, 26, 96–106. [Google Scholar] [CrossRef]
- Ji, P.; Xu, X.; Ma, S.; Fan, J.; Zhou, Q.; Mao, X.; Qiao, C. Novel 2-Carbonylbenzo[b]thiophene 1,1-Dioxide Derivatives as Potent Inhibitors of STAT3 Signaling Pathway. ACS Med. Chem. Lett. 2015, 6, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Li, W.L.; Sun, H.H.; Xu, F.J.; Shuai, W.; Liu, J.; Xu, S.T.; Yao, H.Q.; Ma, C.; Zhu, Z.Y.; Xu, J.Y. Synthesis, molecular properties prediction and biological evaluation of indole-vinyl sulfone derivatives as novel tubulin polymerization inhibitors targeting the colchicine binding site. Bioorg. Chem. 2019, 85, 49–59. [Google Scholar] [CrossRef]
- Chen, X.; Xu, W.; Wang, K.; Mo, M.; Zhang, W.; Du, L.; Yuan, X.; Xu, Y.; Wang, Y.; Shen, J. Discovery of a Novel Series of Imidazo[1,2-a]pyrimidine Derivatives as Potent and Orally Bioavailable Lipoprotein-Associated Phospholipase A2 Inhibitors. J. Med. Chem. 2015, 58, 8529–8541. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Wang, X.; Wang, J.; Liu, J.; Zuo, D.; Jiang, N.; Zeng, T.; Yang, X.; Jing, T.; Gong, P. Discovery and Optimization of Novel 5-Indolyl-7-arylimidazo[1,2-a]pyridine-8-carbonitrile Derivatives as Potent Antitubulin Agents Targeting Colchicine-binding Site. Sci. Rep. 2017, 7, 43398. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zuo, D.; Jing, T.; Guo, M.; Xing, L.; Zhang, W.; Zhao, J.; Shen, J.; Gong, P.; Zhang, D.; et al. Synthesis, biological evaluation and molecular modeling of imidazo[1,2-a]pyridine derivatives as potent antitubulin agents. Biorg. Med. Chem. 2017, 25, 4088–4099. [Google Scholar] [CrossRef]
- Kazan, F.; Yagci, Z.B.; Bai, R.L.; Ozkirimli, E.; Hamel, E.; Ozkirimli, S. Synthesis and biological evaluation of indole-2-carbohydrazides and thiazolidinyl-indole-2-carboxamides as potent tubulin polymerization inhibitors. Comput. Biol. Chem. 2019, 80, 512–523. [Google Scholar] [CrossRef] [PubMed]
- De Martino, G.; La Regina, G.; Coluccia, A.; Edler, M.C.; Barbera, M.C.; Brancale, A.; Wilcox, E.; Hamel, E.; Artico, M.; Silvestri, R. Arylthioindoles, Potent Inhibitors of Tubulin Polymerization. J. Med. Chem. 2004, 47, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Rukkijakan, T.; Ngiwsara, L.; Lirdprapamongkol, K.; Svasti, J.; Phetrak, N.; Chuawong, P. A synthetic 2,3-diarylindole induces cell death via apoptosis and autophagy in A549 lung cancer cells. Bioorg. Med. Chem. Lett. 2016, 26, 2119–2123. [Google Scholar] [CrossRef] [PubMed]
- Thanaussavadate, B.; Ngiwsara, L.; Lirdprapamongkol, K.; Svasti, J.; Chuawong, P. A synthetic 2,3-diarylindole induces microtubule destabilization and G2/M cell cycle arrest in lung cancer cells. Bioorg. Med. Chem. Lett. 2020, 30, 126777. [Google Scholar] [CrossRef]
- Yan, J.; Hu, J.H.; An, B.J.; Huang, L.; Li, X.S. Design, synthesis, and biological evaluation of cyclic-indole derivatives as anti-tumor agents via the inhibition of tubulin polymerization. Eur. J. Med. Chem. 2017, 125, 663–675. [Google Scholar] [CrossRef]
- Yu, H.; Jin, H.W.; Gong, W.Z.; Wang, Z.L.; Liang, H.P. Pharmacological Actions of Multi-Target-Directed Evodiamine. Molecules 2013, 18, 1826–1843. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fang, K.; Cheng, J.F.; Li, Y.; Huang, Y.H.; Chen, S.Q.; Dong, G.Q.; Wu, S.C.; Sheng, C.Q. Scaffold Hopping of Natural Product Evodiamine: Discovery of a Novel Antitumor Scaffold with Excellent Potency against Colon Cancer. J. Med. Chem. 2020, 63, 696–713. [Google Scholar] [CrossRef]
- Schmidt, A.W.; Reddy, K.R.; Knolker, H.J. Occurrence, Biogenesis, and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef]
- Zhang, D.; Song, H.; Qin, Y. Total Synthesis of Indoline Alkaloids: A Cyclopropanation Strategy. Acc. Chem. Res. 2011, 44, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.Y.; Xiao, Y.Y.; Tian, F.L.; Han, L.M.; Gu, Y.L.; Zhu, N. Progress in Annulative Transformations from Indoles to Carbazoles: State of the Art. Chin. J. Org. Chem. 2021, 41, 521–528. [Google Scholar] [CrossRef]
- Tachibana, Y.; Kikuzaki, H.; Lajis, N.H.; Nakatani, N. Antioxidative activity of carbazoles from Murraya koenigii leaves. J. Agric. Food. Chem. 2001, 49, 5589–5594. [Google Scholar] [CrossRef]
- Caruso, A.; Voisin-Chiret, A.S.; Lancelot, J.C.; Sinicropi, M.S.; Garofalo, A.; Rault, S. Efficient and simple synthesis of 6-Aryl-1,4-dimethyl-9H-carbazoles. Molecules 2008, 13, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.; Saha, P.; Panda, D.; Dash, J.; Schwalbe, H. Insights from Binding on Quadruplex Selective Carbazole Ligands. Chem. Eur. J. 2021, 27, 12726–12736. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. Buchwald-Hartwig reaction: An overview. J. Organomet. Chem. 2018, 861, 17–104. [Google Scholar] [CrossRef]
- Roy, M.K.; Thalang, V.N.; Trakoontivakorn, G.; Nakahara, K. Mahanine, a carbazole alkaloid from Micromelum minutum, inhibits cell growth and induces apoptosis in U937 cells through a mitochondrial dependent pathway. Br. J. Pharmacol. 2005, 145, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; Smith, A.F.; Horning, E.C. Alkaloids of Ochrosia elliptica Labill.1. J. Am. Chem. Soc. 1959, 81, 1903–1908. [Google Scholar] [CrossRef]
- Wang, Y.M.; Hu, L.X.; Liu, Z.M.; You, X.F.; Zhang, S.H.; Qu, J.R.; Li, Z.R.; Li, Y.; Kong, W.J.; He, H.W.; et al. N-(2,6-Dimethoxypyridine-3-yl)-9-Methylcarbazole-3-Sulfonamide as a Novel Tubulin Ligand against Human Cancer. Clin. Cancer Res. 2008, 14, 6218–6227. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Wu, Y.B.; Sun, L.Q.; Gu, Y.X.; Hu, L.X. Synthesis and structure-activity relationship study of water-soluble carbazole sulfonamide derivatives as new anticancer agents. Eur. J. Med. Chem. 2020, 191, 112181. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, Y.; Liu, Y.; Chen, X.; Hu, L. Novel carbazole sulfonamide derivatives of antitumor agent: Synthesis, antiproliferative activity and aqueous solubility. Bioorg. Med. Chem. Lett. 2017, 27, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Yaqub, G.; Hussain, E.A.; Rehman, M.A.; Mateen, B. Advancements in Syntheses of Carbazole and Its Dervatives. Asian J. Chem. 2009, 21, 2485–2520. [Google Scholar]
- Faheem; Kumar, B.K.; Sekhar, K.; Kunjiappan, S.; Jamalis, J.; Balana-Fouce, R.; Sankaranarayanan, M. Recent Update on the Anti-infective Potential of beta-carboline Analogs. Mini-Rev. Med. Chem. 2021, 21, 398–425. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Chang-Hong, W. Strictosidine synthase, an indispensable enzyme involved in the biosynthesis of terpenoid indole and beta-carboline alkaloids. Chin. J. Nat. Med. 2021, 19, 591–607. [Google Scholar] [CrossRef]
- Shen, Y.C.; Chen, C.Y.; Hsieh, P.W.; Duh, C.Y.; Lin, Y.M.; Ko, C.L. The preparation and evaluation of 1-substituted 1,2,3,4-tetrahydro- and 3,4-dihydro-beta-carboline derivatives as potential antitumor agents. Chem. Pharm. Bull. 2005, 53, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Soni, J.P.; Yeole, Y.; Shankaraiah, N. beta-Carboline-based molecular hybrids as anticancer agents: A brief sketch. RSC Med. Chem. 2021, 12, 730–750. [Google Scholar] [CrossRef]
- Formagio, A.S.; Tonin, L.T.; Foglio, M.A.; Madjarof, C.; de Carvalho, J.E.; da Costa, W.F.; Cardoso, F.P.; Sarragiotto, M.H. Synthesis and antitumoral activity of novel 3-(2-substituted-1,3,4-oxadiazol-5-yl) and 3-(5-substituted-1,2,4-triazol-3-yl) beta-carboline derivatives. Bioorg. Med. Chem. 2008, 16, 9660–9667. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, M.; Qian, K.; Lee, K.H.; Morris-Natschke, S.; Peng, S. Novel N-(3-carboxyl-9-benzyl-beta-carboline-1-yl)ethylamino acids: Synthesis, anti-tumor evaluation, intercalating determination, 3D QSAR analysis and docking investigation. Eur. J. Med. Chem. 2009, 44, 4153–4161. [Google Scholar] [CrossRef]
- Samundeeswari, S.; Kulkarni, M.V.; Joshi, S.D.; Dixit, S.R.; Jayakumar, S.; Ezhilarasi, R.M. Synthesis and Human Anticancer Cell Line Studies on Coumarin-β-carboline Hybrids as Possible Antimitotic Agents. ChemistrySelect 2016, 1, 5019–5024. [Google Scholar] [CrossRef]
- Alvarez, M.; Salas, M.; Joule, J.A. ChemInform Abstract: Marine, Nitrogen-Containing Heterocyclic Natural Products. Structures and Syntheses of Compounds Containing Indole Units. ChemInform 1991, 22, 759–794. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Yang, M.R.; Qin, Y.J.; Chen, C.; Zhang, Y.L.; Li, B.Y.; Liu, T.B.; Gong, H.B.; Wang, B.Z.; Zhu, H.L. Synthesis, biological evaluation and molecular docking studies of novel 1-(4,5-dihydro-1H-pyrazol-1-yl)ethanone-containing 1-methylindol derivatives as potential tubulin assembling inhibitors. RSC Adv. 2016, 6, 30412–30424. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Qin, Y.J.; Tang, D.J.; Yang, M.R.; Li, B.Y.; Wang, Y.T.; Cai, H.Y.; Wang, B.Z.; Zhu, H.L. Synthesis and Biological Evaluation of 1-Methyl-1H-indole-Pyrazoline Hybrids as Potential Tubulin Polymerization Inhibitors. Chemmedchem 2016, 11, 1446–1458. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.L.; Fan, J.; Ma, X.; Qin, Y.J.; Zhu, H.L. Novel nicotinoyl pyrazoline derivates bearing N-methyl indole moiety as antitumor agents: Design, synthesis and evaluation. Eur. J. Med. Chem. 2018, 156, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, N.; Kukuła-Koch, W.; Głowniak, K. Modern industrial and pharmacological applications of indigo dye and its derivatives—A review. Acta Pol. Pharm.-Drug Res. 2014, 71, 215–221. [Google Scholar]
- Mohan, L.; Raghav, D.; Ashraf, S.M.; Sebastian, J.; Rathinasamy, K. Indirubin, a bis-indole alkaloid binds to tubulin and exhibits antimitotic activity against HeLa cells in synergism with vinblastine. Biomed. Pharmacother. 2018, 105, 506–517. [Google Scholar] [CrossRef]
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Das, A.; Ganguli, A.; Datta, S.; Kumar, D.; Chakrabarti, G. Development of Novel Bis(indolyl)-hydrazide Hydrazone Derivatives as Potent Microtubule-Targeting Cytotoxic Agents against A549 Lung Cancer Cells. Biochemistry 2016, 55, 3020–3035. [Google Scholar] [CrossRef]
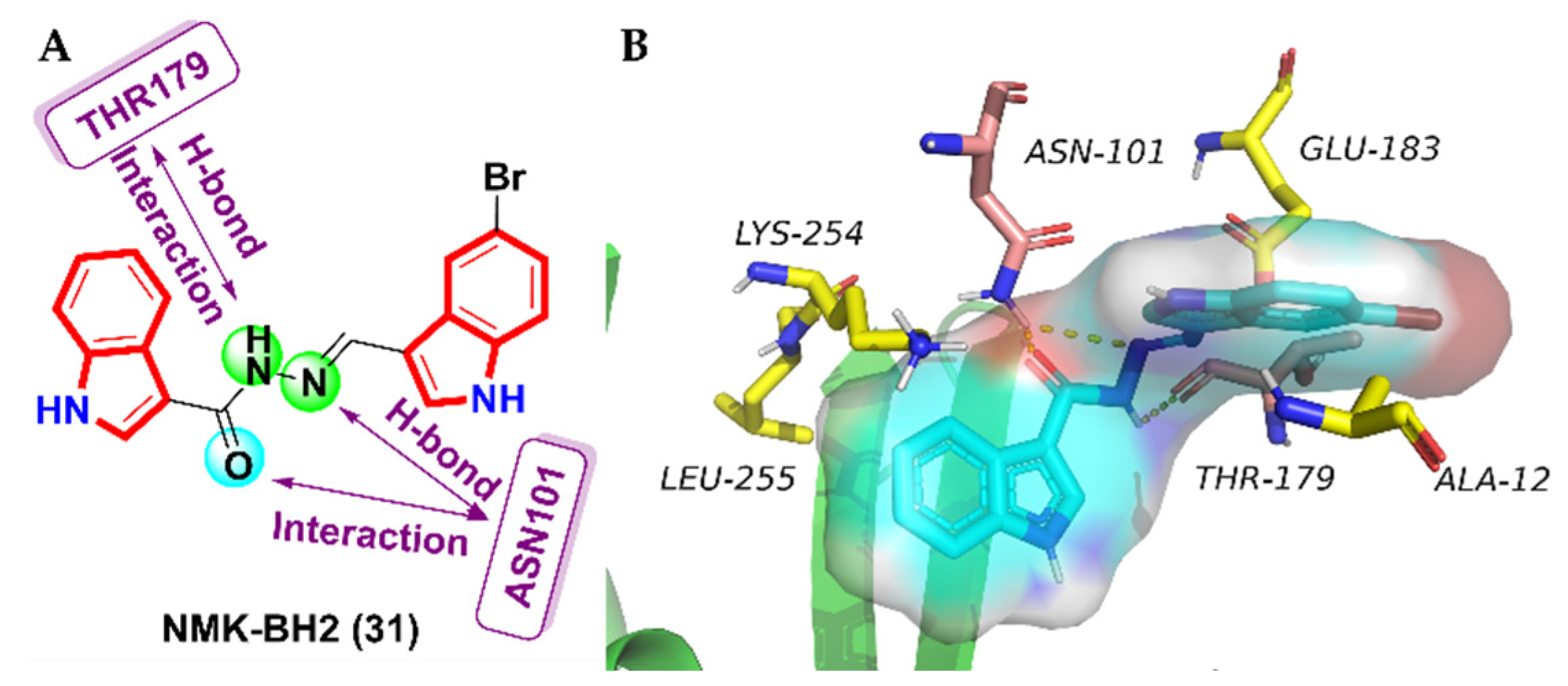
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Datta, S.; Ghosh Dastidar, D.; Kumar, D.; Chakrabarti, G. NMK-BH2, a novel microtubule-depolymerising bis (indolyl)-hydrazide-hydrazone, induces apoptotic and autophagic cell death in cervical cancer cells by binding to tubulin at colchicine—Site. Biochim. Biophys. Acta 2020, 1867, 118762. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.; Herald, D.L.; Verdier-Pinard, P.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic Agents. 379. Synthesis of Phenstatin Phosphate1a. J. Med. Chem. 1998, 41, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
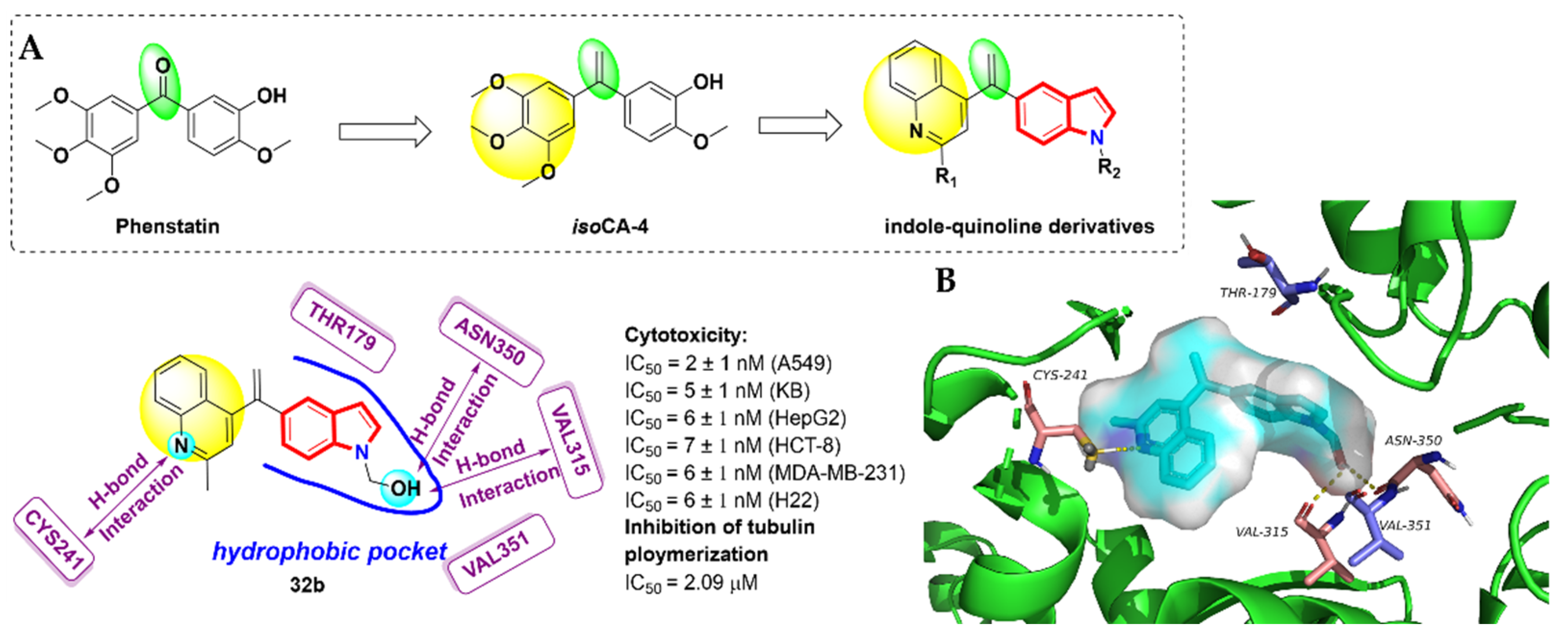
- Messaoudi, S.; Tréguier, B.; Hamze, A.; Provot, O.; Peyrat, J.-F.; De Losada, J.R.; Liu, J.-M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins A versus Combretastatins A: The Forgotten isoCA-4 Isomer as a Highly Promising Cytotoxic and Antitubulin Agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Shuai, W.; Sun, H.H.; Xu, F.J.; Bi, Y.; Xu, J.Y.; Ma, C.; Yao, H.Q.; Zhu, Z.Y.; Xu, S.T. Design, synthesis and biological evaluation of quinoline-indole derivatives as anti-tubulin agents targeting the colchicine binding site. Eur. J. Med. Chem. 2019, 163, 428–442. [Google Scholar] [CrossRef] [PubMed]
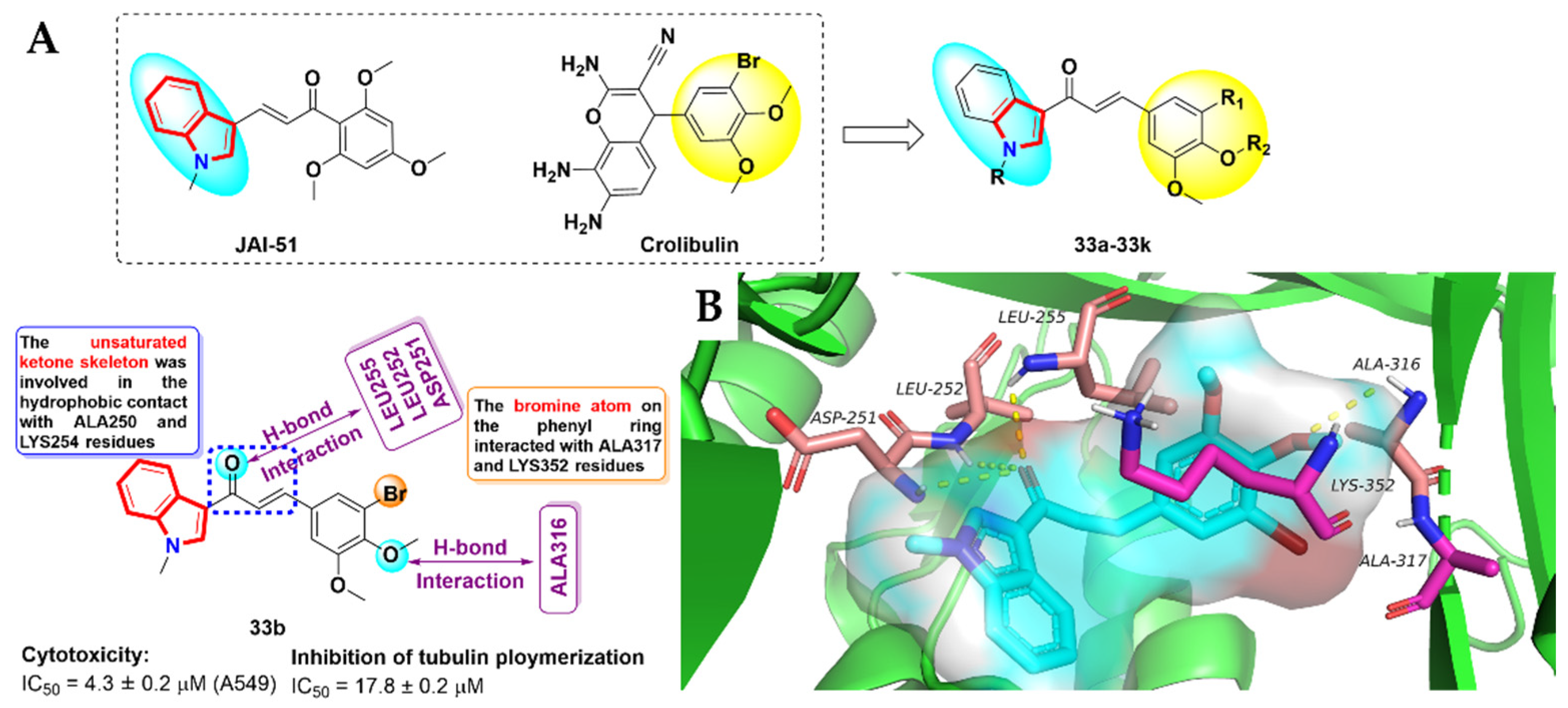
- Boumendjel, A.; McLeer-Florin, A.; Champelovier, P.; Allegro, D.; Muhammad, D.; Souard, F.; Derouazi, M.; Peyrot, V.; Toussaint, B.; Boutonnat, J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivoglioblastoma models. BMC Cancer 2009, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.X. Small molecule vascular disrupting agents: Potential new drugs for cancer treatment. Recent Pat. Anticancer Drug Discov. 2007, 2, 79–101. [Google Scholar] [CrossRef]
- Mirzaei, H.; Shokrzadeh, M.; Modanloo, M.; Ziar, A.; Riazi, G.H.; Emami, S. New indole-based chalconoids as tubulin-targeting antiproliferative agentsd. Bioorg. Chem. 2017, 75, 86–98. [Google Scholar] [CrossRef]
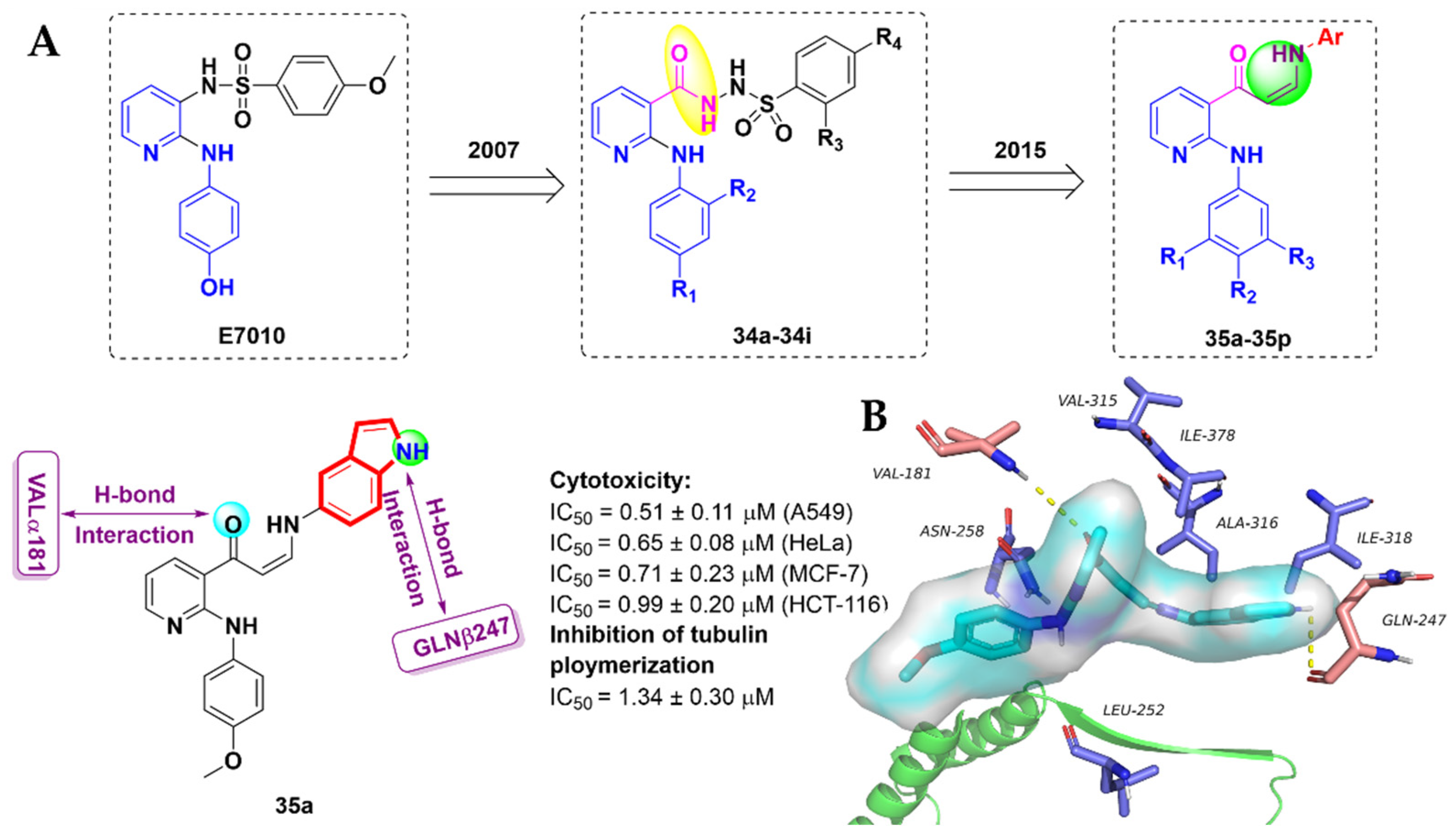
- Kamal, A.; Khan, M.N.A.; Srinivasa Reddy, K.; Rohini, K. Synthesis of a new class of 2-anilino substituted nicotinyl arylsulfonylhydrazides as potential anticancer and antibacterial agents. Biorg. Med. Chem. 2007, 15, 1004–1013. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy, V.S.; Vishnuvardhan, M.; Kumar, G.B.; Shaik, A.B.; Chourasiya, S.S.; Reddy, M.K.; Bin Sayeed, I.; Adiyala, P.R.; Jain, N. Synthesis of 2-anilinopyridine-arylpropenone conjugates as tubulin inhibitors and apoptotic inducers. RSC Adv. 2015, 5, 97367–97380. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name (Company) | Target | Indications | Year (FDA) |
|---|---|---|---|
| Vincristine (Eli Lilly) | Tubulin | Hodgkin lymphoma | 1963 |
| Vinorelbine (Pierre Fabre) | Tubulin | Hodgkin lymphoma | 1994 |
| Alectinib (Roche) | ALK | NSCLC | 2015 |
| Panobinostat (Novartis) | HDACs | Multiple myeloma | 2015 |
| Sunitinib (Pfizer) | VEGFR | Gastrointestinal stromal tumor | 2006 |
| Osimertinib (AstraZeneca) | EGFR | NSCLC | 2015 |
| Comp. | IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| HepG2 | A549 | K562 | HCT-8 | H22 | Bel-7402 | LO2 | |
| 14e | 0.075 ± 0.005 | 0.305 ± 0.026 | 0.055 ± 0.005 | 0.287 ± 0.034 | 0.060 ± 0.015 | 0.080 ± 0.023 | 0.240 ± 0.090 |
| Comp. | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| HeLa | MCF-7 | MDA | PA-1 | HepG2 | PBMC | WI38 | HEK | |
| 31 | 1.5 ± 0.25 | 5.25 ± 0.31 | 8 ± 0.87 | 24.7 ± 2.61 | 29.7 ± 3.25 | 50 ± 4.8 | 41.5 ± 4.2 | 46.5 ± 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Zhou, Z.; Jiang, Z.; Zhu, W.; Qiao, D. Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations. Molecules 2022, 27, 1587. https://doi.org/10.3390/molecules27051587
Tang S, Zhou Z, Jiang Z, Zhu W, Qiao D. Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations. Molecules. 2022; 27(5):1587. https://doi.org/10.3390/molecules27051587
Chicago/Turabian StyleTang, Sheng, Zhihui Zhou, Zhiyan Jiang, Wufu Zhu, and Dan Qiao. 2022. "Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations" Molecules 27, no. 5: 1587. https://doi.org/10.3390/molecules27051587