
Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity


Abstract
1. Introduction
1.1. Isothiocyanates—General Properties
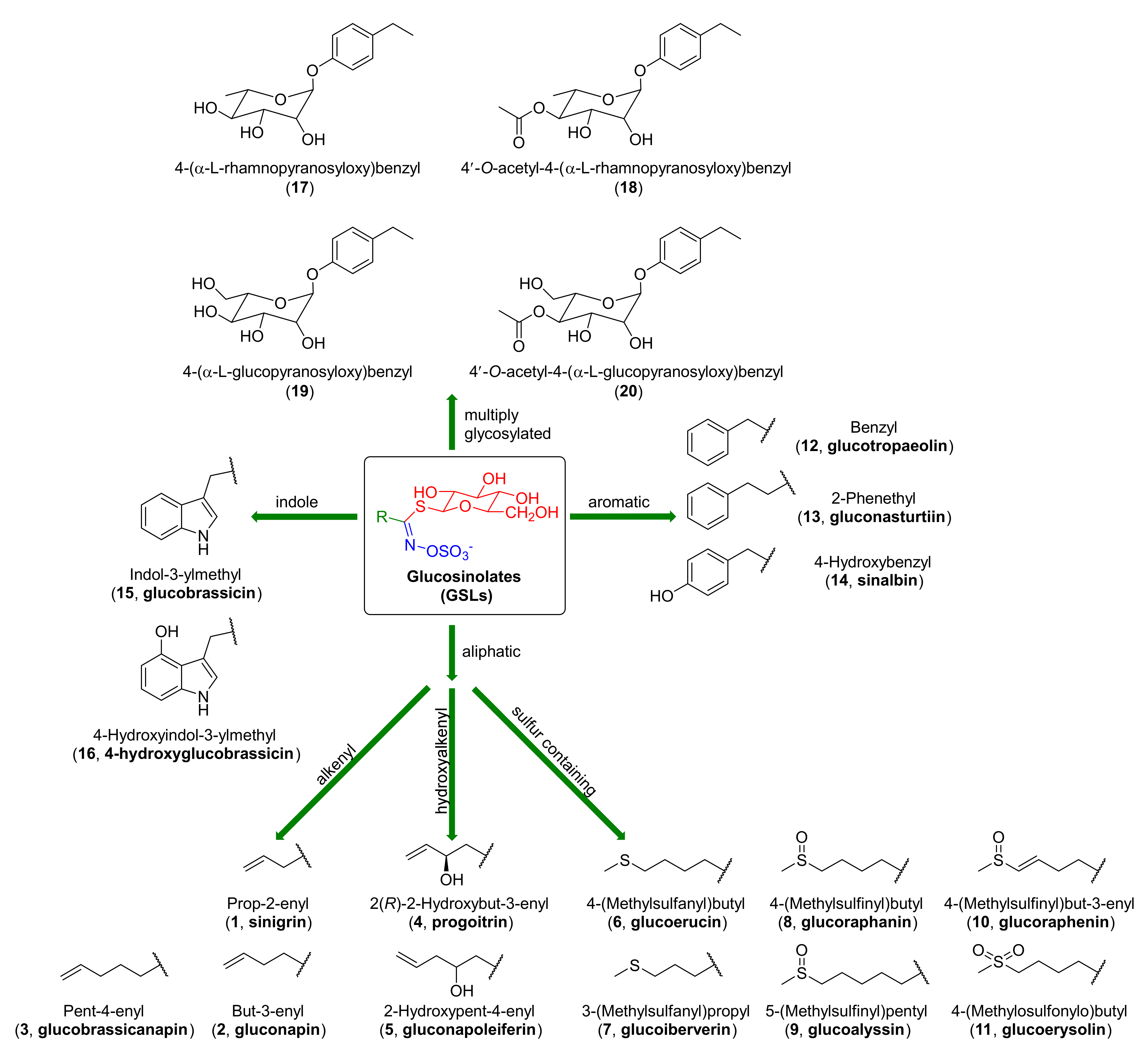
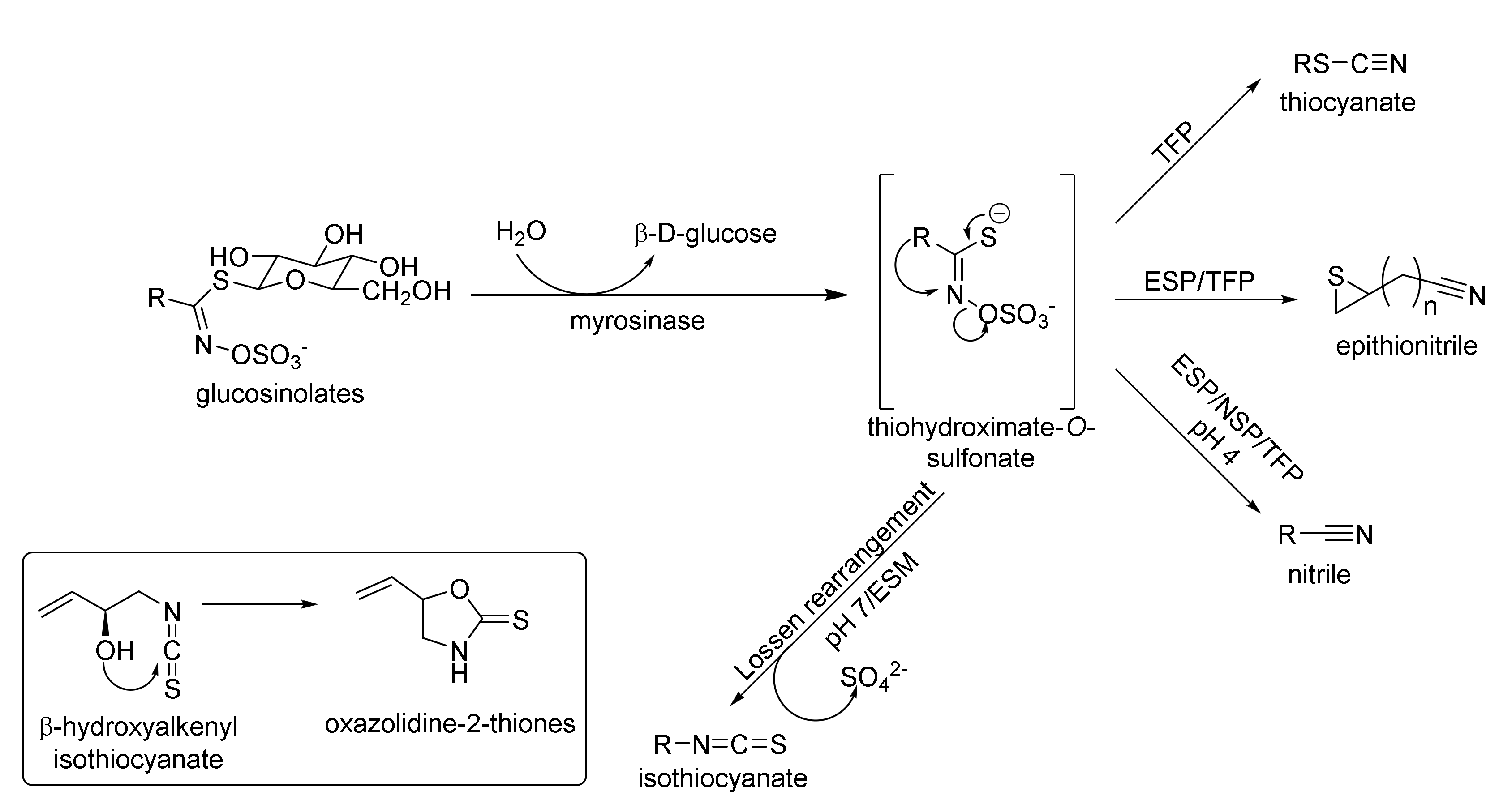
1.1.1. Glucosinolates
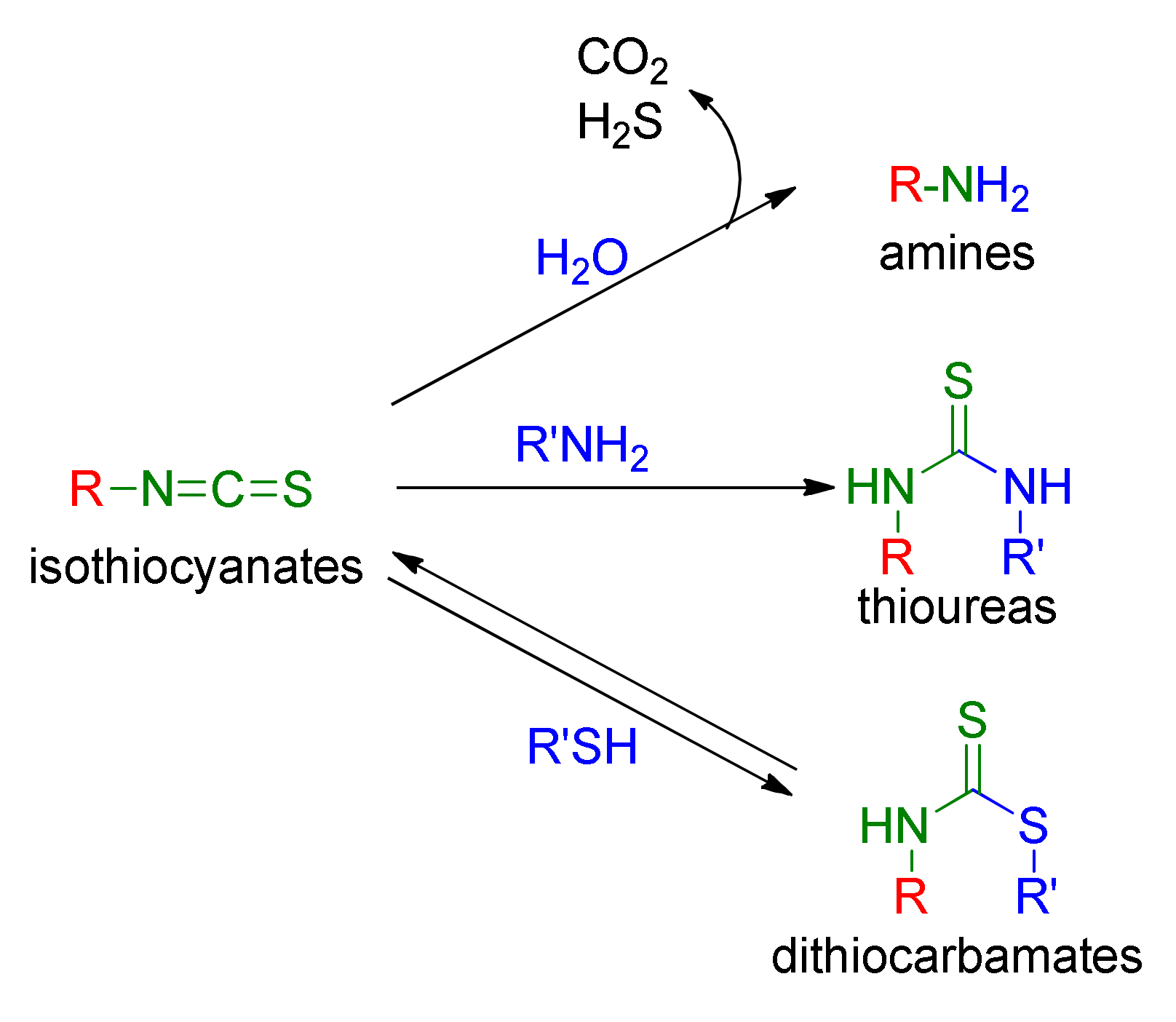
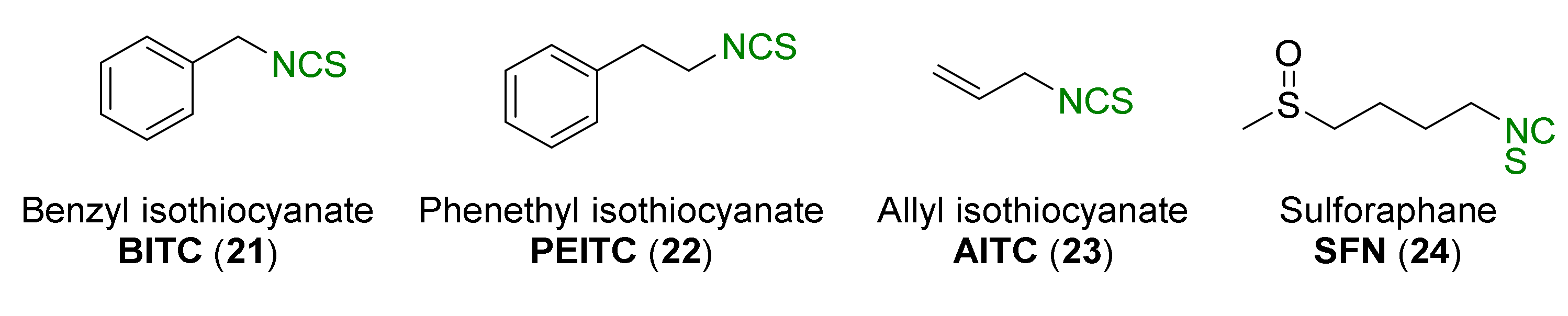
1.1.2. Isothiocyanates
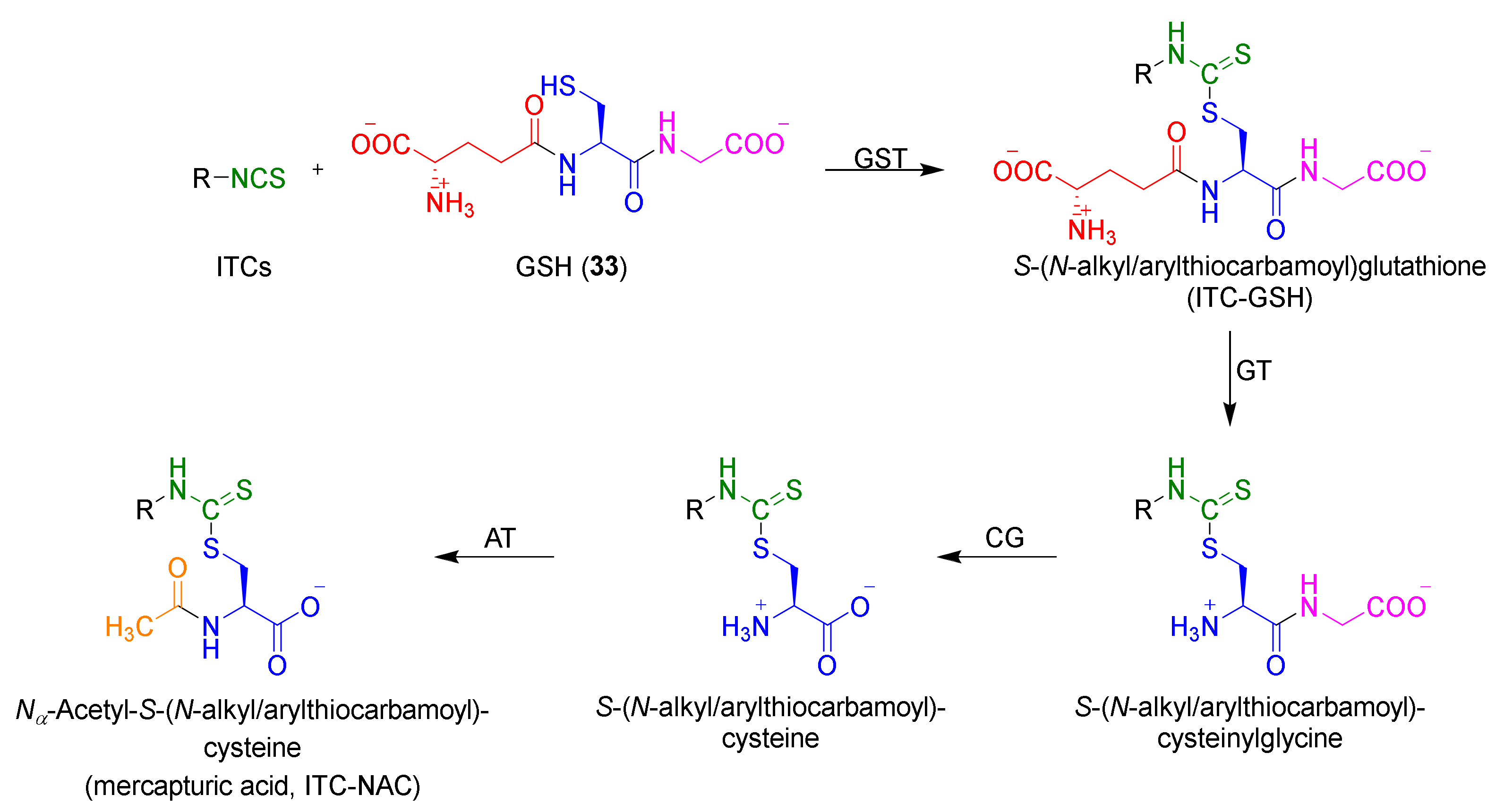
1.1.3. Mercapturic Aid
2. Mechanism Determining Biological Activity of ITCs
2.1. The Mechanism of Anticancer Activity
2.1.1. Initiation Stage
Inhibition Phase I Enzymes and Activation Phase II Enzymes
2.1.2. Promotion Stage
Inhibition of the Cell Cycle
Inducing Apoptosis
Inhibiting Histone Deacetylase (HDAC)
2.1.3. Progression Stage
Inhibition of Angiogenesis and Metastasis
2.2. Antibacterial Activity
Mechanism of Antibacterial Activity
2.3. Clinical Trials of SFN
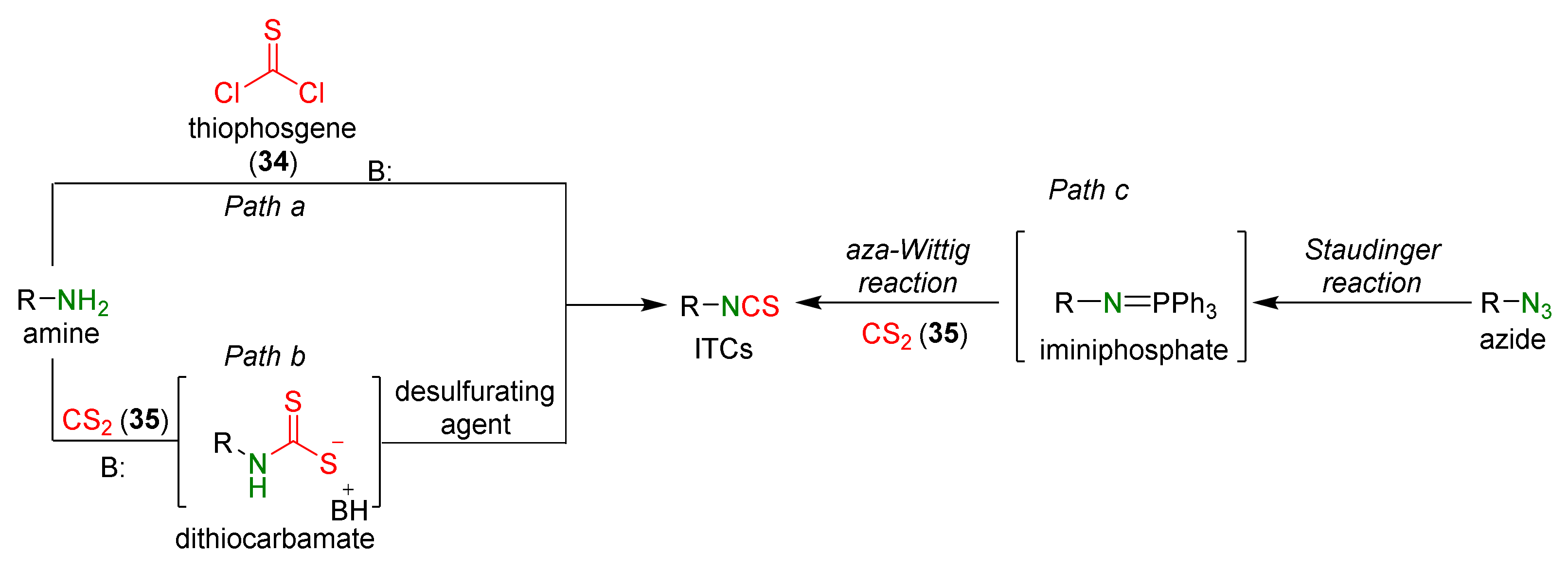
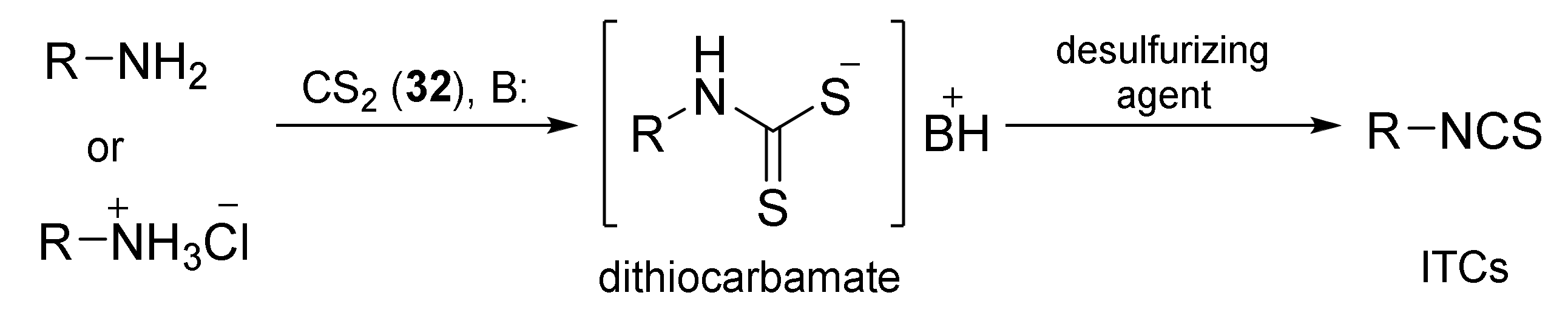
3. Methods of Synthesis of ITCs
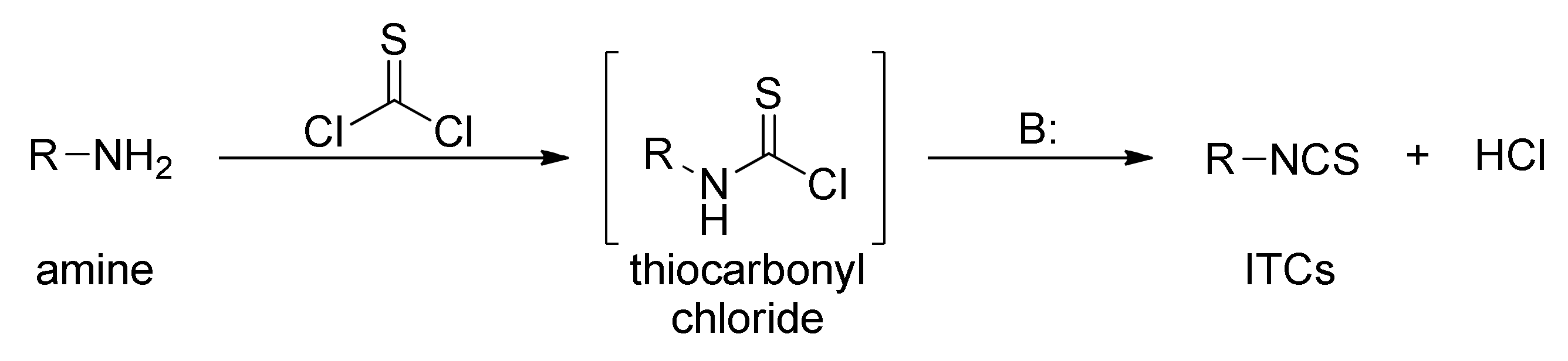
3.1. Synthesis ITCs Using Thiophosgene and Its Substitutes
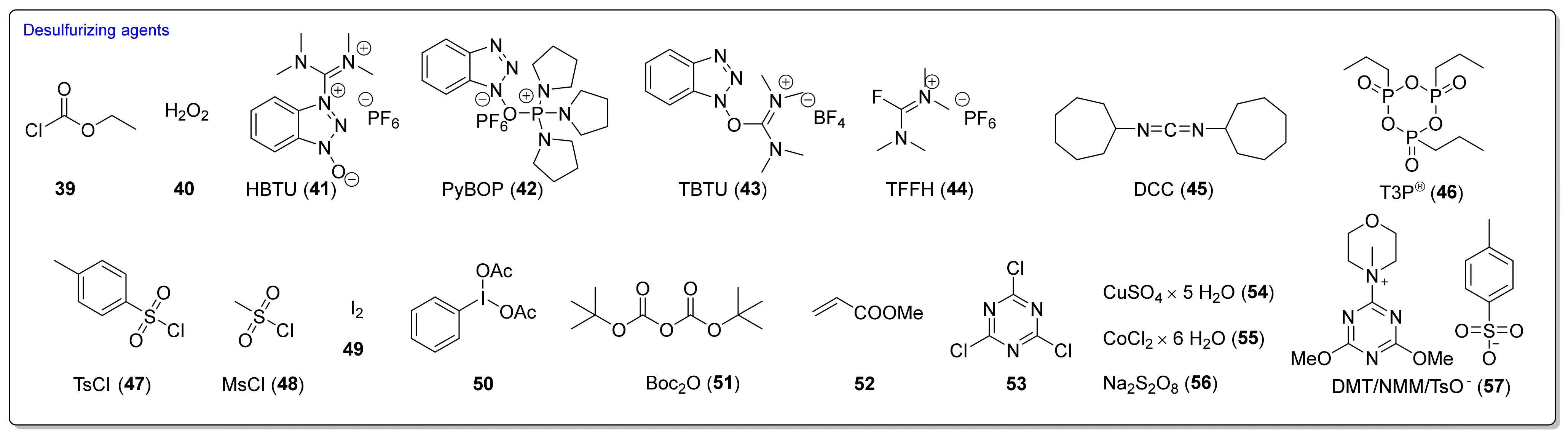
3.2. Synthesis ITCs with a Desulfurizing Agent
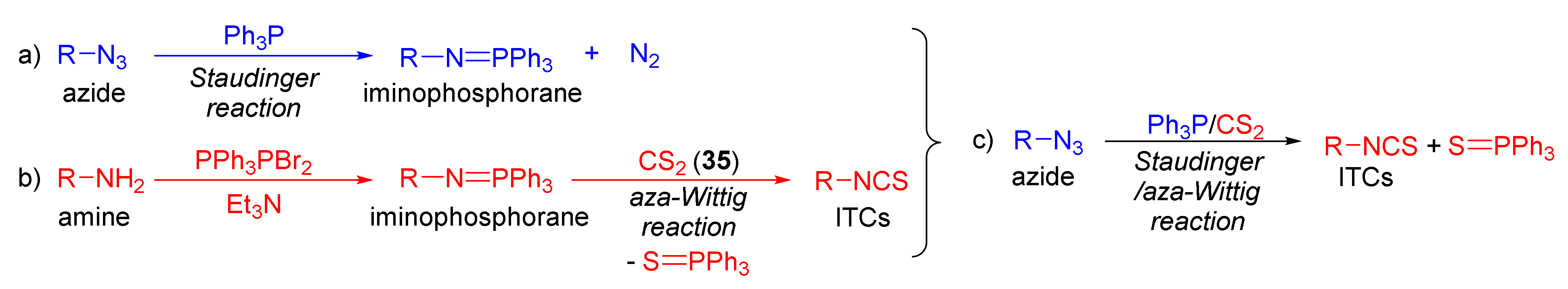
3.3. Synthesis ITCs via the Tandem Staudinger/aza-Wittig Reaction
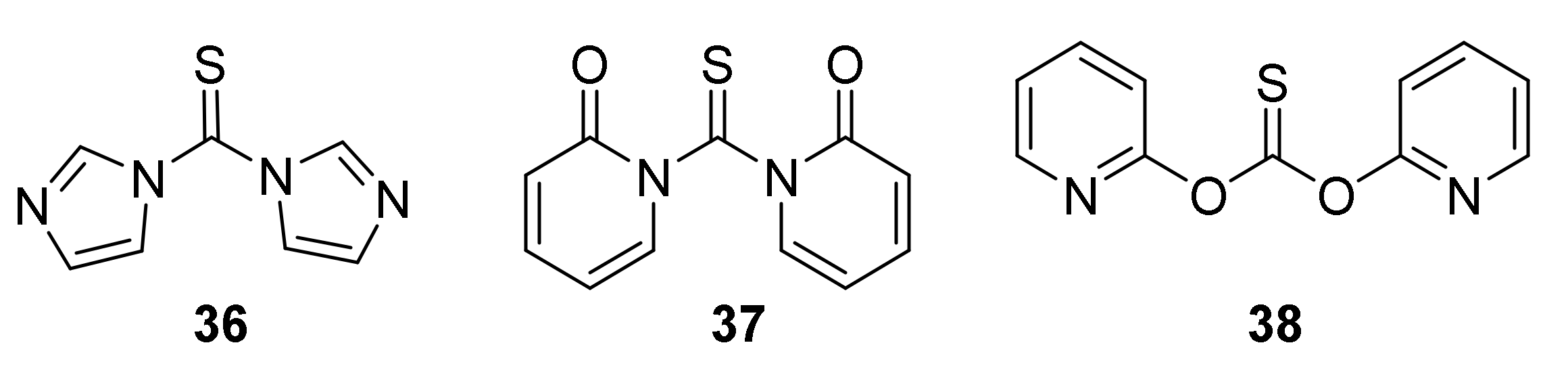
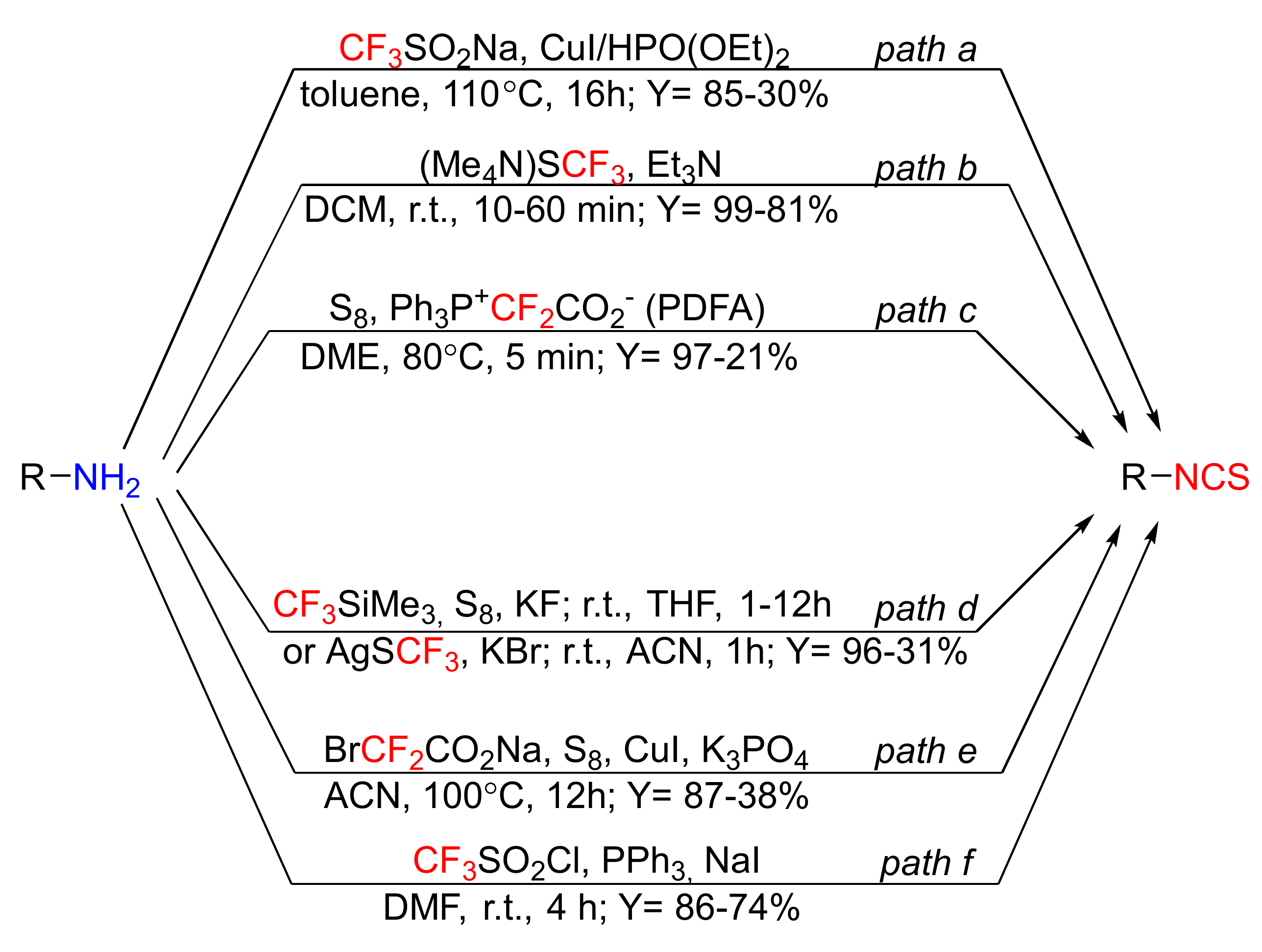
3.4. The Latest Approaches to ITC Syntheses
4. Synthesis of Bifunctional Analogs of Sulforaphane and Their Properties
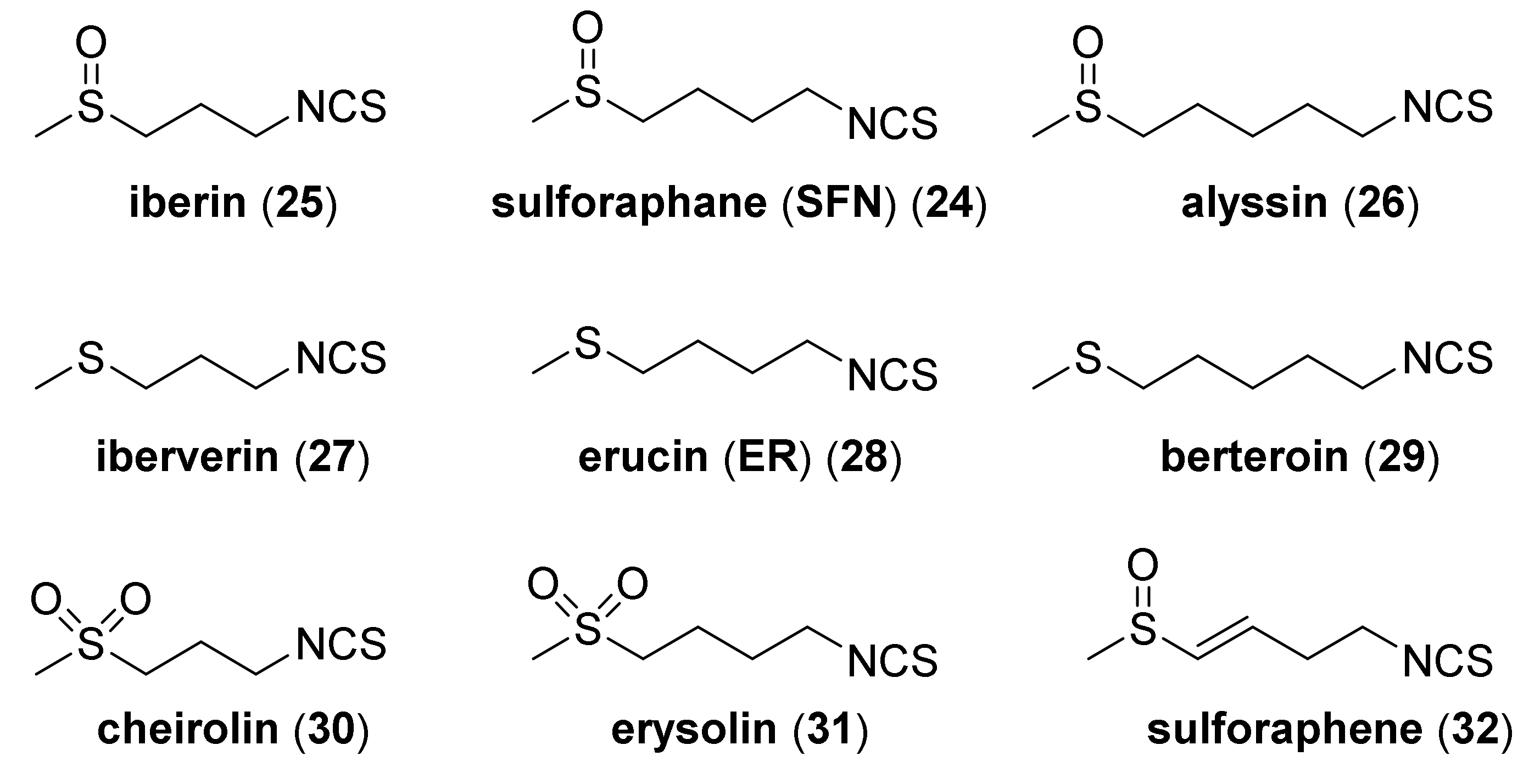
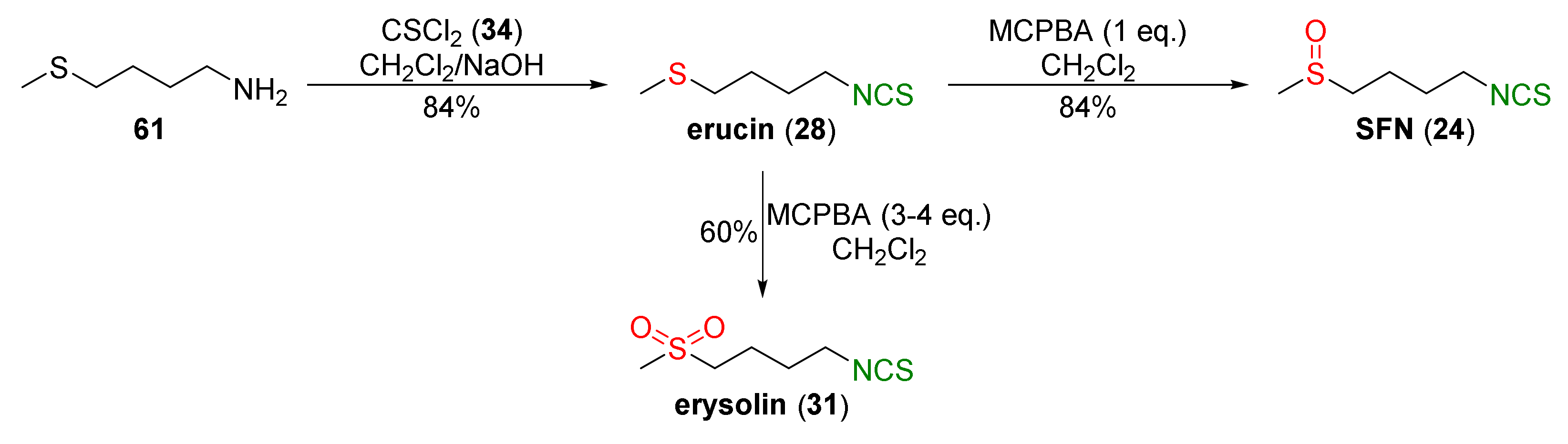
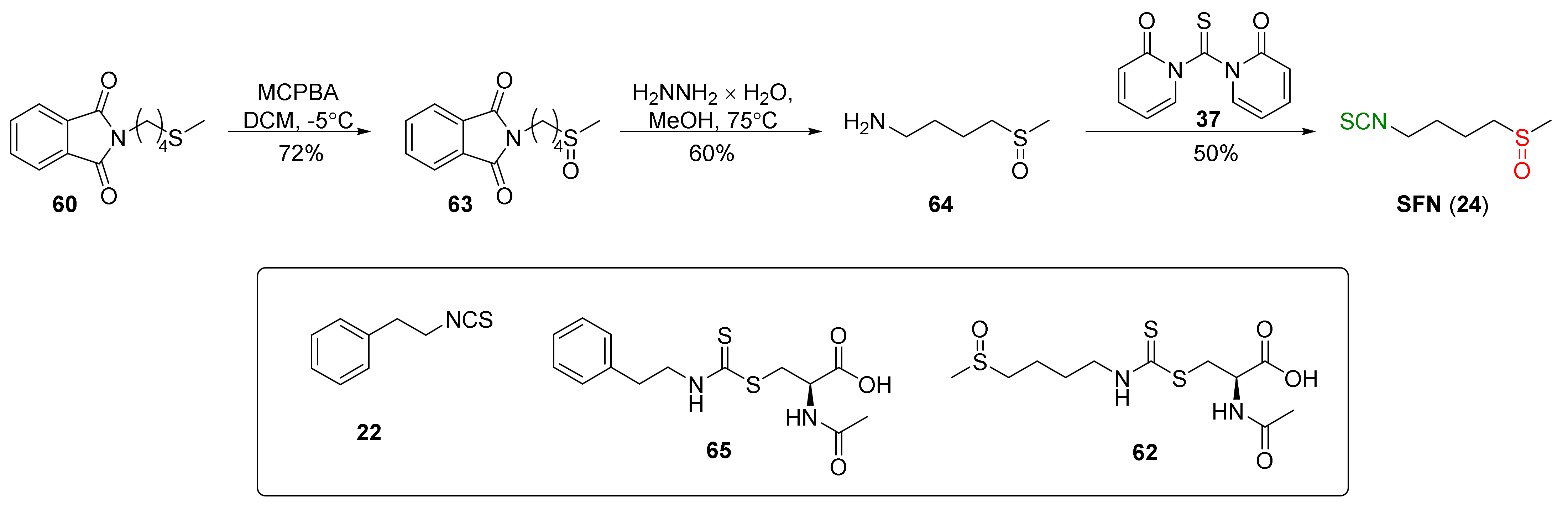
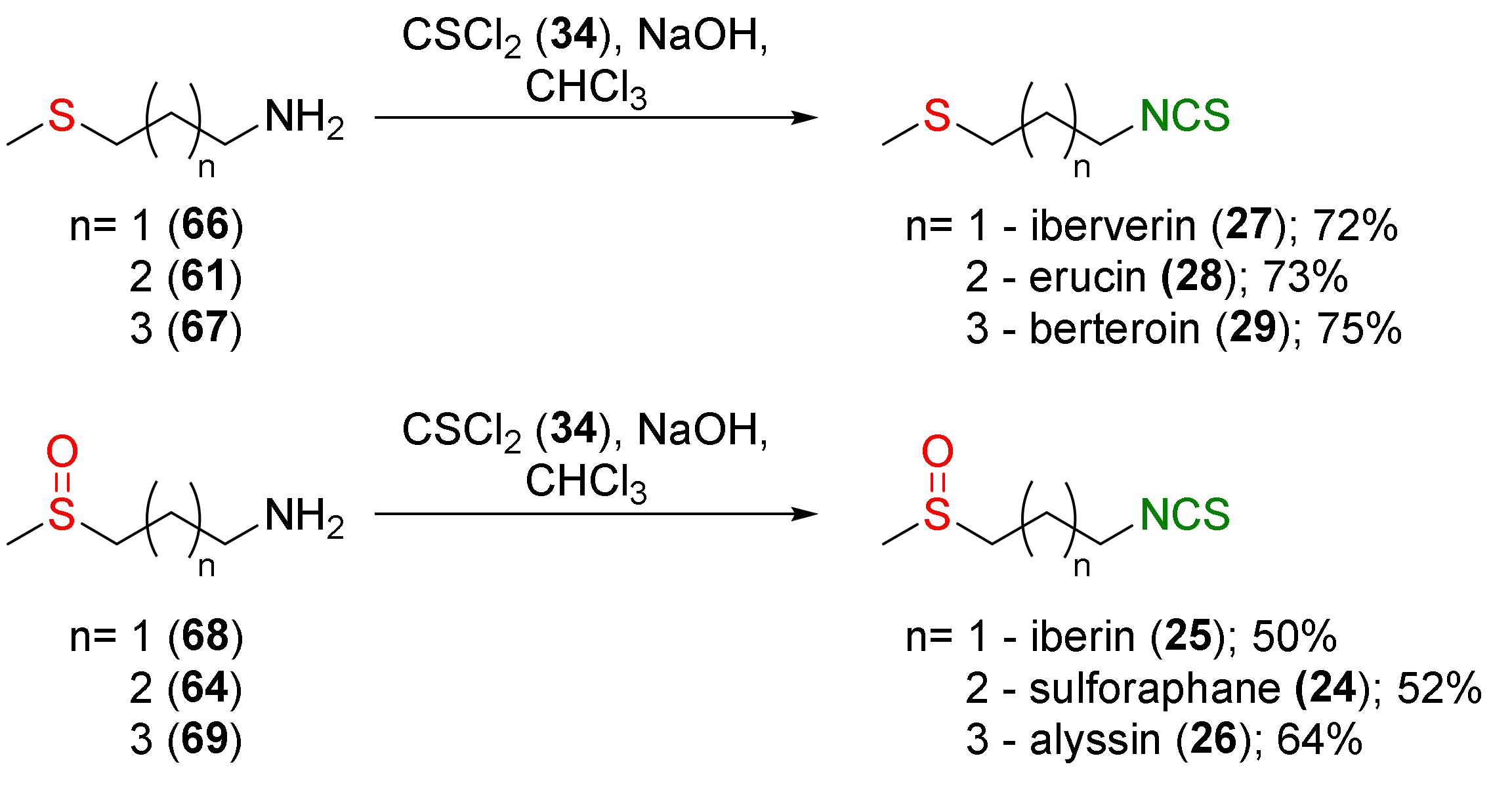
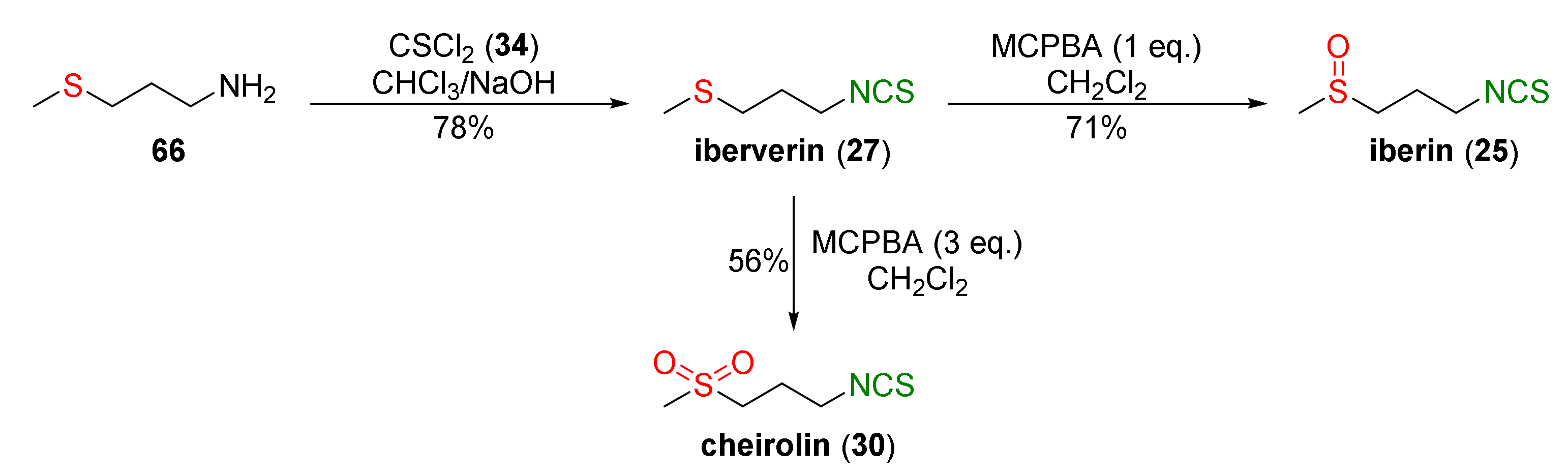
4.1. Synthesis of Sulforaphane and Its Sulfur Analogues and Their Properties
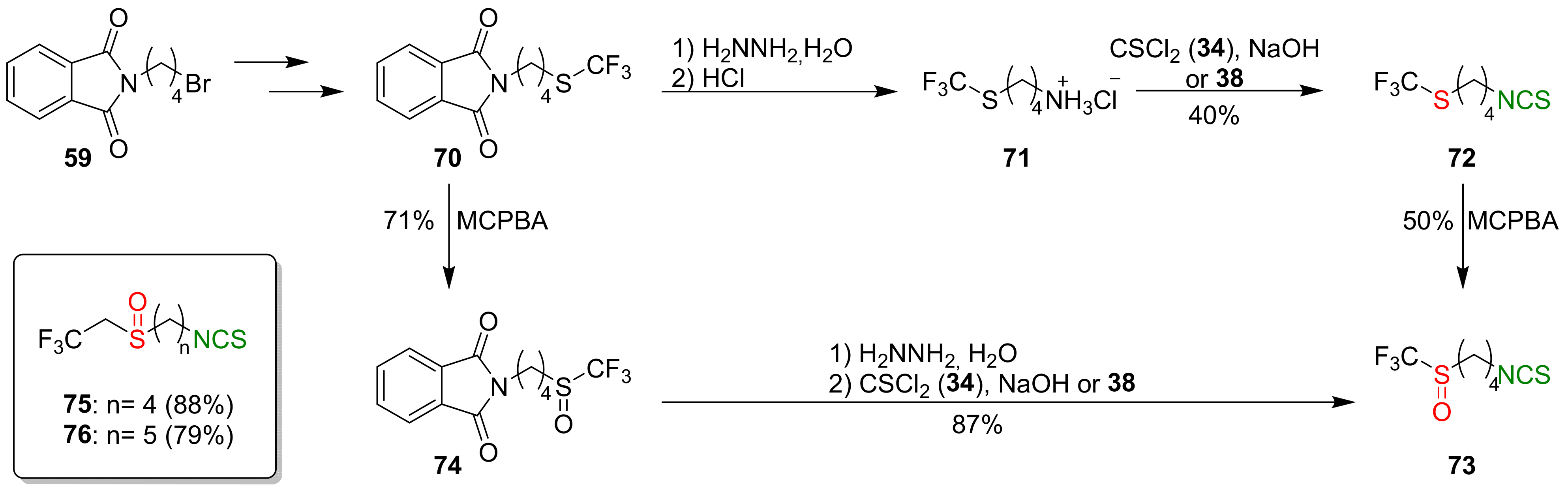
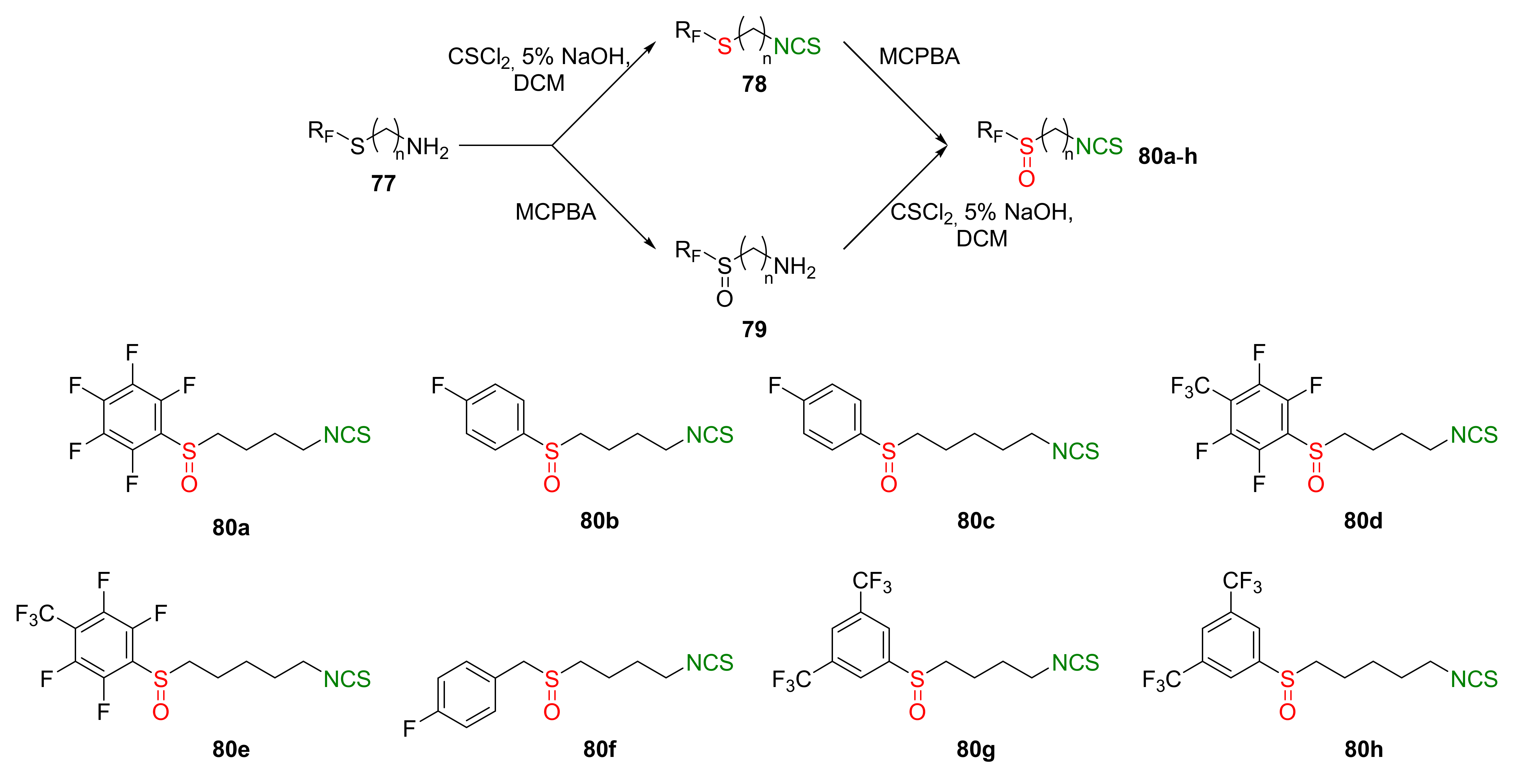
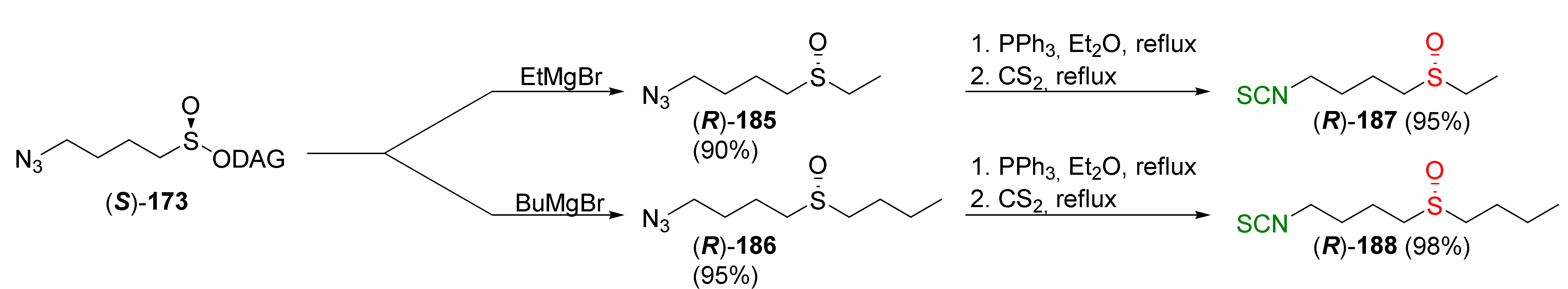
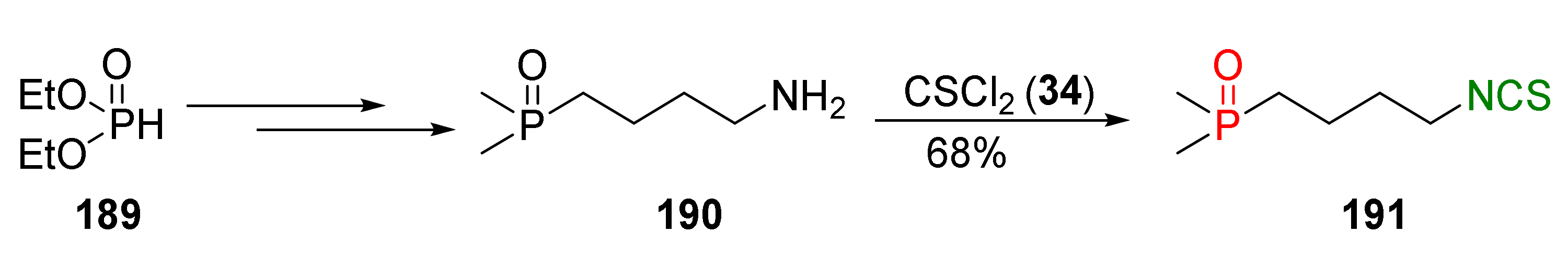
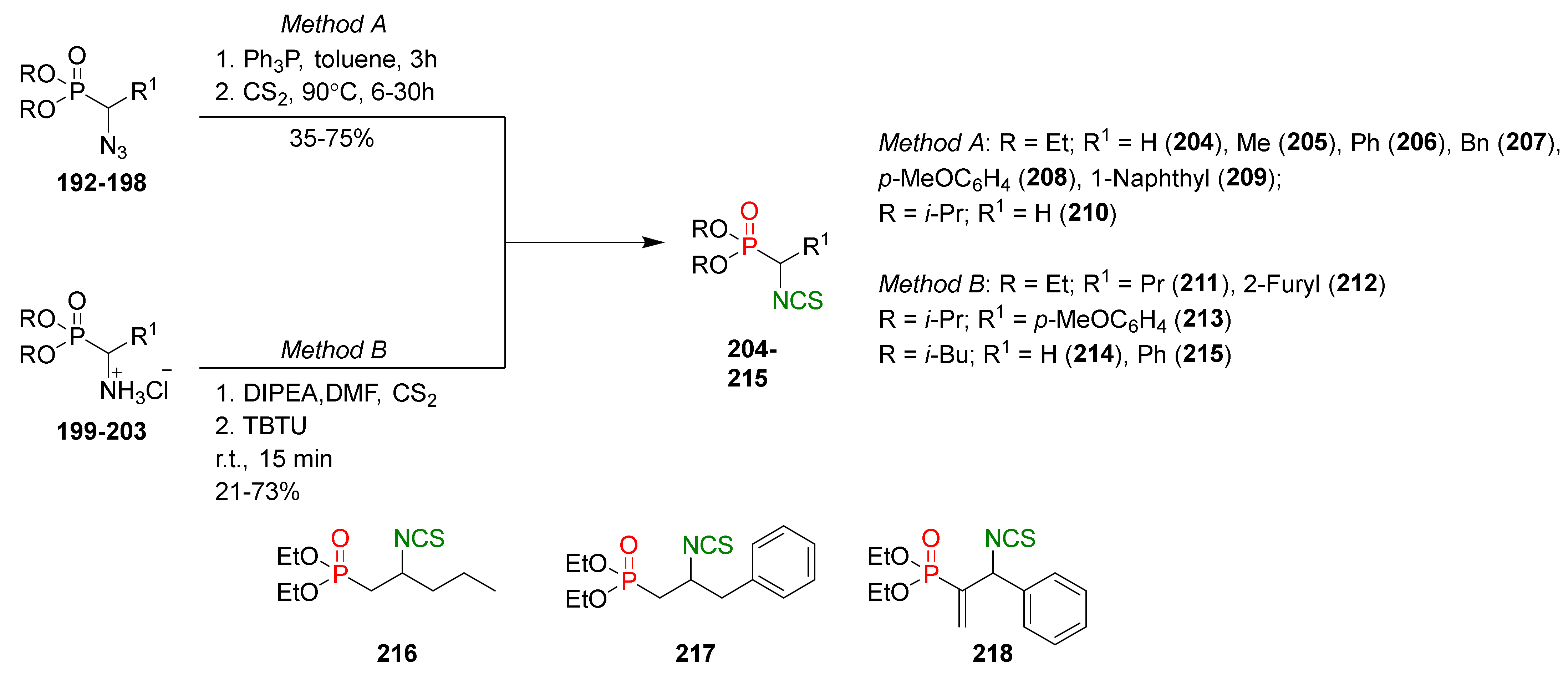
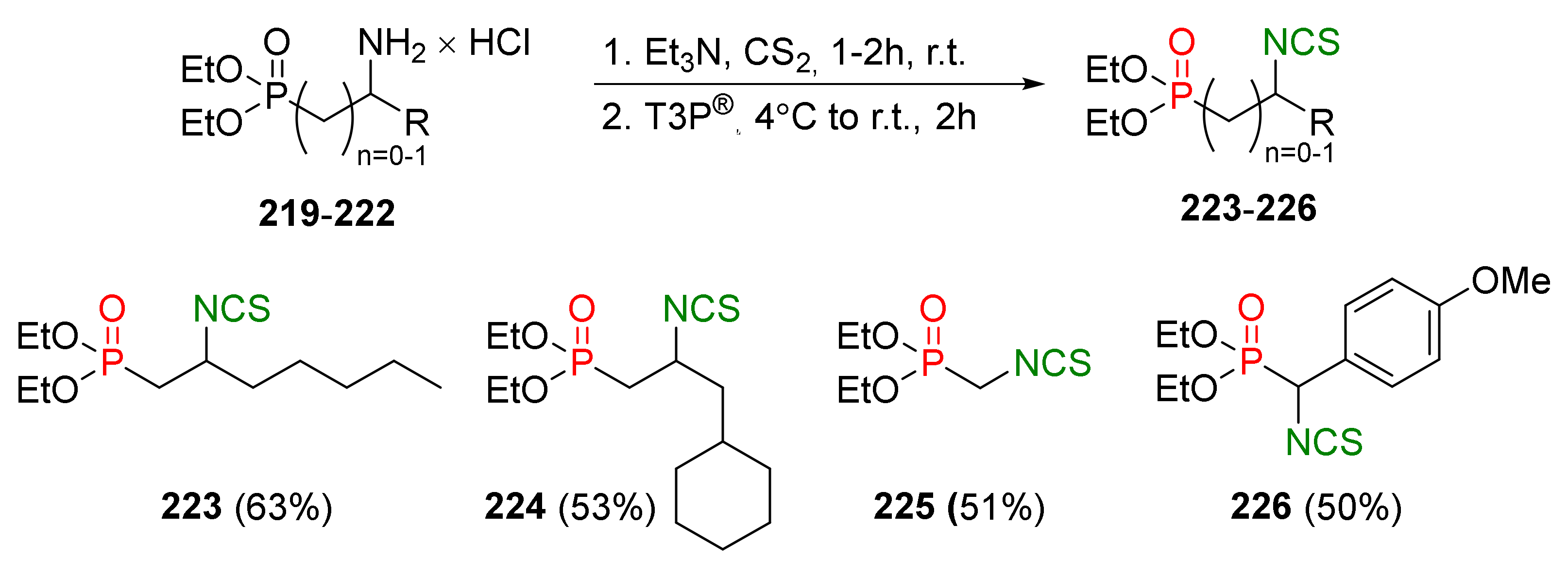
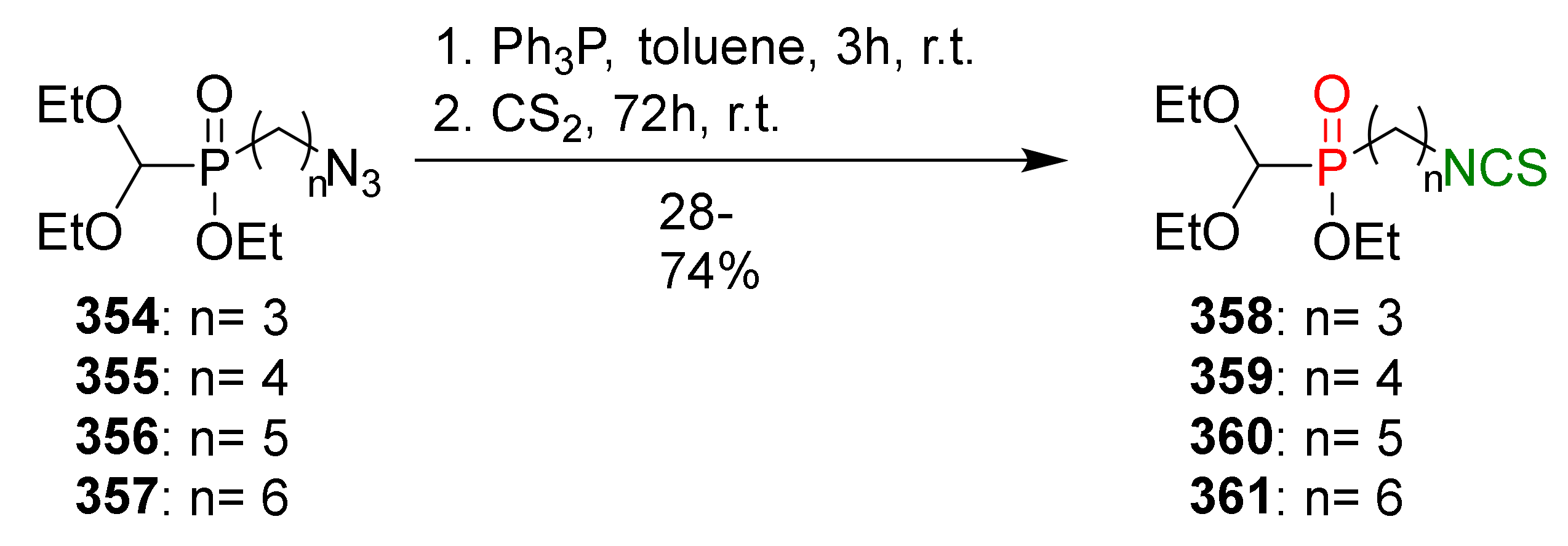
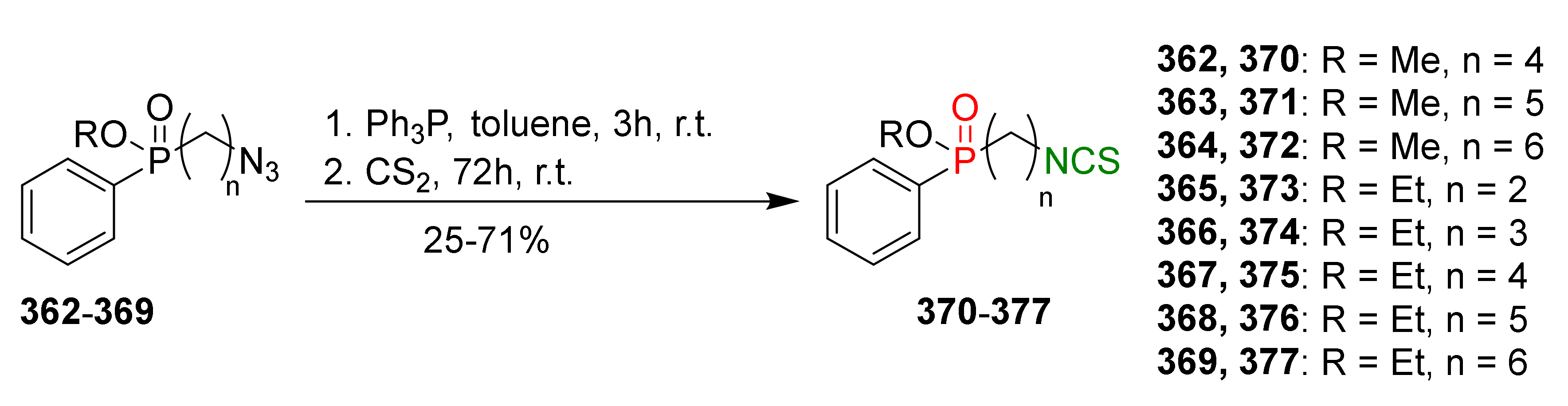
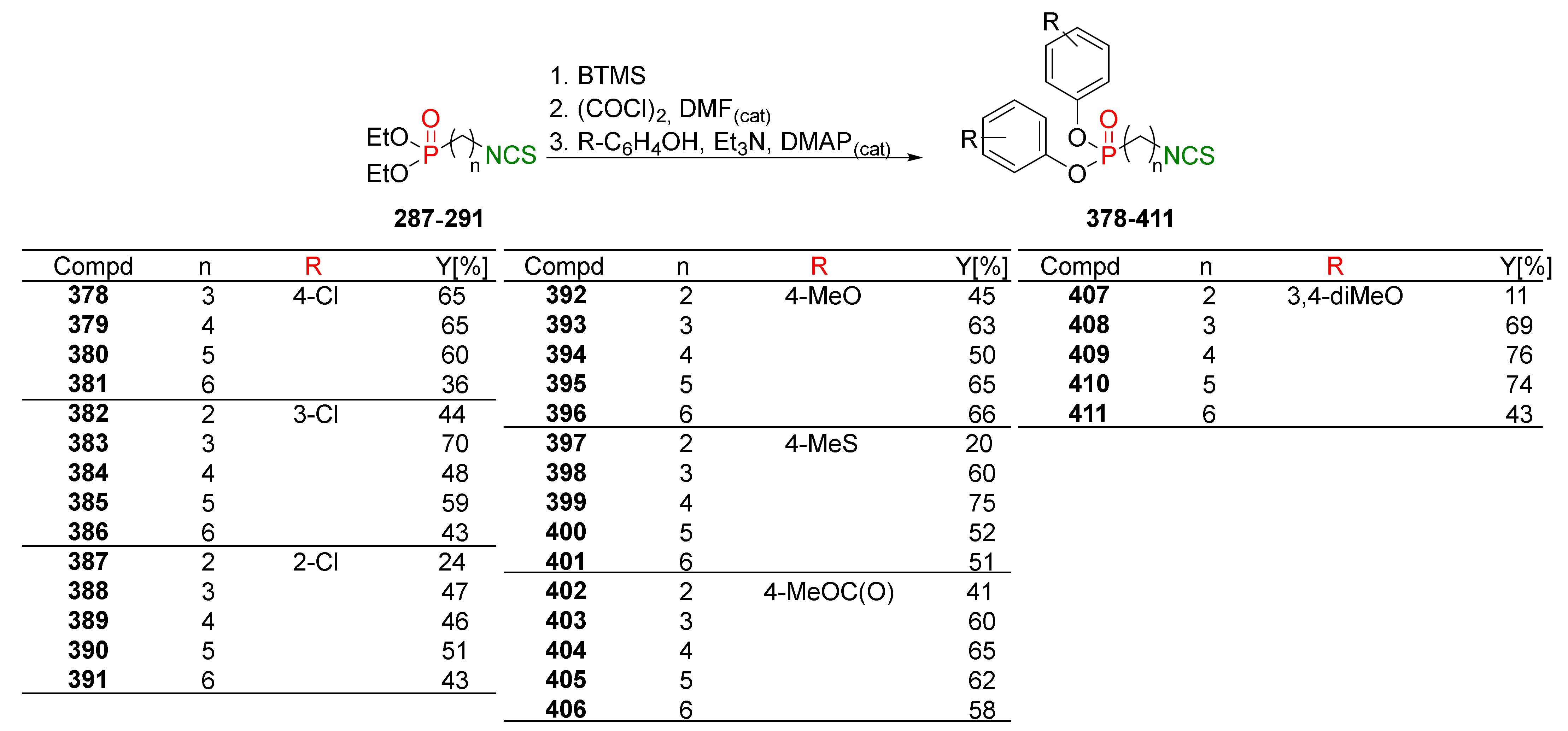
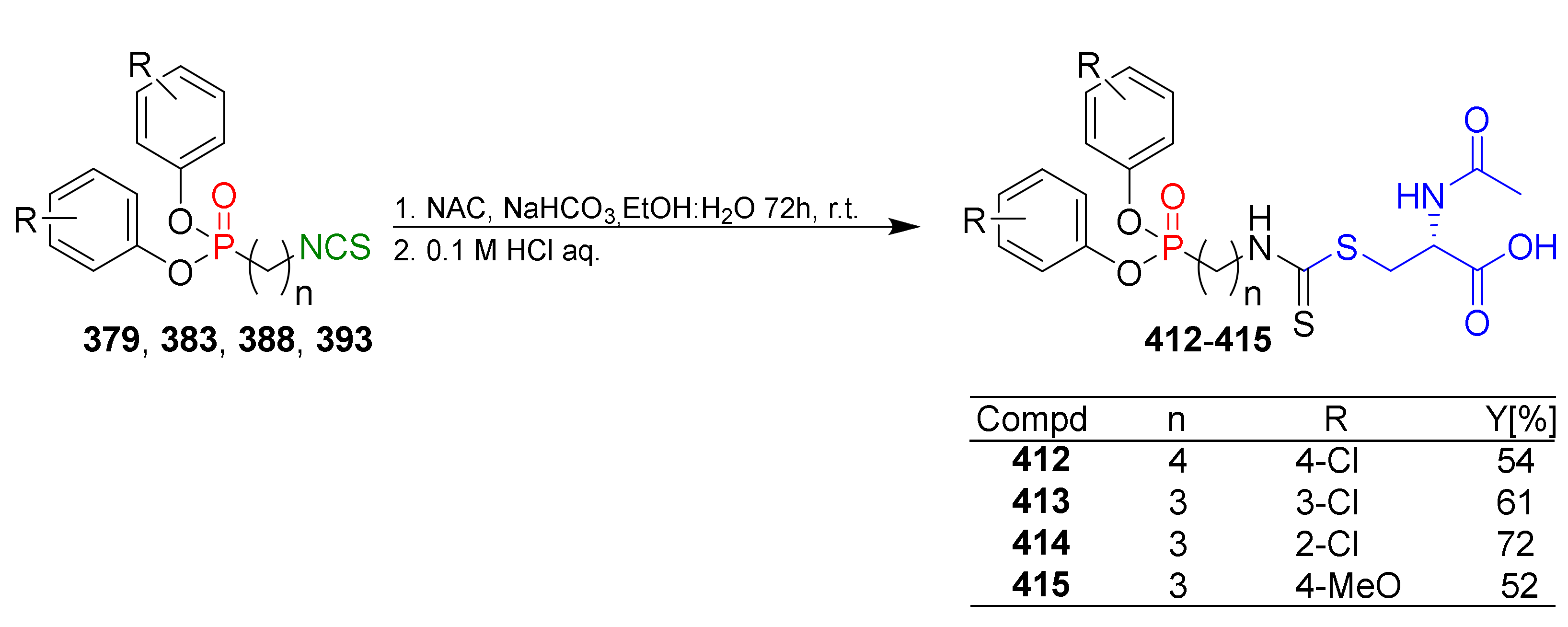
4.2. Synthesis of Phosphorus Analogues of Sulforaphane and Their Properties
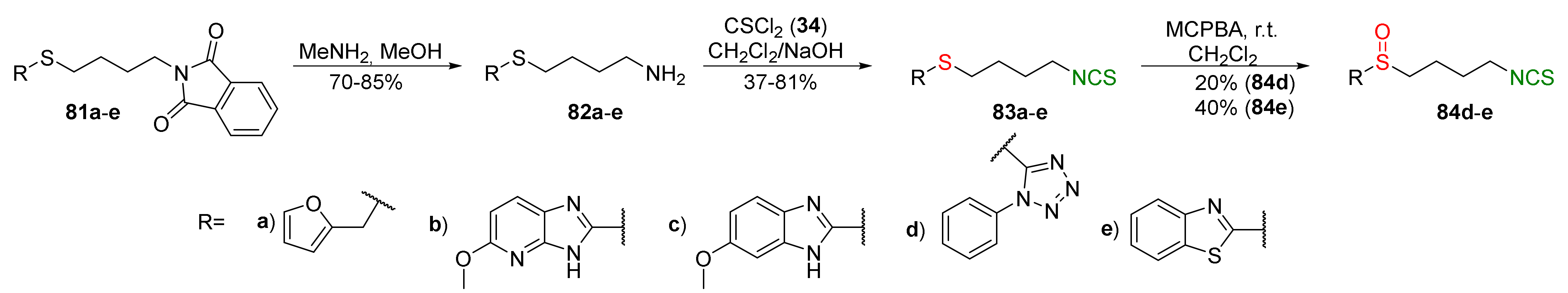
4.3. Synthesis of Carbonyl and Amide Analogues of Sulforaphane and Their Properties
4.4. Synthesis of Ether-Linked Analogues of Sulforaphane and Their Properties
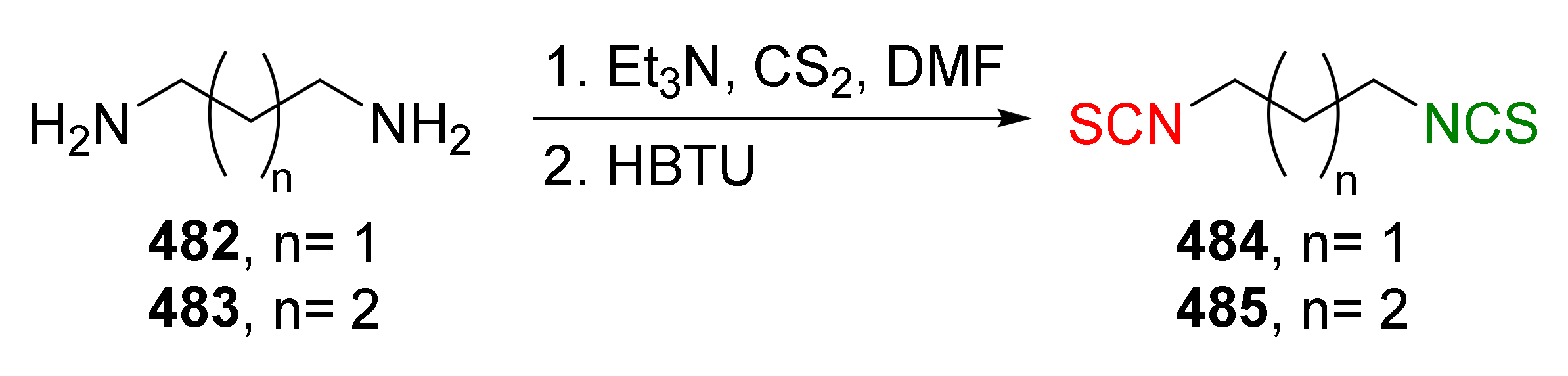
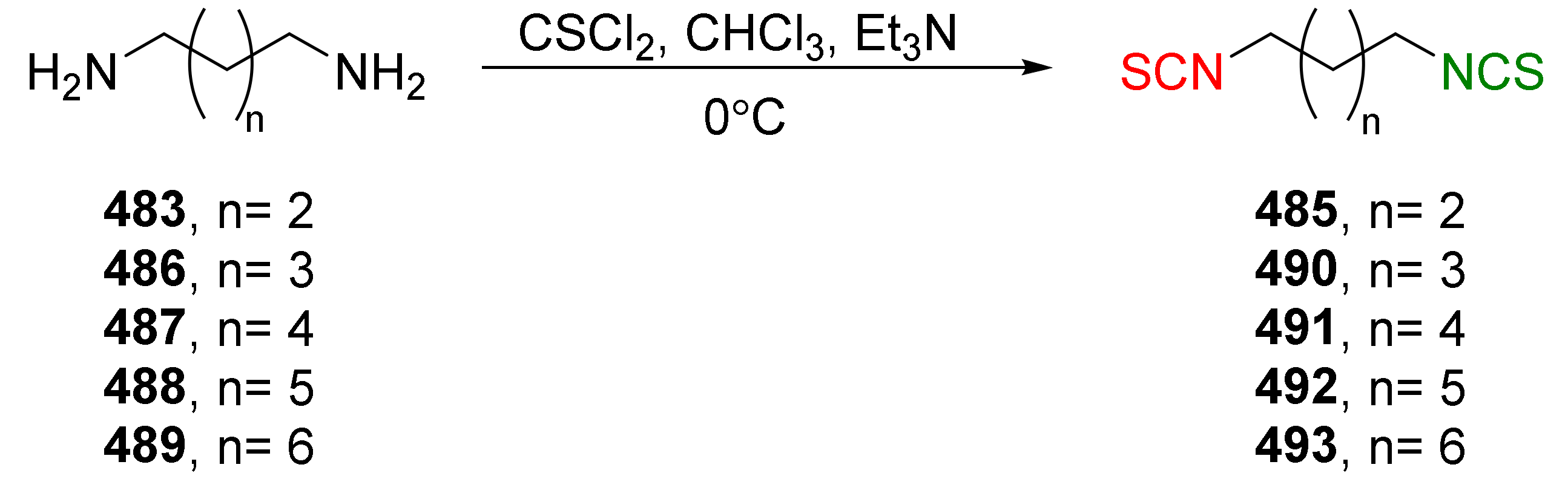
4.5. Synthesis of Diisothiocyanates
4.6. Summary of the Synthetic Routs of SFN and Its Bifunctional Analogs and Their Biological Activity
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Avato, P.; Argentieri, M.P. Brassicaceae: A rich source of health improving phytochemicals. Phytochem. Rev. 2015, 14, 1019–1033. [Google Scholar] [CrossRef]
- Verhoeven, D.T.H.; Verhagen, H.; Goldbohm, R.A.; van den Brandt, P.A.; van Poppel, G. A review of mechanism underlying anticarcinogenicity by brassica vegetables. Chem. Biol. Interact. 1997, 103, 79–129. [Google Scholar] [CrossRef]
- Jeffery, E.H.; Araya, M. Physiological effects of broccoli consumption. Phytochem. Rev. 2009, 8, 283–298. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; McCann, S.E.; Freudenheim, J.L.; Marshall, J.R.; Zhang, Y.; Shields, P.G. Breast cancer risk in premenopausal women is inversely associated with consumption of broccoli, a source of isothiocyanates, but is not modified by GST genotype. J. Nutr. 2004, 134, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.; Hsu, C.C.; Moullan, N.; Szeszenia-Dabrowska, N.; Lissowska, J.; Zaridze, D.; Rudnai, P.; Fabianova, E.; Mates, D.; Bencko, V.; et al. Effect of cruciferous vegetables on lung cancer in patients stratified by genetic status: A mendelian randomisation approach. Lancet 2005, 366, 1558–1560. [Google Scholar] [CrossRef]
- Wang, L.I.; Giovannucci, E.L.; Hunter, D.; Neuberg, D.; Su, L.; Christiani, D.C. Dietary intake of Cruciferous vegetables, Glutathione S-transferase (GST) polymorphisms and lung cancer risk in a Caucasian population. Cancer Causes Control 2004, 15, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Fowke, J.H.; Chung, F.L.; Jin, F.; Qi, D.; Cai, Q.; Conaway, C.; Cheng, J.R.; Shu, X.O.; Gao, Y.T.; Zheng, W. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003, 63, 3980–3986. [Google Scholar] [PubMed]
- Lin, H.J.; Probst-Hensch, N.M.; Louie, A.D.; Kau, I.H.; Witte, J.S.; Ingles, S.A.; Frankl, H.D.; Lee, E.R.; Haile, R.W. Glutathione transferase null genotype, broccoli, and lower prevalence of colorectal adenomas. Cancer Epidemiol. Biomark. Prev. 1998, 7, 647–652. [Google Scholar]
- Joseph, M.A.; Moysich, K.B.; Freudenheim, J.L.; Shields, P.G.; Bowman, E.D.; Zhang, Y.; Marshall, J.R.; Ambrosone, C.B. Cruciferous vegetables, genetic polymorphisms in glutathione S-transferases M1 and T1, and prostate cancer risk. Nutr. Cancer 2004, 50, 206–213. [Google Scholar] [CrossRef]
- Verhoeven, D.T.H.; Goldbohm, R.A.; van Poppel, G.; Verhagen, H.; van den Brandt, P.A. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidermiol. Biomark. Prev. 1996, 5, 733–748. [Google Scholar]
- Rungapamestry, V.; Duncan, A.J.; Fuller, Z.; Ratcliffe, B. Effect of cooking brassica vegetables on the subsequent hydrolysis and metabolic fate of glucosinolates. Proc. Nutr. Soc. 2007, 66, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends. Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Hara, M.; Fukino, N.; Kakizaki, T.; Morimitsu, Y. Glucosinolate metabolism, functionality and breeding for the improvement of Brassicaceae vegetables. Breed. Sci. 2014, 64, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouët, A. Glucosinolates: The synthetic approach. C. R. Chimie 2011, 14, 194–210. [Google Scholar] [CrossRef]
- Bell, L.; Wagstaff, C. Glucosinolates, myrosinase hydrolysis products, and flavonols found in rocket (Eruca sativa and Diplotaxis tenuifolia). J. Agric. Food Chem. 2014, 62, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Mithen, R. Glucosinolates, isothiocyanates and human health. Phytochem. Rev. 2009, 8, 269–282. [Google Scholar] [CrossRef]
- Ettlinger, M.G.; Lundeen, A.J. The structures of sinigrin and sinalbin; an enzymatic rearrangement. J. Am. Chem. Soc. 1956, 78, 4172–4173. [Google Scholar] [CrossRef]
- Ettlinger, M.G.; Lundeen, A.J. First synthesis of a mustard oil glucoside; the enzymatic lossen rearrangement. J. Am. Chem. Soc. 1957, 79, 1764–1765. [Google Scholar] [CrossRef]
- Hanschen, F.S.; Lamy, E.; Schreiner, M.; Rohn, S. Reactivity and stability of glucosinolates and their breakdown products in foods. Angew. Chem. Int. Ed. 2014, 53, 11430–11450. [Google Scholar] [CrossRef]
- Fahey, J.W.; Olson, M.E.; Stephenson, K.K.; Wade, K.L.; Chodur, G.M.; Odee, D.; Nouman, W.; Massiah, M.; Alt, J.; Egner, P.A.; et al. The diversity of chemoprotective glucosinolates in Moringaceae (Moringa spp.). Sci. Rep. 2018, 8, 7994. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W. Moringa oleifera: A review of the medical evidence for its nutritional, therapeutic, and prophylactic properties. Trees Life J. 2015, 1, 5. [Google Scholar]
- Olson, M.E.; Fahey, J.W. Moringa oleifera: A multipurpose tree for the dry tropics. Rev. Mex. Biodivers. 2011, 82, 1071–1082. [Google Scholar]
- Kissen, R.; Rossiter, J.; Bones, A.M. The ‘mustard oil bomb’: Not so easy to assemble?! Localization, expression and distribution of the components of the myrosinase enzyme system. Phytochem. Rev. 2009, 8, 69–86. [Google Scholar] [CrossRef]
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.R.; van Dam, N.M.; van Loon, J.J.A. Role of glucosinolates in insect-plant relationships and multitrophic interactions. Annu. Rev. Entomol. 2009, 54, 57–83. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotective glucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Uda, Y.; Kurata, T.; Arakawa, N. Effects of pH and ferrous ion on the degradation of glucosinolates by myrosinase. Agric. Biol. Chem. 1986, 50, 2735–2740. [Google Scholar]
- Foo, H.L.; Grønning, M.; Goodenough, L.; Bones, A.M.; Danielsen, B.E.; Whiting, D.A.; Rossiter, J.T. Purification and characterisation of epithiospecifier protein from Brassica napus: Enzymic intramolecular sulphur addition within alkenyl thiohydroximates derived from alkenyl glucosinolate hydrolysis. FEBS Lett. 2000, 468, 243–246. [Google Scholar] [CrossRef]
- Burow, M.; Losansky, A.; Müller, R.; Plock, A.; Kliebenstein, D.J.; Wittstock, U. The genetic basis of constitutive and herbivore-induced ESP-independent nitrile formation in Arabidopsis. Plant Physiol. 2009, 149, 561–574. [Google Scholar] [CrossRef]
- Burow, M.; Bergner, A.; Gershenzone, J.; Wittstock, U. Glucosinolate hydrolysis in Lepidium sativum-identification of the thiocyanate-forming protein. Plant Mol. Biol. 2007, 63, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wentzell, A.M.; Kliebestein, D.J. Genotype, age, tissue, and environment regulate the structural outcome of glucosinolate activation. Plant Physiol. 2008, 147, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Burow, M.; Wittstock, U. Regulation and function of specifier proteins in plants. Phytochem. Rev. 2009, 8, 87–99. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Koutsidis, G.; Mavroudis, N.; Trafalis, D.T.; Botaitis, S.; Franco, R.; Zoumpourlis, V.; Amery, T.; Galanis, A.; Pappa, A.; et al. The role of isothiocyanates as cancer chemo-preventive, chemo-therapeutic and anti-melanoma agents. Antioxidants 2019, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Palliyaguru, D.L.; Yuan, J.M.; Kensler, T.W.; Fahey, J.W. Isothiocyanates: Translating the power of plants to people. Mol. Nutr. Food Res. 2018, 62, e1700965. [Google Scholar] [CrossRef]
- Fimognari, C.; Turrini, E.; Ferruzzi, L.; Lenzi, M.; Hrelia, P. Natural isothiocyanates: Genotoxic potential versus chemoprevention. Mutat. Res. 2012, 750, 107–131. [Google Scholar] [CrossRef]
- Oliviero, T.; Verkerk, R.; Dekker, M. Isothiocyanates from brassica vegetables—Effects of processing, cooking, mastication, and digestion. Mol. Nutr. Food Res. 2018, 62, e1701069. [Google Scholar] [CrossRef]
- Bell, L.; Oloyede, O.O.; Lignou, S.; Wagstaff, C.; Methven, L. Taste and flavor perceptions of glucosinolates, isothiocyanates, and related compounds. Mol. Nutr. Food Res. 2018, 62, e170990. [Google Scholar] [CrossRef]
- Drobnica, L.; Kristian, P.; Augustin, J. The chemistry of the -NCS group. In Chemistry of Cyanates and Their Thio Derivatives; John Wiley & Sons: Chichester, UK, 1977; Volume 2, p. 1003. [Google Scholar]
- Wu, X.; Zhou, Q.H.; Xu, K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol. Sin. 2009, 30, 501–512. [Google Scholar] [CrossRef]
- Rao, C.V. Benzyl isothiocyanate: Double trouble for breast cancer cells. Cancer Prev. Res. 2013, 6, 760–763. [Google Scholar] [CrossRef]
- Gupta, P.; Wright, S.E.; Kim, S.H.; Srivastava, S.K. Phenethyl isothiocyanate: A comprehensive review of anti-cancer mechanism. Biochim. Biophys. Acta 2014, 1846, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.Z.; Zhang, X.; Wu, L.X.; Wen, C.J.; Hu, L.; Lv, Q.L.; Shen, D.Y.; Zhou, H.H. Advances in molecular signaling mechanisms of β-phenethyl isothiocyanate antitumor effects. J. Agric. Food Chem. 2015, 63, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Chiao, J.W. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (Review). Int. J. Oncol. 2010, 37, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Allyl isothiocyanate as a cancer chemopreventive phytochemical. Mol. Nutr. Food Res. 2010, 54, 127–135. [Google Scholar] [CrossRef]
- Myzak, M.C.; Dashwood, R.H. Chemoprotection by sulforaphane: Keep one eye beyond Keap1. Cancer Lett. 2006, 233, 208–218. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Briones-Herrera, A.; Eugenio-Pérez, D.; Reyes-Ocampo, J.G.; Rivera-Mancía, S.; Pedraza-Chaverri, J. New highlights on the health-improving effects of sulforaphane. Food Funct. 2018, 9, 2589–2606. [Google Scholar] [CrossRef]
- Mi, L.; Di Pasqua, A.J.; Chung, F.L. Proteins as binding targets of isothiocyanates in cancer prevention. Carcinogenesis 2011, 32, 1405–1413. [Google Scholar] [CrossRef]
- Brown, K.K.; Hampton, M.B. Biological targets of isothiocyanates. Biochim. Biophys. Acta 2011, 1810, 888–894. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef]
- Hecht, S.S. Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 2000, 32, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Dufour, V.; Stahl, M.; Baysse, C. The antibacterial properties of isothiocyanates. Microbiology 2015, 161, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Romeo, L.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. Isothiocyanates: An overview of their antimicrobial activity against human infections. Molecules 2018, 23, 624. [Google Scholar] [CrossRef]
- Kurepina, N.; Kreiswirth, B.N.; Mustaev, A. Growth-inhibitory activity of natural and synthetic isothiocyanates against representative human microbial pathogens. J. Appl. Microbiol. 2013, 115, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, A.K.; Ashare, R. Isothiocyanates in the chemistry of heterocycles. Chem. Rev. 1991, 91, 1–24. [Google Scholar] [CrossRef]
- Brandsma, L.; Nedolya, N.A.; Tarasova, O.A.; Trofimov, B.A. Synthesis of heterocyclic compounds from metallated unsaturated compounds and isothiocyanates. (Review). Chem. Heterocycl. Compd. 2000, 36, 1241–1260. [Google Scholar] [CrossRef]
- Pace, V.; Monticelli, S.; de la Vega-Hernández, K.; Castoldi, L. Isocyanates and isothiocyanates as versatile platforms for accessing (thio)amide-type compounds. Org. Biomol. Chem. 2016, 14, 7848–7854. [Google Scholar] [CrossRef]
- Koutoulogenis, G.; Kaplaneris, N.; Kokotos, C.G. (Thio)urea-mediated synthesis of functionalized six-membered rings with multiple chiral centers. Beilstein J. Org. Chem. 2016, 12, 462–495. [Google Scholar] [CrossRef]
- Fu, Y.; Mi, L.; Sanda, M.; Silverstein, S.; Aggarwal, M.; Wang, D.; Gupta, P.; Goldman, R.; Appella, D.H.; Chung, F.L. A click chemistry approach to identify protein targets of cancer chemopreventive phenethyl isothiocyanate. RSC Adv. 2014, 4, 3920–3923. [Google Scholar] [CrossRef]
- Clulow, J.A.; Strock, E.M.; Lanyonn-Hogg, T.; Kalesh, K.A.; Jones, L.H.; Tate, E.W. Competition-based, quantitative chemical proteomics in breast cancer cells identifies new target profiles for sulforaphane. Chem. Commun. 2017, 53, 5182–5185. [Google Scholar] [CrossRef]
- Schmid, H.; Karrer, P. Synthese der racemischen und der optisch aktiven formen des Sulforaphans. Helv. Chim. Acta 1948, 31, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.H.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Kushad, M.M.; Brown, A.F.; Kurilich, A.C.; Juvik, J.A.; Klein, B.P.; Wallig, M.A.; Jeffery, E.H. Variation of glucosinolates in vegetable crops of Brassica oleracea. J. Agric. Food Chem. 1999, 47, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Information Based on Web of Science Datebase of 3 January 2022.
- Abdull Razis, A.F.; Iori, R.; Ioannides, C. The natural chemopreventive phytochemical R-sulforaphane is a far more potent inducer of the carcinogen-detoxifying enzyme systems in rat liver and lung than the S-isomer. Int. J. Cancer 2011, 128, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopnicki, S. Bardxolone metyl displays detrimental effects on endothelial bioenergetics, suppresses endothelial ET-1 release, and increases endothelial permeability in human microvascular endothelium. Oxid. Med. Cell. Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Crowell, J.A.; Steele, V.E.; Lubet, R.A.; Malone, W.A.; Boone, C.W.; Kopelovich, L.; Hawk, E.T.; Lieberman, R.; Lawrence, J.A.; et al. Progress in cancer chemoprevention: Development of diet-derived chemopreventive agents. J. Nutr. 2000, 130, 467–471. [Google Scholar] [CrossRef]
- Karrer, P.; Scheitlin, E.; Siegrist, H. Über homologe des sulforaphans und über ω-aminoalkyl-sulfoxyde. Helv. Chim. Acta 1950, 33, 1237–1241. [Google Scholar] [CrossRef]
- Kjær, A.; Gmelin, R.; Larsen, I. Isothiocyanates XII. 3-Methylthiopropyl isothiocyanate (Iberverin), a new naturally occurring mustard oil. Acta Chem. Scand. 1955, 9, 1143–1147. [Google Scholar] [CrossRef]
- Kjær, A.; Gmelin, R. Isothiocyanates XI. 4-Methylthiobutyl isothiocyanate, a new naturally occurring mustard Oil. Acta Chem. Scand. 1955, 9, 542–544. [Google Scholar] [CrossRef]
- Kjær, A.; Larsen, I.; Gmelin, R.; Prydz, H. Isothiocyanates XIV. 5-Methylthiopentyl isothiocyanate, a new mustard oil present in nature as a glucoside (glucoberteroin). Acta Chem. Scand. 1955, 9, 1311–1316. [Google Scholar] [CrossRef]
- Schneider, W. Zur Kenntnis des schwefelhaitigen Alkaloid aus dem Goldlack-Samen. Chem. Ber. 1908, 41, 4469. [Google Scholar] [CrossRef]
- Schneider, W.; Kaufmann, H. Untersuchungen über Senföle. II. Erysolin, ein Sulfonsenföl aus Erysimum perowskianum. Justus Liebig’s Ann. Chem. 1912, 392, 1–15. [Google Scholar] [CrossRef][Green Version]
- Balenović, K.; Deljac, A.; Monković, I.; Štefanac, Z. Synthesis of (±) sulphoraphene. Tetrahedron 1966, 22, 2139–2143. [Google Scholar] [CrossRef]
- Bilska, A.; Kryczyk, A.; Włodek, L. Różne oblicza biologicznej roli glutationu. The different aspects of the biological role of glutathione. Postepy Hig. Med. Dosw. 2007, 61, 438–453. [Google Scholar]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Cotgreave, I.A.; Gerdes, R.G. Recent trends in glutathione biochemistry—Glutathione–protein interactions: A molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 1998, 242, 1–9. [Google Scholar] [CrossRef]
- Ghibelli, L.; Fanelli, C.; Rotilio, G.; Lafavia, E.; Coppola, S.; Colussi, C.; Civitareale, P.; Ciriolo, M.R. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998, 12, 479–486. [Google Scholar] [CrossRef]
- Poot, M.; Teubert, H.; Rabinovitch, P.S.; Kavanagh, T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol. 1995, 163, 555–560. [Google Scholar] [CrossRef]
- Hall, A.G. Glutathione and the regulation of cell death. Adv. Exp. Med. Biol. 1999, 457, 199–203. [Google Scholar]
- Chung, F.L.; Jiao, D.; Getahun, S.M.; Yu, M.C. A urinary biomarker for uptake of dietary isothiocyanates in humans. Cancer Epidemiol. Biomark. Prev. 1998, 7, 103–108. [Google Scholar]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Kensler, T.W.; Chen, J.G.; Gange, S.J.; Groopman, J.D.; Friesen, M.D. Quantification of sulforaphane mercapturic acid pathway conjugates in human urine by high-performance liquid chromatography and isotope-dilution tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Lamy, E.; Scholtes, C.; Herz, C.; Mersch-Sundermann, V. Pharmacokinetics and pharmacodynamics of isothiocyanates. Drug Metab. Rev. 2011, 43, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Cavell, B.E.; Syed Alwi, S.S.; Donlevy, A.; Packham, G. Anti-angiogenic effects of dietary isothiocyanates: Mechanisms of action and implications for human health. Biochem. Pharmacol. 2011, 81, 327–336. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary agents as histone deacetylase inhibitors: Sulforaphane and structurally related isothiocyanates. Nutr. Rev. 2008, 66, S36–S38. [Google Scholar] [CrossRef]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef]
- Milelli, A.; Fimognari, C.; Ticchi, N.; Neviani, P.; Minarini, A.; Tumiatti, V. Isothiocyanate synthetic analogs: Biological activities, structure-activity relationships and synthetic strategies. Mini Rev. Med. Chem. 2014, 14, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Tuli, H.S.; Mittal, S.; Shandilya, J.K.; Tiwari, A.; Sandhu, S.S. Isothiocyanates: A class of bioactive metabolites with chemopreventive potential. Tumor Biol. 2015, 36, 4005–4016. [Google Scholar] [CrossRef] [PubMed]
- Gründemann, C.; Huber, R. Chemoprevention with isothiocyanates—From bench to bedside. Cancer Lett. 2018, 414, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanism of action of isothiocyanates in cancer chemoprevention: An update. Food Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-target prevention of cancer by sulforaphane. Cancer. Res. 2008, 269, 291–304. [Google Scholar]
- Tomczyk, J.; Olejnik, A. Sulforafan—Potencjalny czynnik w prewencji i terapii chorób nowotworowych. Sulforaphane—A possibile agent in prevention and therapy of cancer. Postepy Hig. Med. Dosw. 2010, 64, 590–603. [Google Scholar]
- Jiang, X.; Liu, Y.; Ma, L.; Ji, R.; Qu, Y.; Xin, Y.; Lv, G. Chemopreventive activity of sulforaphane. Drug Des. Dev. Ther. 2018, 12, 2905–2913. [Google Scholar] [CrossRef]
- Leone, A.; Diorio, G.; Sexton, W.; Schell, M.; Alexandrow, M.; Fahey, J.W.; Kumar, N.B. Sulforaphane for the chemoprevention of bladder cancer: Molecular mechanism targeted approach. Oncotarget 2017, 8, 35412–35424. [Google Scholar] [CrossRef]
- Yang, C.S.; Smith, T.J.; Hong, J.Y. Cytochrome P-450 enzymes as targets for chemoprevention against chemical carcinogenesis and toxicity: Opportunities and limitations. Cancer. Res. 1994, 54, 1982s–1986s. [Google Scholar]
- Nakajima, M.; Yoshida, R.; Shimada, N.; Yamazaki, H.; Yokoi, T. Inhibition and inactivation of human cytochrome P450 isoforms by phenethyl isothiocyanate. Drug Metab. Dispos. 2001, 29, 1110–1113. [Google Scholar]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond nicotinamide metabolism: Potential role of nicotinamide N-methyltransferase as a biomarker in skin cancer. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, V.; Salvolini, E.; Lucarini, G.; Salvucci, A.; Campagna, R.; Rubini, C.; Sartini, D.; Emanuelli, M. Cancer stem cell enrichment is associated with enhancement of nicotinamide N-methyltransferase expression. IUBMB Life 2020, 72, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J. Nutr. 1999, 129, 768S–774S. [Google Scholar] [CrossRef] [PubMed]
- Bacon, J.R.; Williamson, G.; Garner, R.C.; Lappin, G.; Langouet, S.; Bao, Y. Sulforaphane and quercetin modulate PhIP–DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis 2003, 24, 1903–1911. [Google Scholar] [CrossRef]
- Jiang, Z.Q.; Chen, C.; Yang, B.; Hebbar, V.; Tony Kong, A.N. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003, 72, 2243–2253. [Google Scholar] [CrossRef]
- Basten, G.P.; Bao, Y.; Wiliamson, G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis 2002, 23, 1399–1404. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef]
- Fimognari, C.; Nusse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef]
- Garrett, M.D. Cell cycle control and cancer. Curr. Sci. 2001, 5, 515–522. [Google Scholar]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 2004, 279, 25813–25822. [Google Scholar] [CrossRef]
- Rose, P.; Huang, Q.; Ong, C.N.; Wheiteman, M. Broccoli and watercress suppress matrix metalloproteinase-9 activity and invasiveness of human MDA-MB-231 breast cancer cells. Toxicol. Appl. Pharmacol. 2005, 209, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Khor, T.O.; Yu, S.; Kong, A.N. PEITC induces G1 cell cycle arrest on HT-29 cells through the activation of p38 MAPK signaling pathway. AAPS J. 2008, 10, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Jakubíková, J.; Sedlák, J.; Mithen, M.; Bao, Y. Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane- and erucin-induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco-2 cells. Biochem. Pharmacol. 2005, 69, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Pappa, G.; Bartsch, H.; Gerhauser, C. Biphasic modulation of cell proliferation by sulforaphane at physiologically relevant exposure times in a human colon cancer cell line. Mol. Nutr. Food Res. 2007, 51, 977–984. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, G.Y.; Bae, S.J.; Yoo, Y.H.; Choi, Y.H. Induction of apoptosis by isothiocyanate sulforaphane in human cervical carcinoma HeLa and hepatocarcinoma HepG2 cells through activation of caspase-3. Oncol. Rep. 2007, 18, 181–187. [Google Scholar] [CrossRef]
- Choi, S.; Lew, K.L.; Xiao, H.; Herman-Antosiewicz, A.; Xiao, D.; Brown, C.K.; Singh, S.V. D,L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis 2007, 28, 151–162. [Google Scholar] [CrossRef]
- Basu, A.; Halder, S. Dietary isothiocyanate mediated apoptosis of human cancer cells is associated with Bcl-XL phosphorylation. Int. J. Oncol. 2008, 33, 657–663. [Google Scholar]
- Xu, K.; Thornalley, P.J. Signal transduction activated by the cancer chemopreventive isothiocyanates: Cleavage of BID protein, tyrosine phosphorylation and activation of JNK. Br. J. Cancer 2001, 84, 670–673. [Google Scholar] [CrossRef]
- Xiao, D.; Srivastava, S.K.; Lew, K.L.; Zeng, Y.; Hershberger, P.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003, 24, 891–897. [Google Scholar] [CrossRef]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L.; et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef]
- Marek, Ł. Rola apoptosomu w aktywacji prokaspazy 9. The role of the apoptosome in the activation of procaspase-9. Postepy Hig. Med. Dosw. 2013, 67, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramirez, M.; Rocchini, C.; Ausio, J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 1995, 270, 17923–17928. [Google Scholar] [CrossRef] [PubMed]
- Walia, H.; Chen, H.Y.; Sun, J.M.; Holtz, L.T.; Davie, J.R. Histone acetylation is required to maintain the unfolded nucleosome structure associated with transcribing DNA. J. Biol. Chem. 1998, 273, 14516–14522. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef]
- Ho, E.; Dashwood, R.H. Dietary manipulation of histone structure and function. World Rev. Nutr. Diet 2010, 101, 95–102. [Google Scholar]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate–mediated inhibition of histone deacetylase leads to NF-κB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 9, 1596–1608. [Google Scholar] [CrossRef]
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Zielonka, T.M. Angiogeneza—Część 1. Mechanizm powstawania nowych naczyń krwionośnych. Angiogenesis—Part 1. Mechanism of neovascularization. Alerg. Astma Immun. 2003, 8, 169–174. [Google Scholar]
- Wideł, M.S.; Wideł, M. Mechanizmy przerzutowania i molekularne markery progresji nowotworów złośliwych. I. Rak jelita grubego. Mechanisms of metastasis and molecular markers of malignant tumor progression. I. Colorectal cancer. Postepy Hig. Med. Dosw. 2006, 60, 453–470. [Google Scholar]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate inhibits angiogenesis In vitro and Ex vivo. Cancer Res. 2007, 67, 2239–2246. [Google Scholar] [CrossRef]
- Asakage, M.; Tsuno, N.T.; Kitayama, J.; Tsuchiya, T.; Yoneyama, S.; Yamada, J.; Okaji, Y.; Kaisaki, S.; Osada, T.; Takahashi, K.; et al. Sulforaphane induces inhibition of human umbilical vein endothelial cells proliferation by apoptosis. Angiogenesis 2006, 9, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, D.; Han, J.H.; Holley, R.A. Use of mustard flour to inactivate Escherichia coli O157:H7 in ground beef under nitrogen flushed packaging. Int. J. Food Microbiol. 2005, 99, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Delaquis, P.J.; Sholberg, P.L. Antimicrobial activity of gaseous allyl isothiocyanate. J. Food Prot. 1997, 60, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Tajima, H.; Kimoto, H.; Taketo, A. Specific antimicrobial synergism of synthetic hydroxy isothiocyanates with aminoglycoside antibiotics. Biosci. Biotechnol. Biochem. 2001, 65, 1886–1888. [Google Scholar] [CrossRef]
- Saavedra, M.J.; Borges, A.; Dias, C.; Aires, A.; Bennett, R.N.; Rosa, E.S.; Simões, M. Antimicrobial activity of phenolics and glucosinolate hydrolysis products and their synergy with streptomycin against pathogenic bacteria. Med. Chem. 2010, 6, 174–183. [Google Scholar] [CrossRef]
- Tajima, H.; Kimoto, H.; Taketo, A. Paradoxical effect of synthetic hydroxy isothiocyanates on antimicrobial action of aminoglycosides. Biosci. Biotechnol. Biochem. 2003, 67, 1844–1846. [Google Scholar] [CrossRef]
- Bressan, M.; Roncato, M.A.; Bellvert, F.; Comte, G.; Haichar, F.Z.; Achouak, W.; Berge, O. Exogenous glucosinolate produced by Arabidopsis thaliana has an impact on microbes in the rhizosphere and plant roots. ISME J. 2009, 3, 1243–1257. [Google Scholar] [CrossRef]
- Borek, V.; Morra, M.J.; McCaffrey, J.P. Myrosinase activity in soil extracts. Soil. Sci. Soc. Am. J. 1996, 60, 1792–1797. [Google Scholar] [CrossRef]
- Björkholm, N.; Zhukhovitsky, V.; Löfman, C.; Hultén, K.; Enroth, H.; Block, M.; Rigo, R.; Falk, P.; Engstrand, L. Helicobacter pylori entry into human gastric epithelial cells: A potential determinant of virulence, persistence, and treatment failures. Helicobacter 2000, 5, 148–154. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef]
- Ha, N.C.; Oh, S.T.; Sung, J.Y.; Cha, K.A.; Lee, M.H.; Oh, B.H. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 2001, 8, 505–509. [Google Scholar] [CrossRef]
- Fahey, J.W.; Stephenson, K.K.; Wade, K.L.; Talalay, P. Urease from Helicobacter pylori is inactivated by sulforaphane and other isothiocyanates. Biochem. Biophys. Res. Commun. 2013, 435, 1–7. [Google Scholar] [CrossRef]
- Dufour, V.; Alazzam, B.; Ermel, G.; Thepaut, M.; Rossero, A.; Tresse, O.; Baysse, C. Antimicrobial activities of isothiocyanates against Campylobacter jejuni isolates. Front. Cell. Infect. Microbiol. 2012, 2, 53. [Google Scholar] [CrossRef]
- Dufour, V.; Stahl, M.; Rosenfeld, E.; Stintzi, A.; Baysse, C. Insights into the Mode of Action of Benzyl Isothiocyanate on Campylobacter jejuni. Appl. Environ. Microbiol. 2013, 79, 6958–6968. [Google Scholar] [CrossRef]
- Feasey, N.A.; Dougan, G.; Kingsley, R.A.; Heyderman, R.S.; Gordon, M.A. Invasive non-typhoidal salmonella disease: An emerging and neglected tropical disease in Africa. Lancet 2012, 379, 2489–2499. [Google Scholar] [CrossRef]
- Sofrata, A.; Santangelo, E.M.; Azeem, M.; Borg-Karlson, A.K.; Gustafsson, A.; Pütsep, K. Benzyl isothiocyanate, a major component from the roots of Salvadora persica is highly active against Gram-negative bacteria. PLoS ONE 2011, 6, e23045. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef]
- Lin, C.M.; Preston III, J.F.; Wei, C.I. Antibacterial mechanism of allyl isothiocyanate. J. Food Prot. 2000, 63, 727–734. [Google Scholar] [CrossRef]
- Luciano, F.B.; Holley, R.A. Enzymatic inhibition by allyl isothiocyanate and factors affecting its antimicrobial action against Escherichia coli O157:H7. Int. J. Food Microbiol. 2009, 131, 240–245. [Google Scholar] [CrossRef]
- Nowicki, D.; Rodzik, O.; Herman-Antosiewicz, A.; Szalewska-Pałasz, A. Isothiocyanates as effective agents against enterohemorrhagic Escherichia coli: Insight to the mode of action. Sci. Rep. 2016, 6, 22263. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilm formation: A clinically relevant microbiological process. Clin. Infect. Dis. 2001, 33, 1387–1392. [Google Scholar] [CrossRef]
- Defez, C.; Fabbro-Peray, P.; Bouziges, N.; Gouby, A.; Mahamat, A.; Daurès, J.P.; Sotto, A. Risk factors for multidrug-resistant Pseudomonas aeruginosa nosocomial infection. J. Hosp. Infect. 2004, 57, 209–216. [Google Scholar] [CrossRef]
- Bassler, B.L. Small talk. Cell-to-cell communication in bacteria. Cell 2002, 109, 421–424. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Van Delden, C.; Iglewski, B.H. Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg. Infect. Dis. 1998, 4, 551–560. [Google Scholar] [CrossRef]
- Li, Y.H.; Tian, X.L. Quorum sensing and bacterial social interactions in biofilms. Sensors 2012, 12, 2519–2538. [Google Scholar] [CrossRef]
- Whiteley, M.; Lee, K.M.; Greenberg, E.P. Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 13904–13909. [Google Scholar] [CrossRef]
- Kaiser, S.J.; Mutters, N.T.; Blessing, B.; Günther, F. Natural isothiocyanates express antimicrobial activity against developing and mature biofilms of Pseudomonas aeruginosa. Fitoterapia 2017, 119, 57–63. [Google Scholar] [CrossRef]
- Borges, A.; Abreu, A.C.; Ferreira, C.; Saavedra, M.J.; Simões, L.C.; Simões, M. Antibacterial activity and mode of action of selected glucosinolate hydrolysis products against bacterial pathogens. J. Food Sci. Technol. 2015, 52, 4737–4748. [Google Scholar] [CrossRef]
- Ganin, H.; Rayo, J.; Amara, N.; Levy, N.; Krief, P.; Meijler, M.M. Sulforaphane and erucin, natural isothiocyanates from broccoli, inhibit bacterial quorum sensing. Med. Chem. Commun. 2013, 4, 175–179. [Google Scholar] [CrossRef]
- Hall, S.; McDermott, C.; Anoopkumar-Dukie, S.; McFarland, A.J.; Forbes, A.; Perkins, A.V.; Davey, A.K.; Chess-Williams, R.; Kiefel, M.J.; Arora, D.; et al. Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins 2016, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, T.H.; Bragason, S.K.; Phipps, R.K.; Christensen, L.D.; van Gennip, M.; Alhede, M.; Skindersoe, M.; Larsen, T.O.; Hoiby, N.; Bjarnsholt, T.; et al. Food as a source for Quorum Sensing inhibitors: Iberin from horseradish revealed as a Quorum Sensing inhibitor of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2012, 78, 2410–2421. [Google Scholar] [CrossRef]
- Montgomery, C.P.; Daniels, M.; Zhao, F.; Alegre, M.L.; Chong, A.S.; Daum, R.S. Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect. Immun. 2014, 82, 2125–2134. [Google Scholar] [CrossRef]
- Libert, M.; Elkholti, M.; Massaut, J.; Karmali, R.; Mascart, G.; Cherifi, S. Risk factors for meticillin resistance and outcome of Staphylococcus aureus bloodstream infection in a Belgian university hospital. J. Hosp. Infect. 2008, 68, 17–24. [Google Scholar] [CrossRef]
- Aires, A.; Mota, V.R.; Saavedra, M.J.; Rosa, E.A.S.; Bennett, R.N. The antimicrobial effects of glucosinolates and their respective enzymatic hydrolysis products on bacteria isolated from the human intestinal tract. J. Appl. Microbiol. 2009, 106, 2086–2095. [Google Scholar] [CrossRef]
- Lu, Z.J.; Dockery, C.R.; Crosby, M.; Chavarria, K.; Patterson, B.; Giedd, M. Antibacterial activities of wasabi against Escherichia coli O157:H7 and Staphylococcus aureus. Front. Microbiol. 2016, 7, 1403. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isorhiocyanates: A clinical phase I study. Nutr. Cancer. 2006, 55, 53–62. [Google Scholar] [CrossRef]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.-S.A.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogesis 2007, 28, 148–1490. [Google Scholar] [CrossRef]
- Brown, R.H.; Reynolds, C.; Brookre, A.; Talalay, P.; Fahey, J.W. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir. Res. 2015, 16, 106. [Google Scholar] [CrossRef]
- Yanaka, A.; Fahey, J.W.; Fukumoto, A.; Nakayama, M.; Inoue, S.; Zhang, S.; Tauchi, M.; Suzuki, H.; Hyodo, I.; Yamamoto, M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev. Res. 2009, 2, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A. Sulforaphane: Its “coming of age” as a clinically relevant nutraceutical in the prevention and treatment of chronic disease. Oxid. Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or sulforaphane: Is it the source or dose that matters? Moleclues 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.W.; Peterson, S. Brassica, biotransformation and cancer risk: Genetic polymorphism after the preventive effects of cruciferous vegetables. J. Nutr. 2002, 132, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Seow, A.; Shi, C.Y.; Chung, F.L.; Jiao, D.; Hankin, J.H.; Lee, H.P.; Coetzee, G.A.; Yu, M.C. Urinary total isothiocyanate (ITC) in a population-based sample of middle-aged and older Chinese in Singapore: Relationship with dietary total ITC and glutathione S-transferase M1/T1/P1 genotypes. Cancer Epidemiol. Biomark. Prev. 1998, 7, 775–781. [Google Scholar]
- Knight, J.G. Science of Synthesis, 18: Houben-Weyl Methods of Molecular Transformation, Compounds with Four and Three Carbon Heteroatom Bond; Georg Thieme Verlag: Stuttgart, Germany, 2005; Volume 18.2.8, pp. 189–243. [Google Scholar]
- Wentrup, C.; Finnerty, J.J.; Koch, R. Amino-, alkoxy-, and alkylthio-isocyanates and -isothiocyanates, RX-NCY, their isomers RX-YCN and RX-CNY, and their rearrangements. Curr. Org. Chem. 2011, 15, 1745–1759. [Google Scholar] [CrossRef]
- Bedane, K.G.; Singh, G.S. Reactivity and diverse synthetic applications of acyl isothiocyanates. Arkivoc 2015, 6, 206–245. [Google Scholar] [CrossRef]
- Rathke, A. Die Chemie auf der 45. Versammlung deutscher Naturforscher und Aerzte (12.—18. August). Ber. Dtsch. Chem. Ges. 1872, 5, 799. [Google Scholar]
- Rathke, A. Ueber Chlorschwefelkohlenstoffe. Ann. Chim. 1873, 167, 218. [Google Scholar] [CrossRef]
- Dyson, G.M.; George, H.J. CCXX.—The reactions of thiocarbonyl chloride. Part I. Reaction with aromatic primary aminocompounds. J. Chem. Soc. 1924, 1702–1708. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Monn, J.A.; Mattson, M.V.; George, C.; Jacobson, A.E.; Rice, K.C. Synthesis and binding properties of MK-801 isothiocyanates; (+)-3-isothiocyanato-5-methyl-10,11-dihydro-5H dibenzo[a,d]cyclohepten-5,10-imine hydrochloride: A new, potent and selective electrophilic affinity ligand for the NMDA receptor-coupled phencyclidine binding site. J. Med. Chem. 1993, 36, 2499–2507. [Google Scholar] [PubMed]
- Kutschy, P.; Dzurilla, M.; Takasugi, M.; Torok, M.; Achbergerova, I.; Homzova, R.; Racova, M. New syntheses of indole phytoalexins and related compounds. Tetrahedron 1998, 54, 3549–3566. [Google Scholar] [CrossRef]
- Kutschy, P.; Achbergerova, I.; Dzurilla, M.; Takasugi, M. Synthesis of indole phytoalexins brassinin and cyclobrassinin via [1-(tert-butoxycarbonyl)indol-3-yl]-methyl isothiocyanate as the Key biomimetic intermediate. Synlett 1997, 289–290. [Google Scholar] [CrossRef]
- Yamada, K.; Rice, K.C.; Flippen-Anderson, J.L.; Eissenstat, M.A.; Ward, S.J.; Johnson, M.R.; Howlett, A.C. (Aminoalkyl)indole isothiocyanates as potential electrophilic affinity ligands for the brain cannabinoid receptor. J. Med. Chem. 1996, 39, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; Cho, C.G.; Green, J.V.; Zhang, Y.; Talalay, P. Design and synthesis of bifunctional isothiocyanate analogs of sulforaphane: Correlation between structure and potency as inducers of anticarcinogenic detoxication enzymes. J. Med. Chem. 1994, 37, 170–176. [Google Scholar] [CrossRef]
- Dyson, G.M.; George, H.J.; Hunter, R.F. CCCCVI.—The interaction of thiocarbonyl chloride and chloro-substituted anilines and the inhibitory action of ortho-substituents. J. Chem. Soc. 1926, 3041–3044. [Google Scholar] [CrossRef]
- Hallenbach, W.; Horner, L. Die Synthese eines fluoreszierenden isothiocyanats als nachweisreagenz für die NH-Funktion. Synthesis 1985, 8, 791. [Google Scholar] [CrossRef]
- Ares, J.J.; Kador, P.F.; Miller, D.D. Synthesis and biological evaluation of irreversible inhibitors of aldose reductase. J. Med. Chem. 1986, 29, 2384–2389. [Google Scholar] [CrossRef]
- Larsen, C.; Harpp, D.N. Thiocarbonyl transfer reagent chemistry. 3. Selective displacements with formaldehyde hydrazones and other nucleophiles. J. Org. Chem. 1981, 46, 2465–2466. [Google Scholar] [CrossRef]
- Kim, S.; Yi, K.Y. 1,1’-Thiocarbonyldi-2,2’-pyridone. A new useful reagent for functional group conversions under essentially neutral conditions. J. Org. Chem. 1986, 56, 2613–2615. [Google Scholar] [CrossRef]
- Kim, S.; Yi, K.Y. Di-2-Pyridyl thionocarbonate. A new reagent for the preparation of isothiocyanates and carbodiimides. Tetrahedron Lett. 1985, 26, 1661–1664. [Google Scholar] [CrossRef]
- Hodgkins, J.E.; Ettlinger, M.G. The synthesis of isothiocyanates from amines. J. Org. Chem. 1956, 21, 404–405. [Google Scholar] [CrossRef]
- Hofmann, A.W. Ueber die dem Senföl entsprechenden Isomeren der Schwefelcyanwasserstoffäther. Chem. Ber. 1868, 1, 170. [Google Scholar] [CrossRef]
- Hodgkins, J.E.; Reeves, W.P. The modified kaluza synthesis. III. The synthesis of some aromatic isothiocyanates. J. Org. Chem. 1964, 29, 3098–3099. [Google Scholar] [CrossRef]
- Li, G.; Tajima, H.; Ohtani, T. An improved procedure for the preparation of isothiocyanates from primary amines by using hydrogen peroxide as the dehydrosulfurization reagent. J. Org. Chem. 1997, 62, 4539–4540. [Google Scholar] [CrossRef]
- Boas, U.; Gertz, H.; Christensen, J.B.; Heegaard, P.M.H. Facile synthesis of aliphatic isothiocyanates and thioureas on solid phase using peptide coupling reagents. Tetrahedron Lett. 2004, 45, 269–272. [Google Scholar] [CrossRef]
- Psurski, M.; Błażewska, K.; Gajda, A.; Gajda, T.; Wietrzyk, J.; Oleksyszyn, J. Synthesis and antiproliferative activity of novel α- and β-dialkoxyphosphoryl isothiocyanates. Bioorg. Med. Chem. Lett. 2011, 21, 4572–4576. [Google Scholar] [CrossRef]
- Boas, U.; Pedersen, B.; Christensen, J.B. Tetramethyl fluoro formamidinium hexafluorophoshate—An improved synthesis and some new uses. Synth. Commun. 1998, 28, 1223–1231. [Google Scholar] [CrossRef]
- Liu, S.; Tang, C.; Ho, B.; Ankersen, M.; Stidsen, C.E.; Crider, A.M. Nonpeptide somatostatin agonists with sst4 selectivity: synthesis and structure–activity relationships of thioureas. J. Med. Chem. 1998, 41, 4693–4705. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Gajda, A.; Frankowski, S.; Goszczyński, T.M.; Gajda, T. T3P®—A benign desulfurating reagent in the synthesis of isothiocyanates. Synthesis 2018, 50, 1141–1151. [Google Scholar]
- Wong, R.; Dolman, S.J. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J. Org. Chem. 2007, 72, 3969–3971. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Sun, X.; Ma, H.; Chen, X.; Ren, J.; Hu, K. New method for the synthesis of sulforaphane and related isothiocyanates. Synthesis 2011, 24, 3991–3996. [Google Scholar] [CrossRef]
- Nath, J.; Ghosh, H.; Yella, R.; Patel, B.K. Molecular iodine mediated preparation of isothiocyanates from dithiocarbamic acid salts. Eur. J. Org. Chem. 2009, 1849–1851. [Google Scholar] [CrossRef]
- Ghosh, H.; Yella, R.; Nath, J.; Patel, B.K. Desulfurization mediated by hypervalent iodine(III): A novel strategy for the construction of heterocycles. Eur. J. Org. Chem. 2008, 6189–6196. [Google Scholar] [CrossRef]
- Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008, 49, 3117–3119. [Google Scholar] [CrossRef]
- Jamir, L.; Ali, A.R.; Ghosh, H.; Chipem, F.A.S.; Patel, B.K. The thiocarbonyl ‘S’ is softer than thiolate ‘S’: A catalyst-free one-pot synthesis of isothiocyanates in water. Org. Biomol. Chem. 2010, 8, 1674–1678. [Google Scholar] [CrossRef]
- Sun, N.; Li, B.; Shao, J.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. A general and facile one-pot process of isothiocyanates from amines under aqueous conditions. Beilstein J. Org. Chem. 2012, 8, 61–70. [Google Scholar] [CrossRef]
- Mandapati, U.; Pinapati, S.; Rudraraju, R. Copper promoted desulfurization towards the synthesis of isothiocyanates. Tetrahedron Lett. 2017, 58, 125–128. [Google Scholar] [CrossRef]
- Seelam, M.; Shaik, B.; Kammela, P.R. Cobalt mediated by desulfurization toward the synthesis of isothiocyanates. Synth. Commun. 2016, 46, 1759–1765. [Google Scholar] [CrossRef]
- Fu, Z.; Yuan, W.; Chen, N.; Yang, Z.; Xu, J. Na2S2O8-mediated efficient synthesis of isothiocyanates from primary amines in water. Green Chem. 2018, 20, 4484–4491. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Kręgiel, D.; Kolesińska, B. Synthesis of isothiocyanates using DMT/NMM/TsO− as a new desulfurization reagent. Molecules 2021, 26, 2740. [Google Scholar] [CrossRef] [PubMed]
- Eschliman, K.; Bossmann, S.H. Synthesis of isothiocyanates: An update. Synthesis 2019, 51, 1746–1752. [Google Scholar] [CrossRef]
- Nath, J.; Jamir, L.; Patel, B.K. Improved procedure for the preparation of isothiocyanates via iodine-mediated desulfurization of dithiocarbamic acid salts. Green Chem. Lett. Rev. 2011, 4, 1–34. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Gajda, A.; Gajda, T. Direct, microwave-assisted synthesis of isothiocyanates. Eur. J. Org. Chem. 2019, 2528–2532. [Google Scholar] [CrossRef]
- Staudinger, H.; Hauser, E. Über neue organische Phosphorverbindungen IV† Phosphinimine. Helv. Chim. Acta 1921, 4, 1353. [Google Scholar] [CrossRef]
- Molina, P.; Alajarin, M.; Arques, A. Convenient improved syntheses of isocyanates or isothiocyanates from amines. Synthesis 1982, 596–597. [Google Scholar] [CrossRef]
- Tsuge, O.; Kanemasa, S.; Matsuda, K. One-pot synthesis of N-[(trimethylsilyl)methyl]imines and (trimethylsilyl)methyl-substituted heterocumulenes from (trimethylsilyl)methyl azide. J. Org. Chem. 1984, 49, 2688–2691. [Google Scholar] [CrossRef]
- Isoda, T.; Hayashi, K.; Tamai, S.; Kumagai, T.; Nagao, Y. Efficient synthesis of isothiocyanates based on the tandem Staudinger/aza-Wittig reactions and mechanistic consideration of the tandem reactions. Chem. Pharm. Bull. 2006, 54, 1616–1619. [Google Scholar] [CrossRef][Green Version]
- Liao, Y.Y.; Deng, J.C.; Ke, Y.P.; Zhong, X.L.; Xu, L.; Tang, R.Y.; Zheng, W. Isothiocyanation of amines using the Langlois reagent. Chem. Commun. 2017, 53, 6073–6076. [Google Scholar] [CrossRef]
- Langlois, B.R.; Laurent, E.; Roidot, N. Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Lett. 1991, 32, 7525–7528. [Google Scholar] [CrossRef]
- Scattolin, T.; Klein, A.; Schoenebeck, F. Synthesis of isothiocyanates and unsymmetrical thioureas with the bench-stable solid reagent (Me4N)SCF3. Org. Lett. 2017, 19, 1831–1833. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lin, J.H.; Xiao, J.C. Reaction of thiocarbonyl fluoride generated from difluorocarbene with amines. Angew. Chem. Int. Ed. 2017, 56, 16669–16673. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Cai, J.; Lin, J.H.; Guo, Y.; Xiao, J.C. Synthesis and decarboxylative Wittig reaction of difluoromethylene phosphobetaine. Chem. Commun. 2013, 49, 7513–7515. [Google Scholar] [CrossRef]
- Zhen, L.; Fan, H.; Wang, X.; Jiang, L. Synthesis of thiocarbamoyl fluorides and isothiocyanates using CF3SiMe3 and elemental sulfur or AgSCF3 and KBr with amines. Org. Lett. 2019, 21, 2106–2110. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Wang, M.; Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic trifluoromethylation and beyond. Chem. Rev. 2015, 115, 683–730. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, X.G. Organophosphine-free copper-catalyzed isothiocyanation of amines with sodium bromodifluoroacetate and sulfur. Chem. Commun. 2019, 55, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.G.; Ábrányi-Balogh, P. Recent advances in the synthesis of isothiocyanates using elemental sulfur. Catalyst 2021, 11, 1081. [Google Scholar] [CrossRef]
- Wei, J.; Liang, S.; Jiang, L.; Yi, W. Synthesis of thiocarbamoyl fluorides and isothiocyanates using amines with CF3SO2Cl. J. Org. Chem. 2020, 85, 12374–12381. [Google Scholar] [CrossRef]
- Hamashima, Y.; Nagi, T.; Shimizu, R.; Tsuchimoto, T.; Sodeoka, M. Catalytic asymmetric α-chlorination of 3-acyloxazolidin-2-one with a trinary catalytic system. Eur. J. Org. Chem. 2011, 2011, 3675–3678. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Jiang, L.; Ding, T.; Yi, W.-b.; Zeng, X.; Zhang, W. Fluoroalkylsulfonyl chlorides promoted vicinal chloro-fluoroalkylthiolation of alkenes and alkynes. Org. Lett. 2018, 20, 2236–2240. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, G.R.; Rollin, P.; Mazzon, E.; Iori, R. Novel gram-scale production of enantiopure R-sulforaphane from Tuscan black kale seeds. Molecules 2014, 19, 6975–6986. [Google Scholar] [CrossRef] [PubMed]
- Iori, R.; Bernardi, R.; Gueyrard, D.; Rollin, P.; Palmieri, S. Formation of glucoraphanin by chemoselective oxidation of natural glucoerucin: A chemoenzymatic route to sulforaphane. Bioorg. Med. Chem. Lett. 1999, 9, 1047–1048. [Google Scholar] [CrossRef]
- Papi, A.; Orlandi, M.; Bartolini, G.; Barillari, J.; Iori, R.; Paolini, M.; Ferroni, F.; Fumo, M.G.; Pedulli, G.F.; Valgimigli, L. Cytotoxic and antioxidant activity of 4-methylthio-3-butenyl isothiocyanate from Raphanus sativus L. (Kaiware Daikon) sprouts. J. Agric. Food Chem. 2008, 56, 875–883. [Google Scholar] [CrossRef]
- Terada, Y.; Masuda, H.; Watanabe, T. Structure−activity relationship study on isothiocyanates: Comparison of TRPA1-activating ability between allyl isothiocyanate and specific flavor components of wasabi, horseradish, and white mustard. J. Nat. Prod. 2015, 78, 1937–1941. [Google Scholar] [CrossRef]
- Vermeulen, M.; Zwanenburg, B.; Chittenden, G.J.F.; Verhagen, H. Synthesis of isothiocyanate-derived mercapturic acids. Eur. J. Med. Chem. 2003, 38, 729–737. [Google Scholar] [CrossRef]
- Mays, J.R.; Roska, R.L.W.; Sarfaraz, S.; Mukhtar, H.; Rajski, S.R. Identification, synthesis, and enzymology of non-natural glucosinolate chemopreventive candidates. ChemBioChem 2008, 9, 729–747. [Google Scholar] [CrossRef]
- Conaway, C.C.; Wang, C.X.; Pittman, B.; Yang, Y.M.; Schwartz, J.E.; Tian, D.; McIntee, E.J.; Hecht, S.S.; Chung, F.L. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005, 65, 8548–8557. [Google Scholar] [CrossRef]
- Moon, J.K.; Kim, J.R.; Ahn, Y.J.; Shibamoto, T. Analysis and anti-Helicobacter activity of sulforaphane and related compounds present in broccoli (Brassica oleracea L.) sprouts. J. Agric. Food Chem. 2010, 58, 6672–6677. [Google Scholar] [CrossRef]
- Ernst, I.M.A.; Palani, K.; Esatbeyoglu, T.; Schwarz, K.; Rimbach, G. Synthesis adn Nrf2-inducing activity of the isothiocyanates iberverin, iberin and cheirolin. Pharmacol. Res. 2013, 70, 155–162. [Google Scholar] [CrossRef]
- Kiełbasiński, P.; Łuczak, J.; Cierpiał, T.; Błaszczyk, J.; Sieroń, L.; Wiktorska, K.; Lubelska, K.; Milczarek, M.; Chilmończyk, Z. New enantiomeric fluorine-containing derivatives of sulforaphane: Synthesis, absolute configurations and biological activity. Eur. J. Med. Chem. 2014, 76, 332–342. [Google Scholar] [CrossRef]
- Cierpiał, T.; Kiełbasiński, P.; Kwiatkowska, M.; Łyżwa, P.; Lubelska, K.; Kuran, D.; Dąbrowska, A.; Kruszewska, H.; Mielczarek, L.; Chilmonczyk, Z.; et al. Fluoroaryl analogs of sulforaphane—A group of compounds of anticancer and antimicrobial activity. Bioorg. Chem. 2020, 94, 103454. [Google Scholar] [CrossRef]
- Shi, Y.H.; Dai, D.F.; Li, J.; Dong, Y.W.; Jiang, Y.; Li, H.G.; Gao, Y.; Chong, C.K.; Li, H.Y.; Chu, X.Q.; et al. Sulforaphane analohues with heterocyclic moieties: Synthesis and inhibitory activities against cancer cell lines. Molecules 2016, 21, 514. [Google Scholar] [CrossRef]
- Setito, S.; Pruccoli, L.; Runfola, M.; Citi, V.; Martelli, A.; Saccomanni, G.; Calderone, V.; Tarozzi, A.; Rapposelli, S. Design and synthesis of H2S-donor hybrids: A new treatment for Alzheimer’s disease? Eur. J. Med. Chem. 2019, 184, 111745. [Google Scholar] [CrossRef]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Hu, K.; Qi, Y.J.; Zhao, J.; Jiang, H.F.; Chen, X.; Ren, J. Synthesis and biological evaluation of sulforaphane derivatives as potential antitumor agents. Eur. J. Med. Chem. 2013, 64, 529–539. [Google Scholar] [CrossRef]
- Khiar, N.; Werner, S.; Mallouk, S.; Lieder, F.; Alcudia, A.; Fernández, I. Enantiopure sulforaphane analogues with various substituents at the sulfinyl sulfur: Asymmetric synthesis and biological activities. J. Org. Chem. 2009, 74, 6002–6009. [Google Scholar] [CrossRef]
- Fernández, I.; Khiar, N.; Llera, J.M.; Alcudia, F. Asymmetric synthesis of alkane- and arenesulfinates of diacetone-D-glucose (DAG): An improved and general route to both enantiomerically pure sulfoxides. J. Org. Chem. 1992, 57, 6789–6796. [Google Scholar] [CrossRef]
- Elhalem, E.; Recio, R.; Werner, S.; Lieder, F.; Calderón-Montaño, J.M.; López-Lázaro, M.; Fernández, I.; Khiar, N. Sulforaphane homologues: Enantiodivergent synthesis of both enantiomers, activation of the Nrf2 transcription factor and selective cytotoxic activity. Eur. J. Med. Chem. 2014, 87, 552–563. [Google Scholar] [CrossRef]
- Psurski, M.; Piguła, M.; Ciekot, J.; Winiarski, Ł.; Wietrzyk, J.; Oleksyszyn, J. Convenient syntheses of novel 1-isothiocyano-alkylphosphonate diphenyl ester derivatives with potential biological activity. Tetrahedron Lett. 2012, 53, 5845–5847. [Google Scholar] [CrossRef]
- Psurski, M.; Janczewski, Ł.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Novel phosphonate analogs of sulforaphane: Synthesis, in vitro and in vivo activity. Eur. J. Med. Chem. 2017, 132, 63–80. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Gajda, A.; Braszczyńska, J.; Gajda, T. Microwave-assisted synthesis of dialkyl ω-azidoalkylphosphonates. Synth. Commun. 2016, 46, 1625–1633. [Google Scholar] [CrossRef]
- Rudzinska-Radecka, M.; Janczewski, Ł.; Gajda, A.; Godlewska, M.; Chmielewska-Krzesinska, M.; Waskowicz, K.; Podlasz, P. The anti-tumoral potential of phosphonate analog of sulforaphane in Zebrafish xenograft model. Cells 2021, 10, 3219. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Psurski, M.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Design, synthesis, and evaluation of ω-(isothiocyanato)alkylphosphinates and phosphine oxides as antiproliferative agents. ChemMedChem 2018, 13, 105–115. [Google Scholar] [CrossRef]
- Janczewski, Ł.; Burchacka, E.; Psurski, M.; Ciekot, J.; Gajda, A.; Gajda, T. New diaryl ω-(isothiocyanato)alkylphosphonates and their mercapturic acids as potential antibacterial agents. Life Sci. 2019, 219, 264–271. [Google Scholar] [CrossRef]
- Psurski, M.; Janczewski, Ł.; Świtalska, M.; Gajda, A.; Goszczyński, T.M.; Ciekot, J.; Winiarski, Ł.; Oleksyszyn, J.; Wietrzyk, J.; Gajda, T. Phosphorus-containing isothiocyanate-derived mercapturic acids as a useful alternative for parental isothiocyanates in experimental oncology. Bioorg. Med. Chem. Lett. 2018, 28, 2611–2615. [Google Scholar] [CrossRef]
- Amara, N.; Mashiach, R.; Amar, D.; Krief, P.; Spieser, S.A.H.; Bottomley, M.J.; Aharoni, A.; Meijler, M.M. Covalent inhibition of bacterial quorum sensing. J. Am. Chem. Soc. 2009, 131, 10610–10619. [Google Scholar] [CrossRef]
- Amara, N.; Gregor, R.; Rayo, J.; Dandela, R.; Daniel, E.; Liubin, N.; Willems, H.M.E.; Ben-Zvi, A.; Krom, B.P.; Meijler, M.M. Fine-tuning covalent inhibition of bacterial quorum sensing. ChemBioChem 2016, 17, 825–835. [Google Scholar] [CrossRef]
- Noshita, T.; Kidachi, Y.; Funayama, H.; Kiyota, K.; Yamaguchi, H.; Ryoyama, K. Anti-nitric oxide production activity of isothiocyanates correlates with their polar surface area rather than their lipophilicity. Eur. J. Med. Chem. 2009, 44, 4931–4936. [Google Scholar] [CrossRef]
- Tarozzi, A.; Marchetti, C.; Nicolini, B.; D’Amico, M.; Ticchi, N.; Pruccoli, L.; Tumiatti, V.; Simoni, E.; Lodola, A.; Mor, M.; et al. Combined inhibition of the EGFR/AKT pathways by a novel conjugate of quinazoline with isothiocyanate. Eur. J. Med. Chem. 2016, 117, 283–291. [Google Scholar] [CrossRef]
- Boehm, J.; Davis, R.; Murar, C.E.; Li, T.; McCleland, B.; Dong, S.; Yan, H.; Kerns, J.; Moody, C.J.; Wilson, A.J.; et al. Discovery of a crystalline sulforaphane analog with good solid-state stability and engagement of the Nrf2 pathway in vitro and in vivo. Bioorg. Med. Chem. 2019, 27, 579–588. [Google Scholar] [CrossRef]
- Wang, X.; Yu, C.; Wang, C.; Ma, Y.; Wang, T.; Li, Y.; Huang, Z.; Zhou, M.; Sun, P.; Zheng, J.; et al. Novel cyclin-dependent kinase 9 (CDK9) inhibitor with suppression of cancer stemness activity against non-small-cell lung cancer. Eur. J. Med. Chem. 2019, 181, 111535. [Google Scholar] [CrossRef]
- O’Brien, K.T.; Noto, J.G.; Nichols-O’Neill, L.; Perez, L.J. Potent irreversible inhibitors of LasR quorum sensing in Pseudomonas aeruginosa. ACS Med. Chem. Lett. 2015, 6, 162–167. [Google Scholar] [CrossRef]
- Fortunato, S.; Lenzi, C.; Granchi, C.; Citi, V.; Martelli, A.; Calderone, V.; Di Pietro, S.; Signore, G.; Di Bussolo, V.; Minutolo, F. First examples of H2S-releasing glycoconjugates: Stereoselective synthesis and anticancer activities. Bioconjugate Chem. 2019, 30, 614–620. [Google Scholar] [CrossRef]
- Nyein, C.M.; Zhong, X.; Lu, J.; Luo, H.; Wang, J.; Rapposelli, S.; Li, M.; Ou-yang, Y.; Pi, R.; He, X. Synthesis and anti-glioblastoma effect of artemisinin-isothiocyanate derivatives. RSC Adv. 2018, 8, 40974–40983. [Google Scholar] [CrossRef]
- Grzywa, R.; Winiarski, Ł.; Psurski, M.; Rudnicka, A.; Wietrzyk, J.; Gajda, T.; Oleksyszyn, J. Synthesis and biological activity of diisothiocyanate-derived mercapturic acids. Bioorg. Med. Chem. Lett. 2016, 26, 667–671. [Google Scholar] [CrossRef]



























































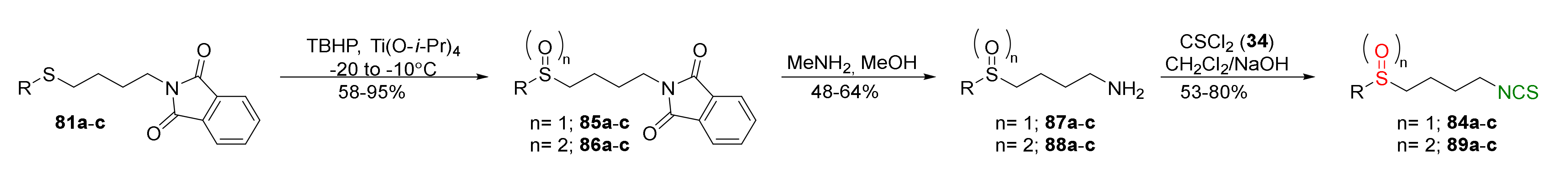
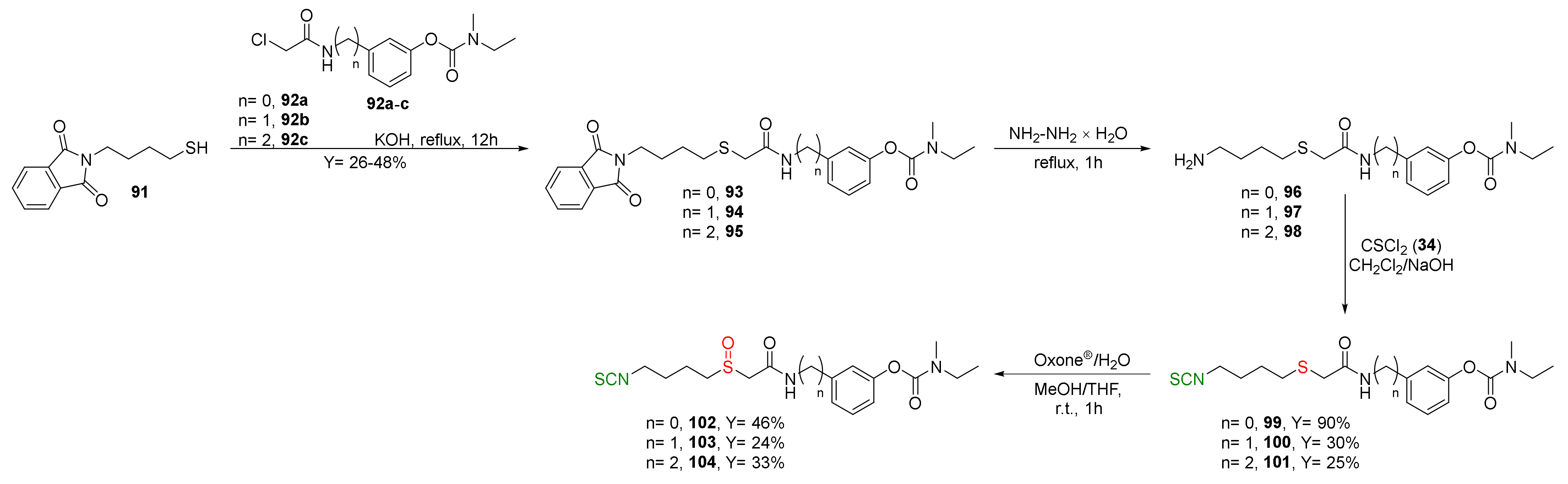
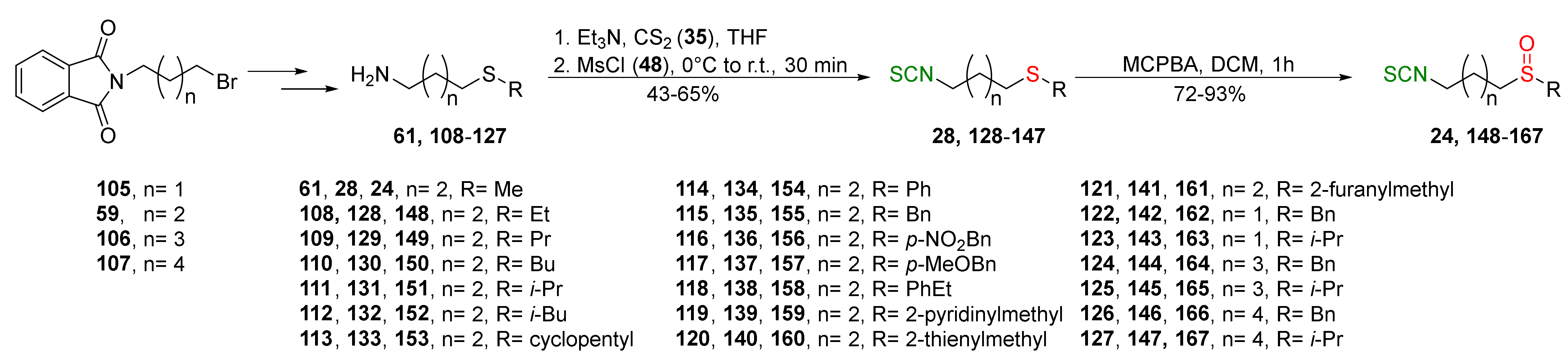
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
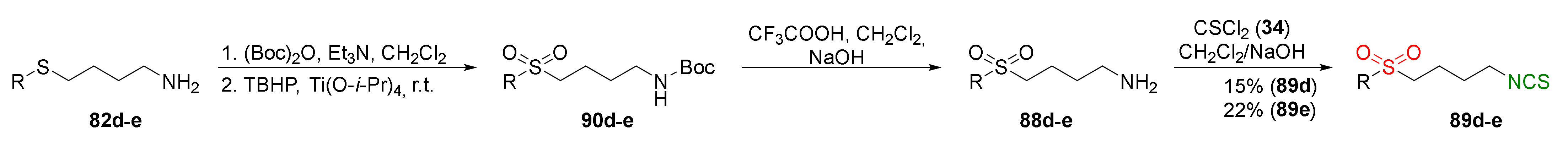{kind=link}
{kind=link}
{kind=link}
{kind=link}
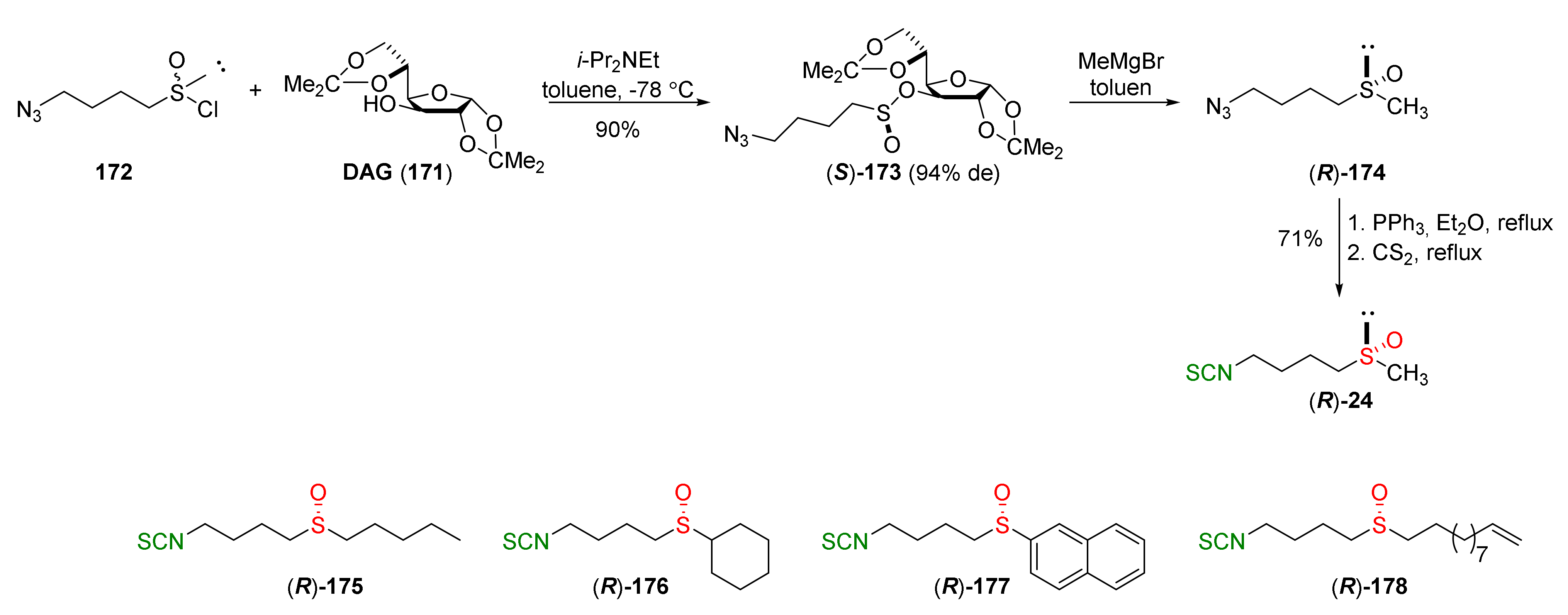{kind=link}
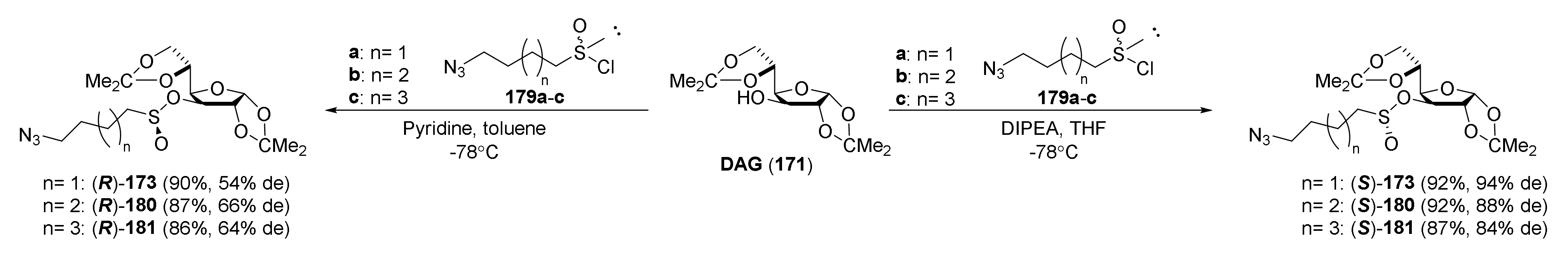{kind=link}
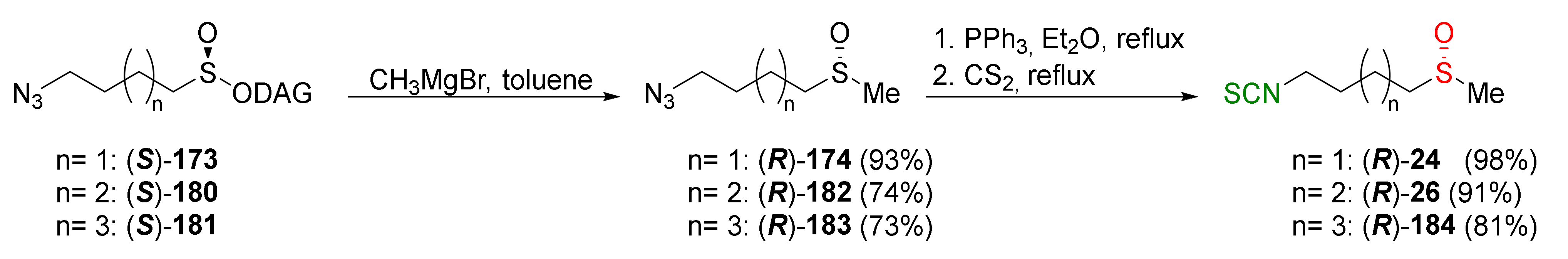{kind=link}
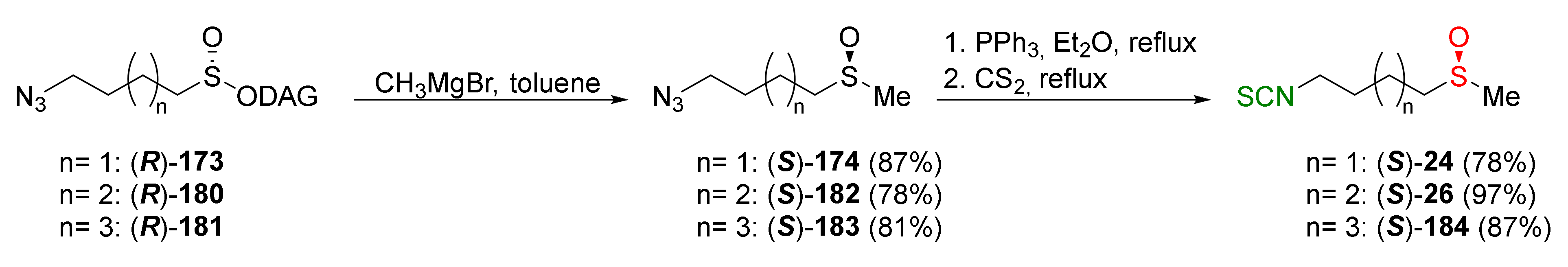{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
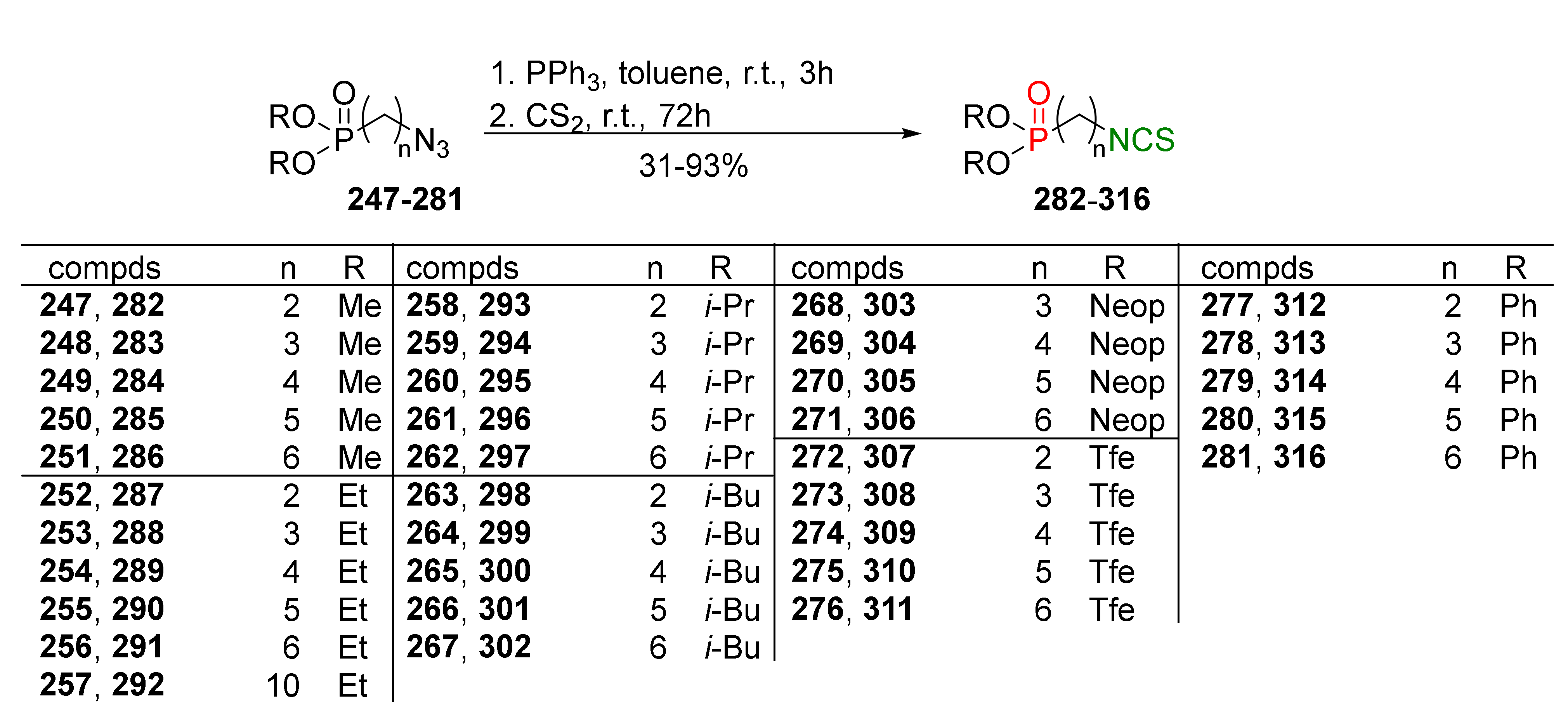{kind=link}
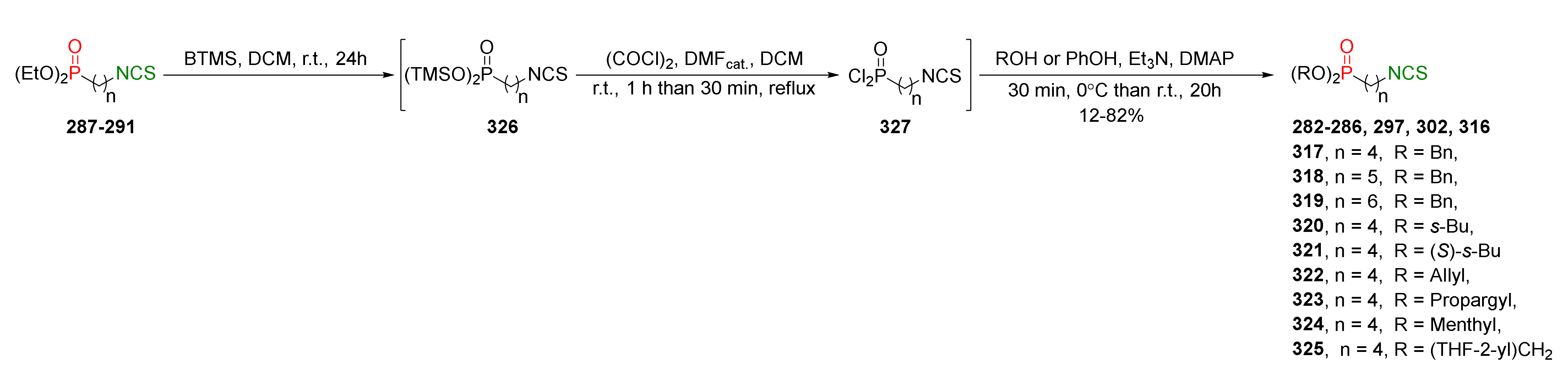{kind=link}
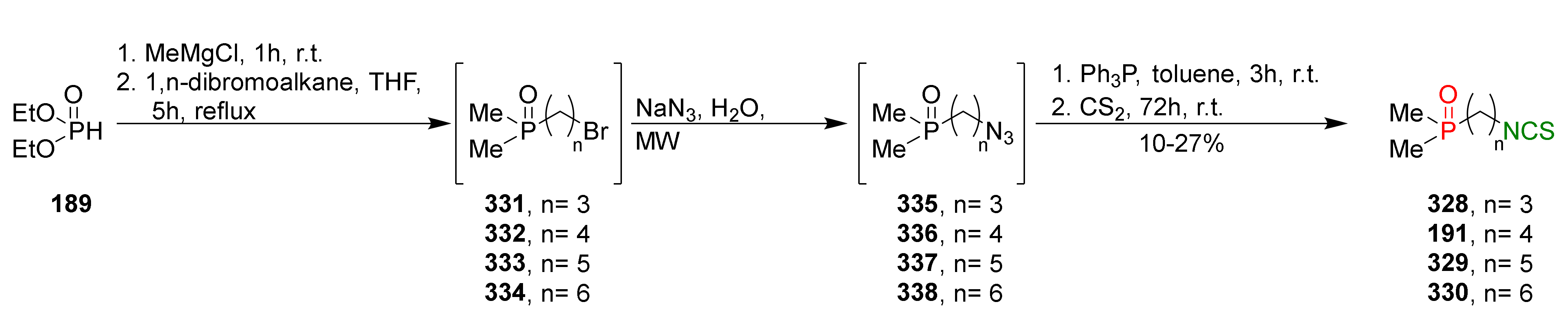{kind=link}
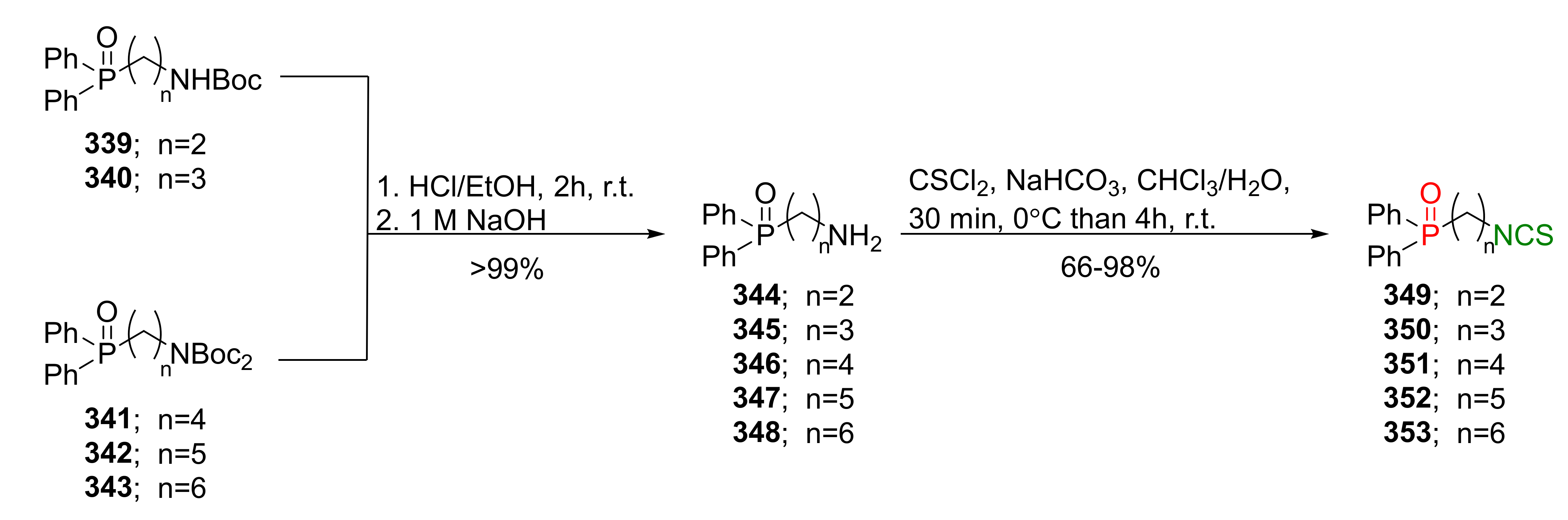{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
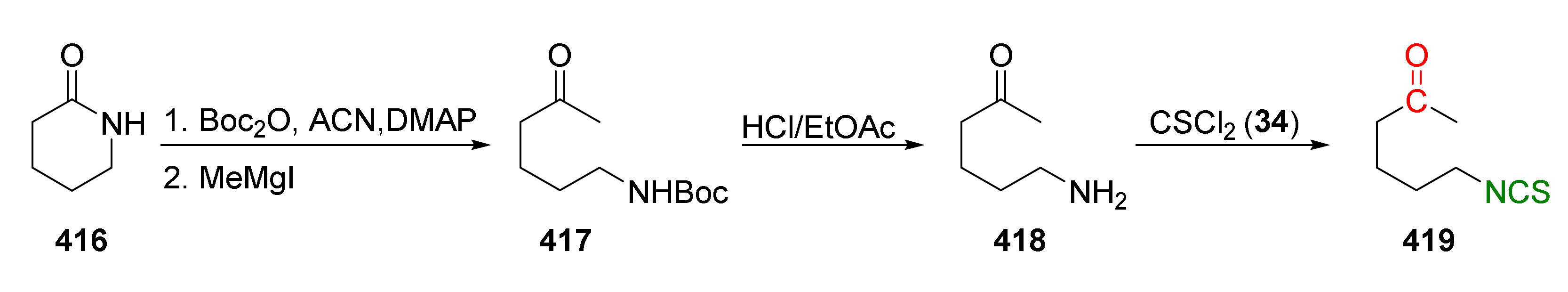{kind=link}
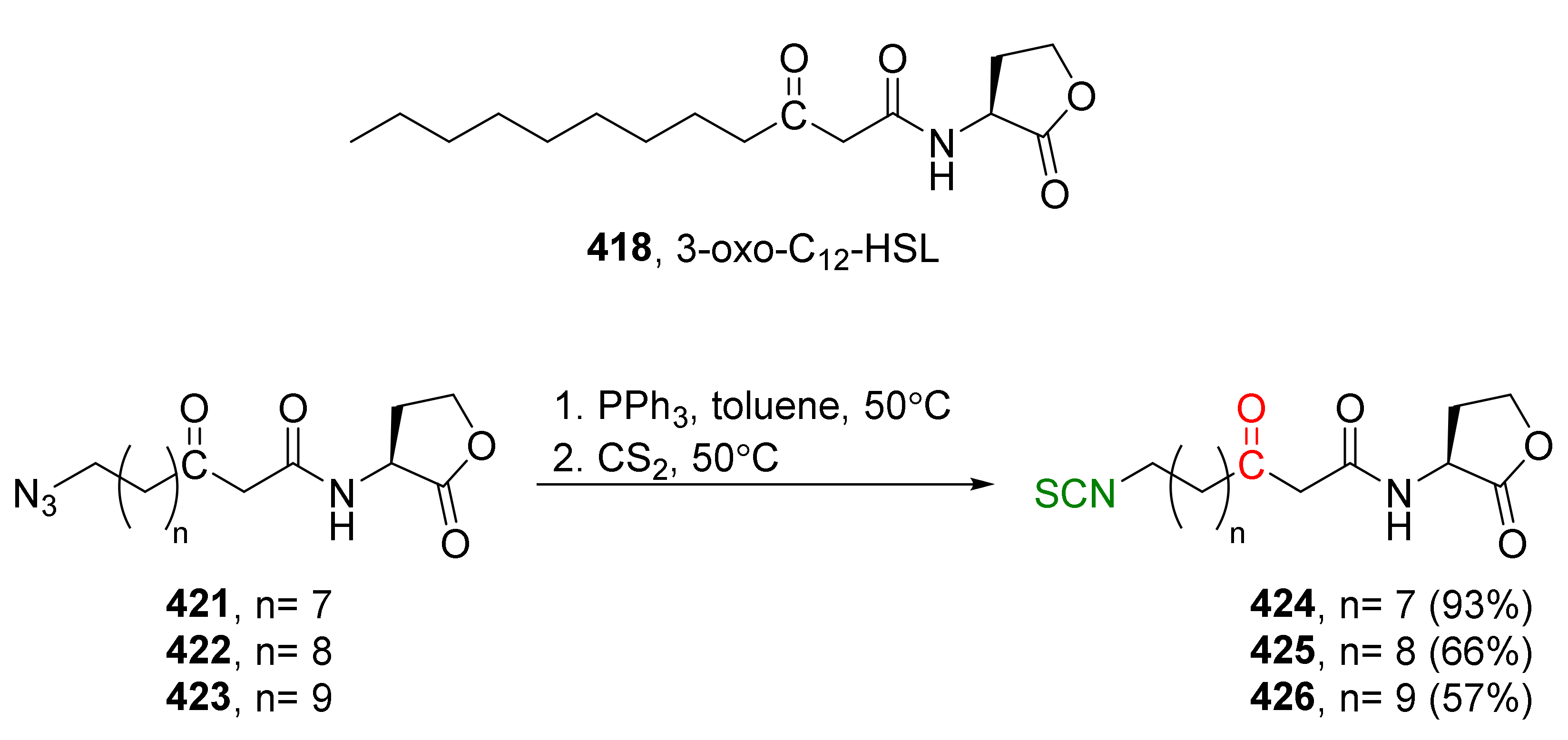{kind=link}
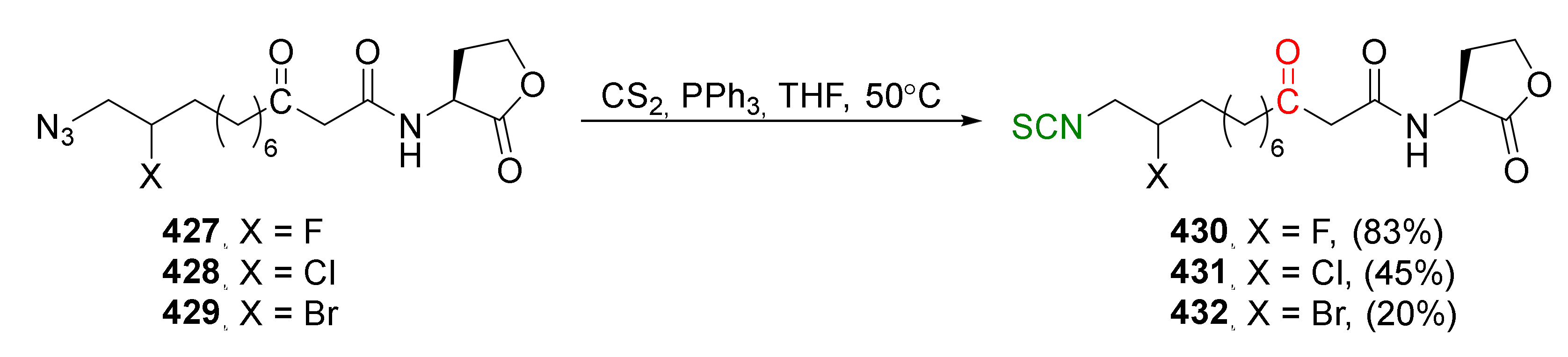{kind=link}
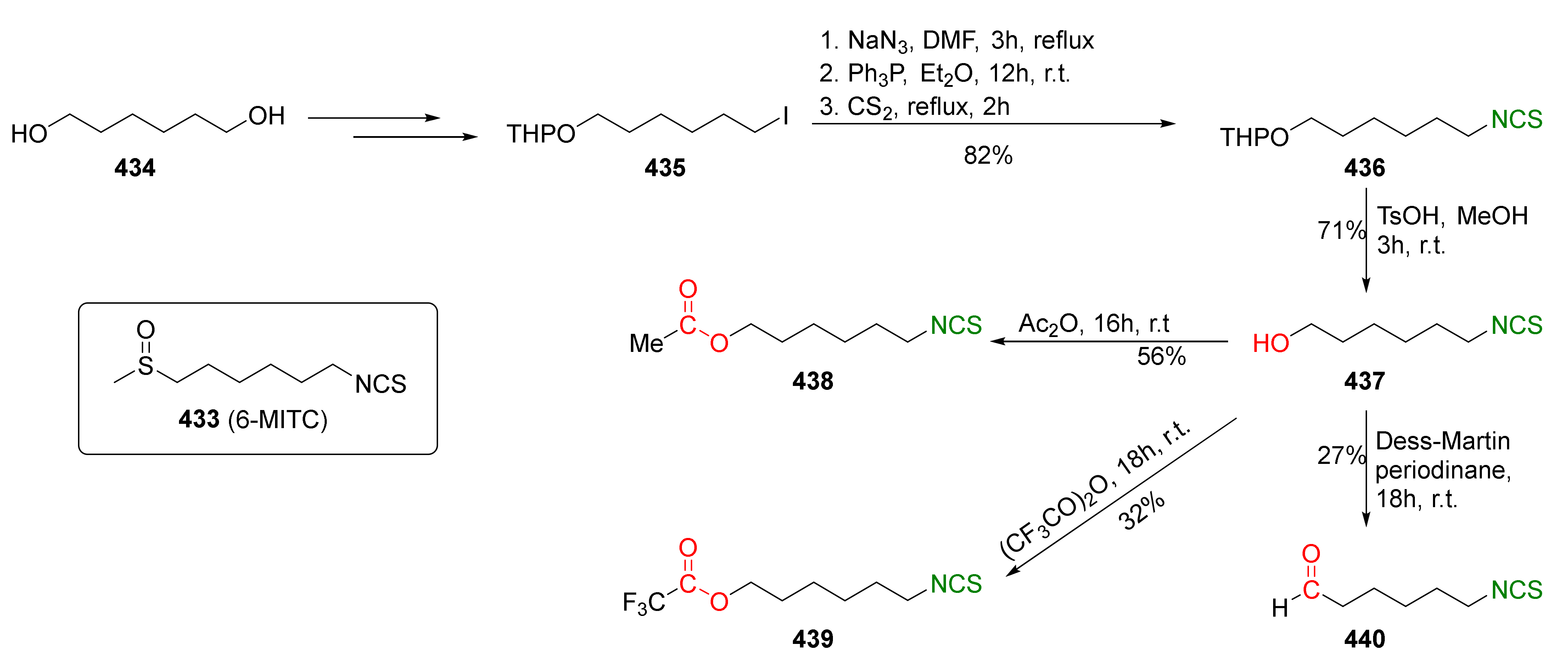{kind=link}
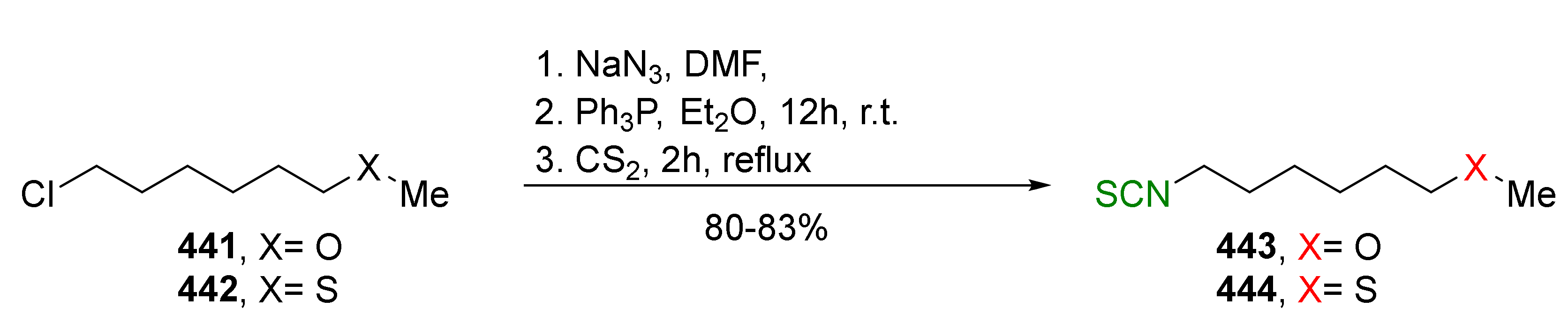{kind=link}
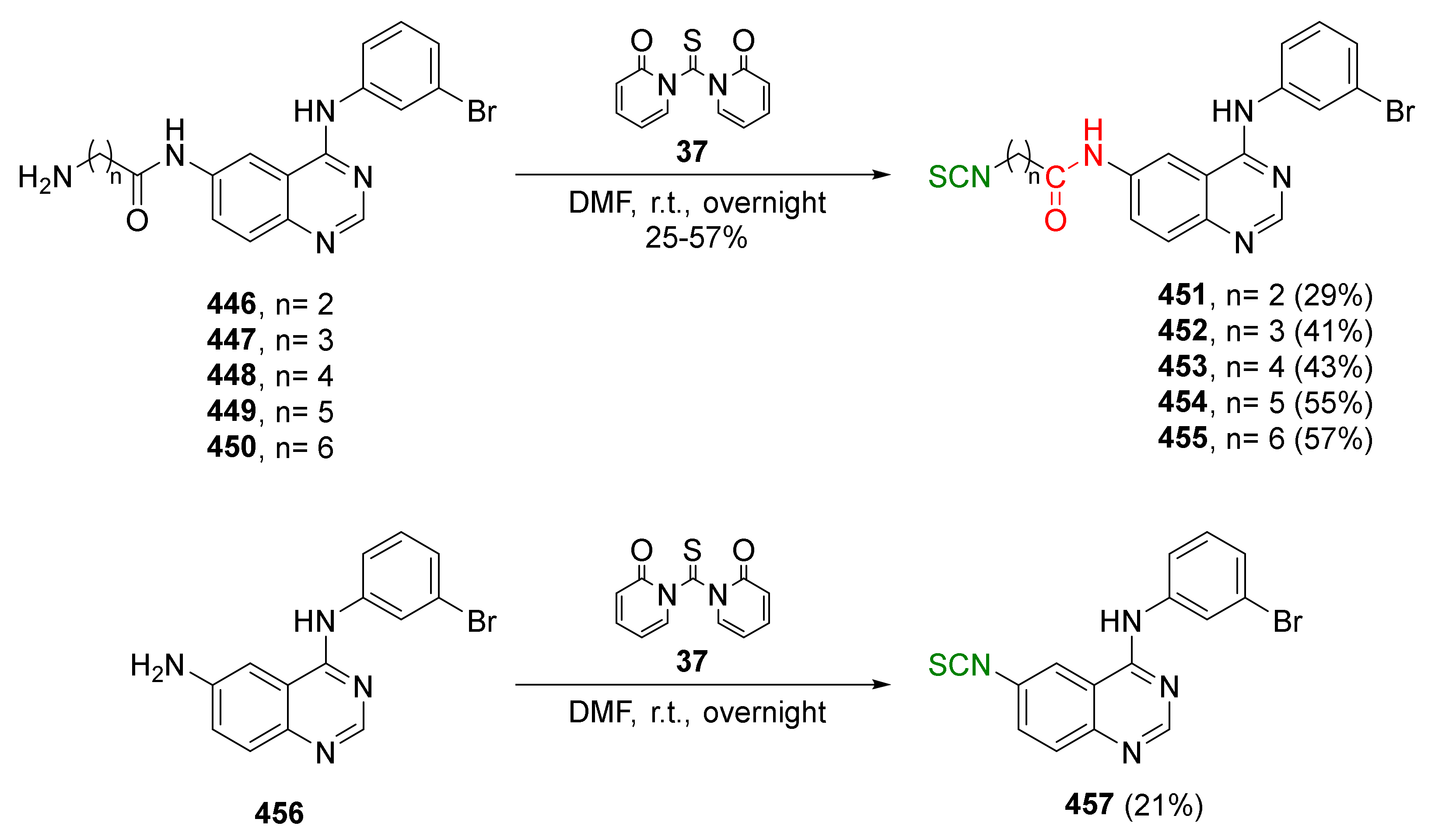{kind=link}
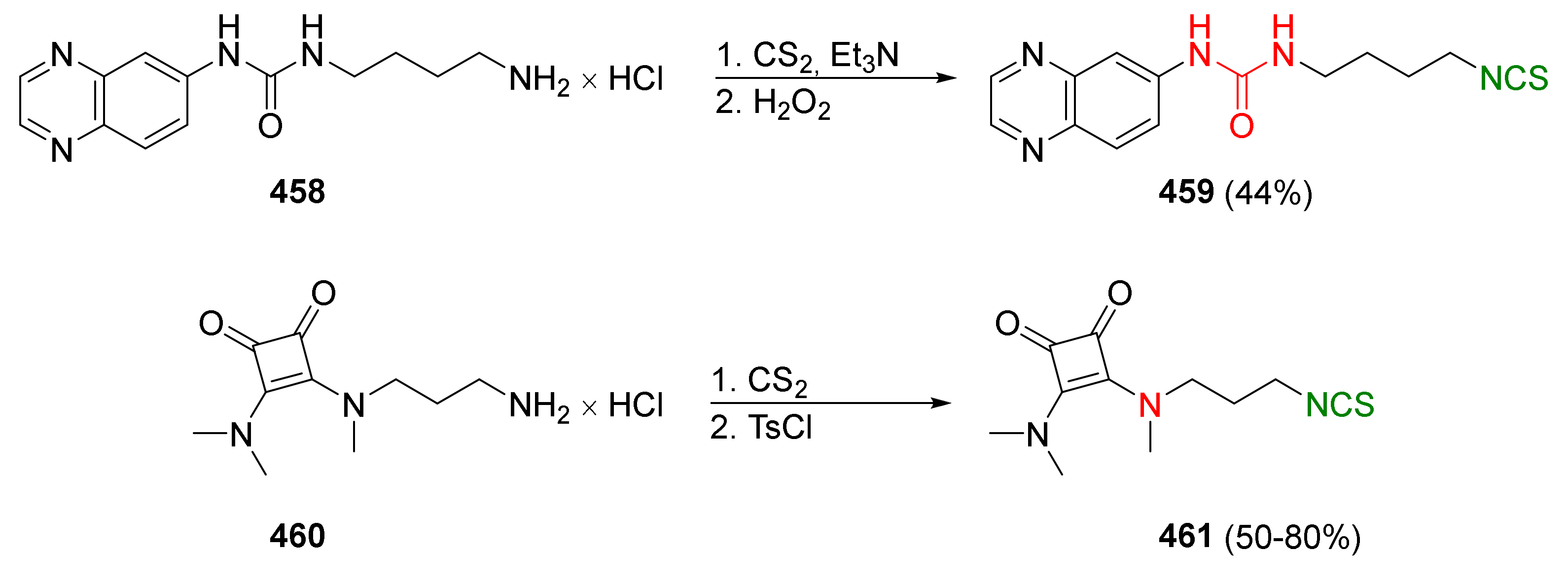{kind=link}
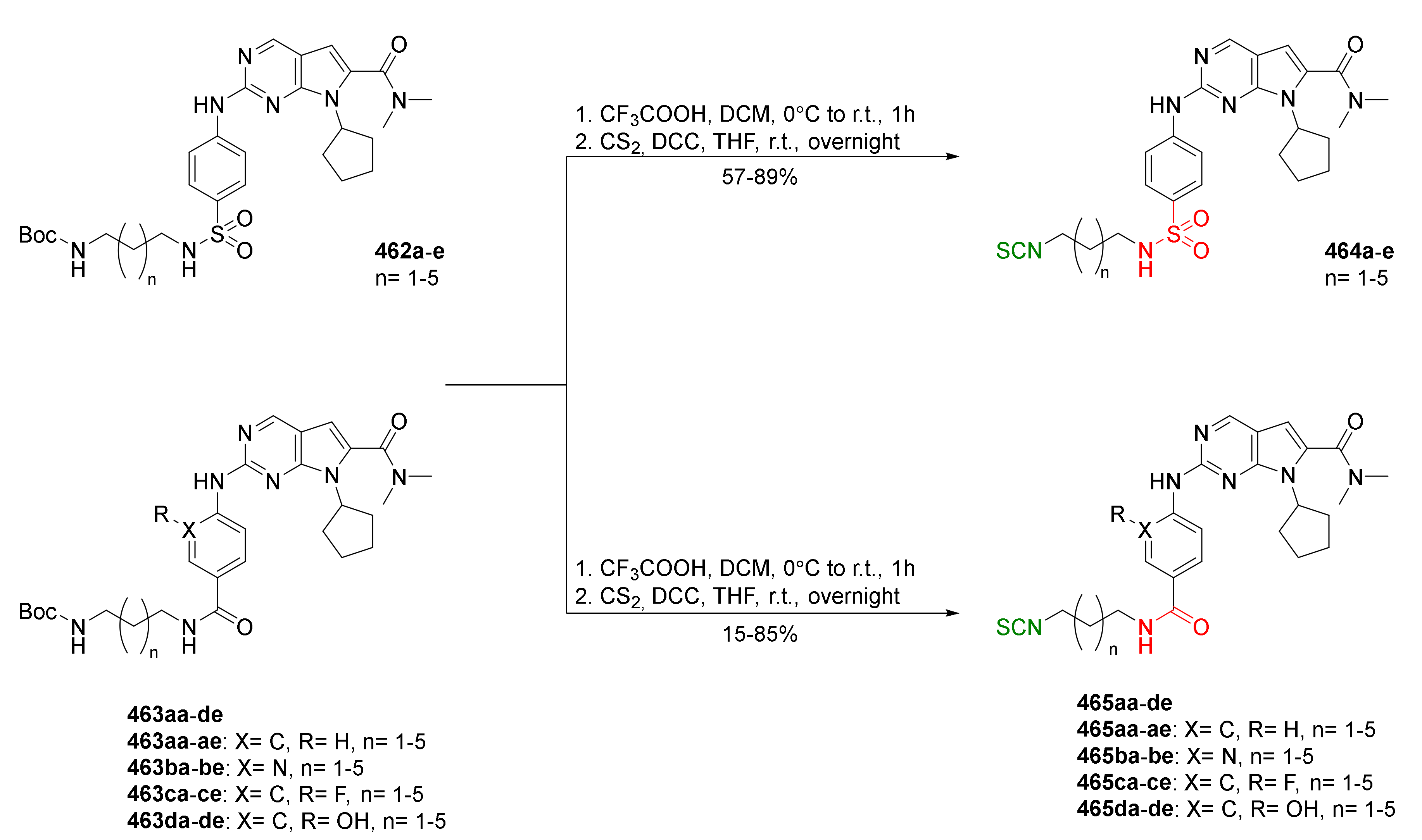{kind=link}
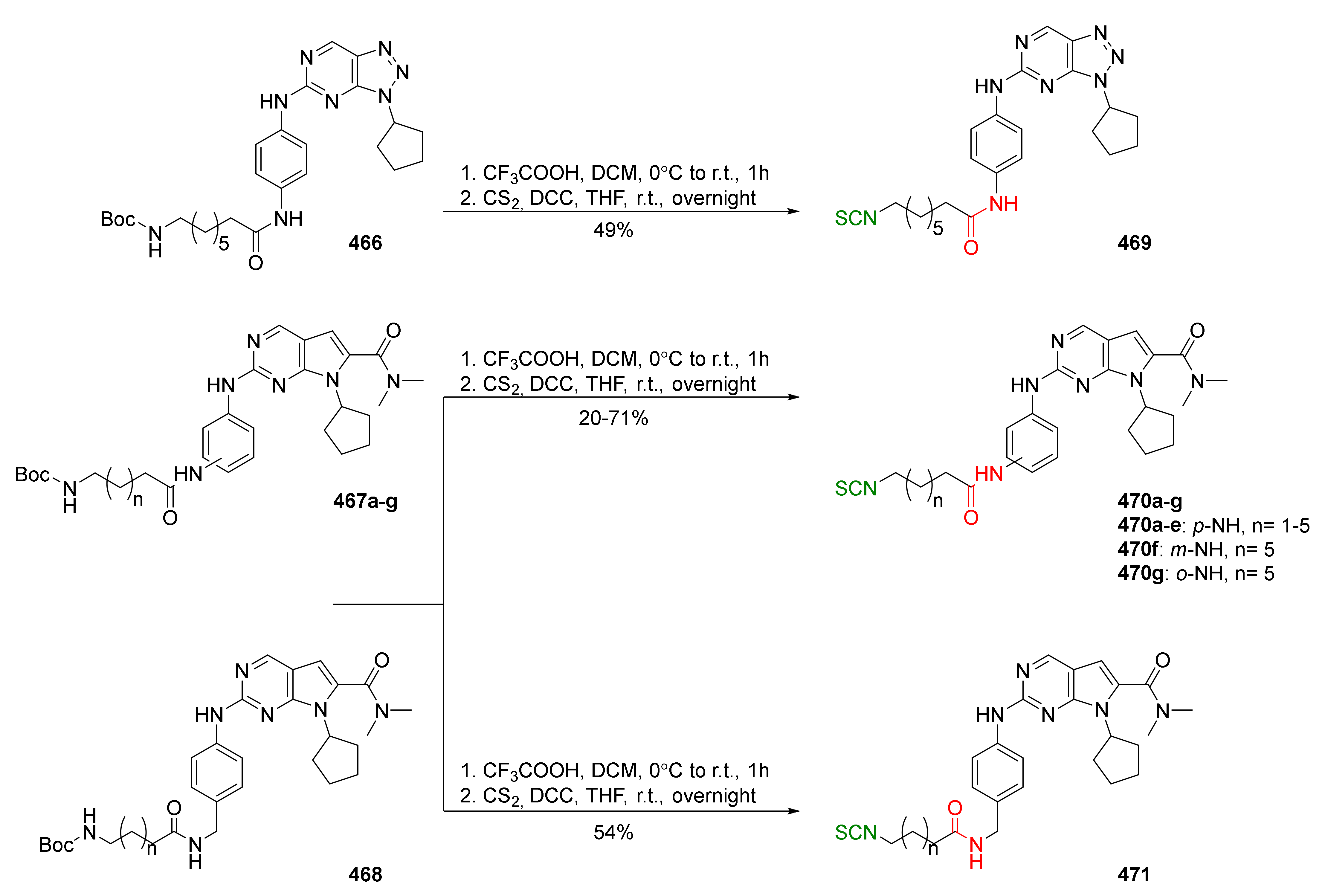{kind=link}
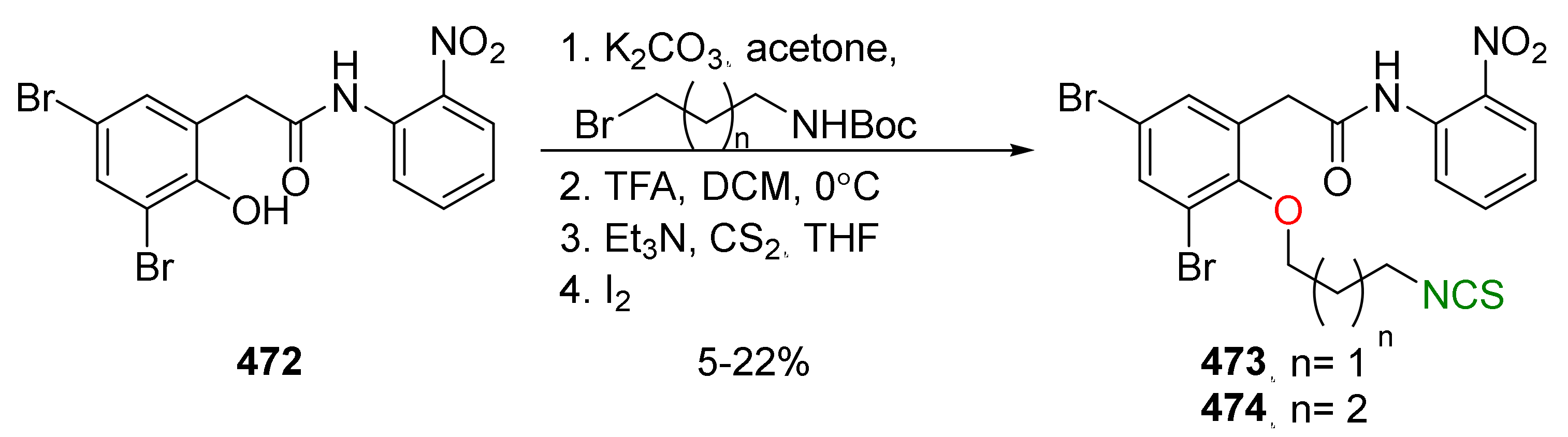{kind=link}
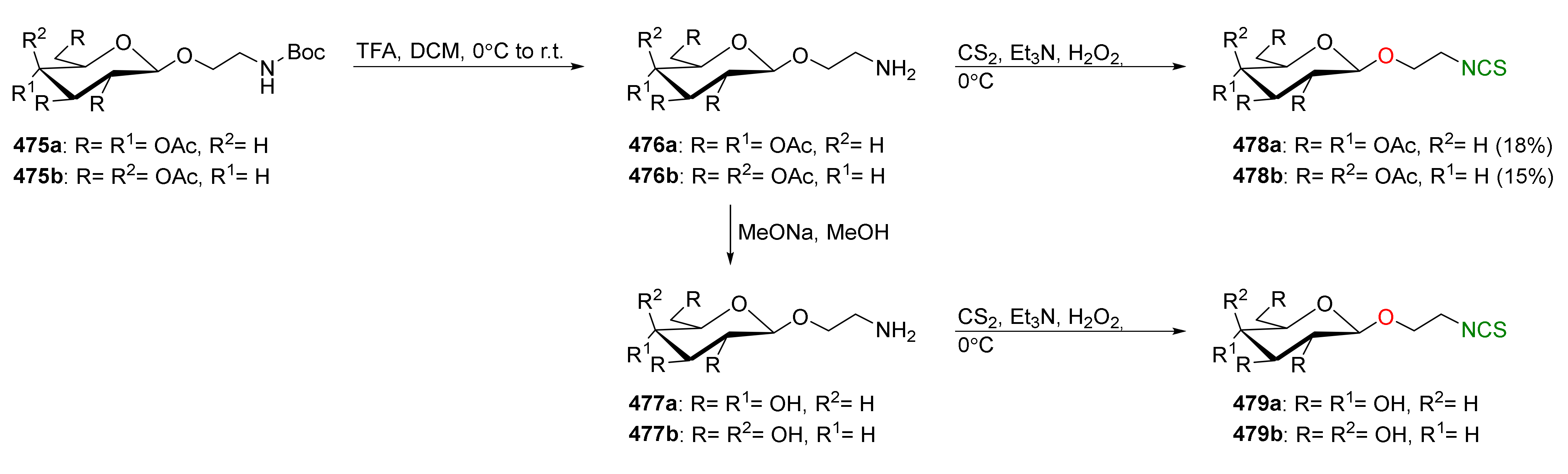{kind=link}
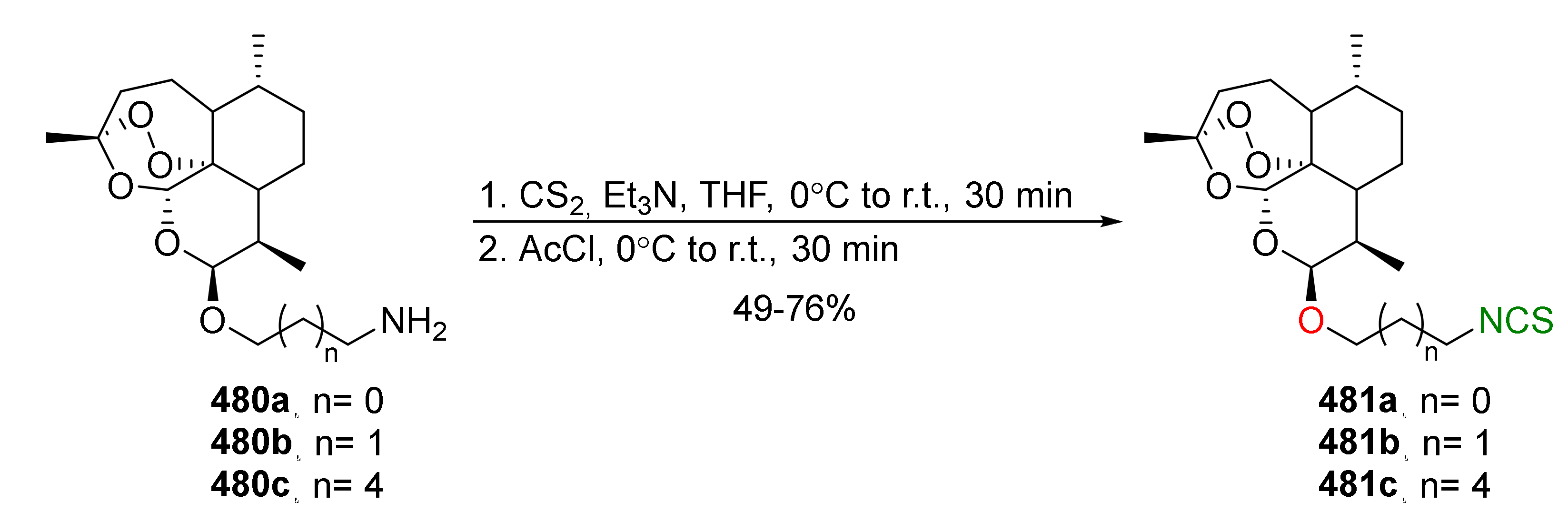{kind=link}
{kind=link}
{kind=link}
| Compound | Low IC50 (μM) | High IC50 (μM) | IC50 (NmuMG) (μM) |
|---|---|---|---|
| SFN (24) | 2.8 ± 0.1 (Hep3B) | 16.5 ± 1.4 (NCI/ADR RES) | 3.2 ± 0.2 |
| Erucin (28) | 8.9 ± 0.4 (SF-268) | 45.3 ± 4.9 (HCI-H460) | 23.5 |
| Erysolin (31) | 2.3 ± 0.8 (MCF-7) | 11.1 ± 0.6 (NCI/ADR RES) | 5.3 ± 0.4 |
| Compound | Malme-3M IC50 (μM) | Malme-3 IC50 (μM) |
|---|---|---|
| (R)-SFN | 25 | 38 |
| (S)-SFN | 30 | 26 |
| (R)-75 | 27 | 37 |
| (S)-75 | 25 | 35 |
| IC50 (μM) ± SD | ||||
|---|---|---|---|---|
| Compound | MALME-3M | HT-29 | MCF-7 | MDA-MB-231 |
| 80d | 2.7 ± 0.7 | 1.2 ± 0.1 | 0.9 ± 0.1 | 0.5 ± 0.1 |
| 80e | 4.3 ± 0.7 | 1.4 ± 0.2 | 0.7 ± 0.1 | 1.2 ± 0.1 |
| SFN | 33.7 ± 0.6 | 11.4 ±0.1 | 11.9 ± 2.0 | 11.3 ± 0.7 |
| Compound | MCF-7 IC50 (μM) | SUM-159 IC50 (μM) | KG-1a IC50 (μM) |
|---|---|---|---|
| SFN | 24.11 ± 6.62 | 7.69 ± 0.92 | 8.24 ± 2.81 |
| 83d | 2.66 ± 0.25 | 1.46 ± 0.19 | 1.52 ± 0.38 |
| 84d | 4.11 ± 0.9 | 1.54 ± 0.29 | 0.51 ± 0.14 |
| 89d | 1.66 ± 0.23 | 2.08 ± 0.24 | 0.88 ± 0.28 |
| Compound | HepG2 IC50 (μM) | A549 IC50 (μM) | MCF-7 IC50 (μM) | HCT-116 IC50 (μM) | SH-SY5Y IC50 (μM) |
|---|---|---|---|---|---|
| SFN (24) | 14.05 | 21.99 | 17.66 | 11.59 | 13.72 |
| 2.05 (151) | 5.64 (161) | 3.3 (155) | 2.06 (152) | 2.79 (157) | |
| 135 | 12.56 | 51.34 | 38.28 | 41.45 | 20.71 |
 | 8.49 | 8.89 | 7.55 | 6.28 | 4.78 |
| Compound | A549 IC50 (μM) | MRC-5 IC50 (μM) |
|---|---|---|
| (S)-SFN ((S)-24) | 19.60 | 46.58 |
| (S)-184 | 7.54 | 17.58 |
| Compound | CD (μM) |
|---|---|
| SFN (24) | 0.2 |
| 191 | 0.4 |
| IC50 (μM) ± SD | |||
|---|---|---|---|
| LoVo | LoVo/DX | A549 | MCF-7 |
| 7 ± 1 | 8 ± 1 | 31 ± 2 | 20 ± 1 |
| Compound | LoVo IC50 (μM) | LoVo/DX IC50 (μM) | Compound | LoVo IC50 (μM) | LoVo/DX IC50 (μM) |
|---|---|---|---|---|---|
| 293 | 2.7 ± 0.4 | 3.6 ± 0.9 | 298 | 1.9 ± 0.4 | 2.6 ± 0.4 |
| 294 | 2.6 ± 0.2 | 3.3 ± 0.9 | 299 | 2.4 ± 0.3 | 5.6 ± 3.2 |
| 295 | 2.5 ± 0.6 | 5.0 ± 3.7 | 300 | 3.3 ± 0.2 | 10.4 ± 1.4 |
| 296 | 2.6 ± 0.1 | 7.9 ± 1.6 | 301 | 2.4 ± 0.5 | 5.1 ± 4.4 |
| 297 | 2.7 ± 0.2 | 9.4 ± 1.6 | 302 | 2.7 ± 0.4 | 9.2 ± 1.4 |
| SFN | 22.9 ± 2.0 | 18.1 ± 3.0 |
| Compound | CD (μM) |
|---|---|
| SFN (24) | 0.2 |
| 419 | 0.2 |
| Compounds | Inhibition (IC50, μM) | |
|---|---|---|
| NO Production | Growth | |
| 433 (6-MITC) | 5.7 ± 0.5 | 8.0 ± 0.6 |
| 437 | 6.0 ± 1.2 | 4.4 ± 0.3 |
| 438 | 6.6 ± 1.2 | 4.1 ± 0.2 |
| 439 | 9.1 ± 1.0 | 5.6 ± 0.4 |
| 443 | 11.5 ± 5.9 | 8.1 ± 0.6 |
 | > 200 | > 200 |
| Reaction | Reference |
|---|---|
| Synthesis of SFN and its sulfur analogues | |
 | [239,240,241,242,243,244,245,246,247] |
 | [249] |
 | [163,250,252] |
| Synthesis of phosphorous analogues of SFN | |
 | [188,257] |
 | [200,203,253] |
 | [200,254,257] |
| Synthesis of carbonyl and amide analogues of SFN | |
 | [188,263] |
 | [264,265] |
 | [260,261,262] |
| Synthesis of ether-linked analogues of SFN | |
 | [266,267,268] |
| Synthesis of diisothiocyanates | |
 | [269] |
 | [55] |
| Compound | Anticancer Activity | Reference | Compound | Anticancer Activity | Reference |
|---|---|---|---|---|---|
 | Malme-3M↑ Malme-3↓ | [244] |  | Malme-3M↑ HT-29↑ MCF-7↑ MDA-MB-231↑ | [245] |
 | MCF-7↑ SUM-159↑ KG-1a↑ | [246] |  | HepG2↑ A549↑ MCF-7↑ HCT-116↑ SH-SY5Y↑ | [249] |
 | CD↔ | [188] |  | LoVo↑ LoVo/DX↑ | [254] |
 | LoVo↑ LoVo/DX↑ | [257] |  | LoVo↑ LoVo/DX↑ | [258] |
 | CD↔ | [188] |  | A431↔ | [263] |
 | A549↑ H1299↑ MCF-7↑ MDA-MB-231↑ HepG2↑ Hela↑ | [264] |  | LoVo↑ LoVo/DX↑ | [269] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janczewski, Ł. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules 2022, 27, 1750. https://doi.org/10.3390/molecules27051750
Janczewski Ł. Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules. 2022; 27(5):1750. https://doi.org/10.3390/molecules27051750
Chicago/Turabian StyleJanczewski, Łukasz. 2022. "Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity" Molecules 27, no. 5: 1750. https://doi.org/10.3390/molecules27051750
APA StyleJanczewski, Ł. (2022). Sulforaphane and Its Bifunctional Analogs: Synthesis and Biological Activity. Molecules, 27(5), 1750. https://doi.org/10.3390/molecules27051750