
Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis

 ,
,
Abstract
:1. Introduction
2. Results
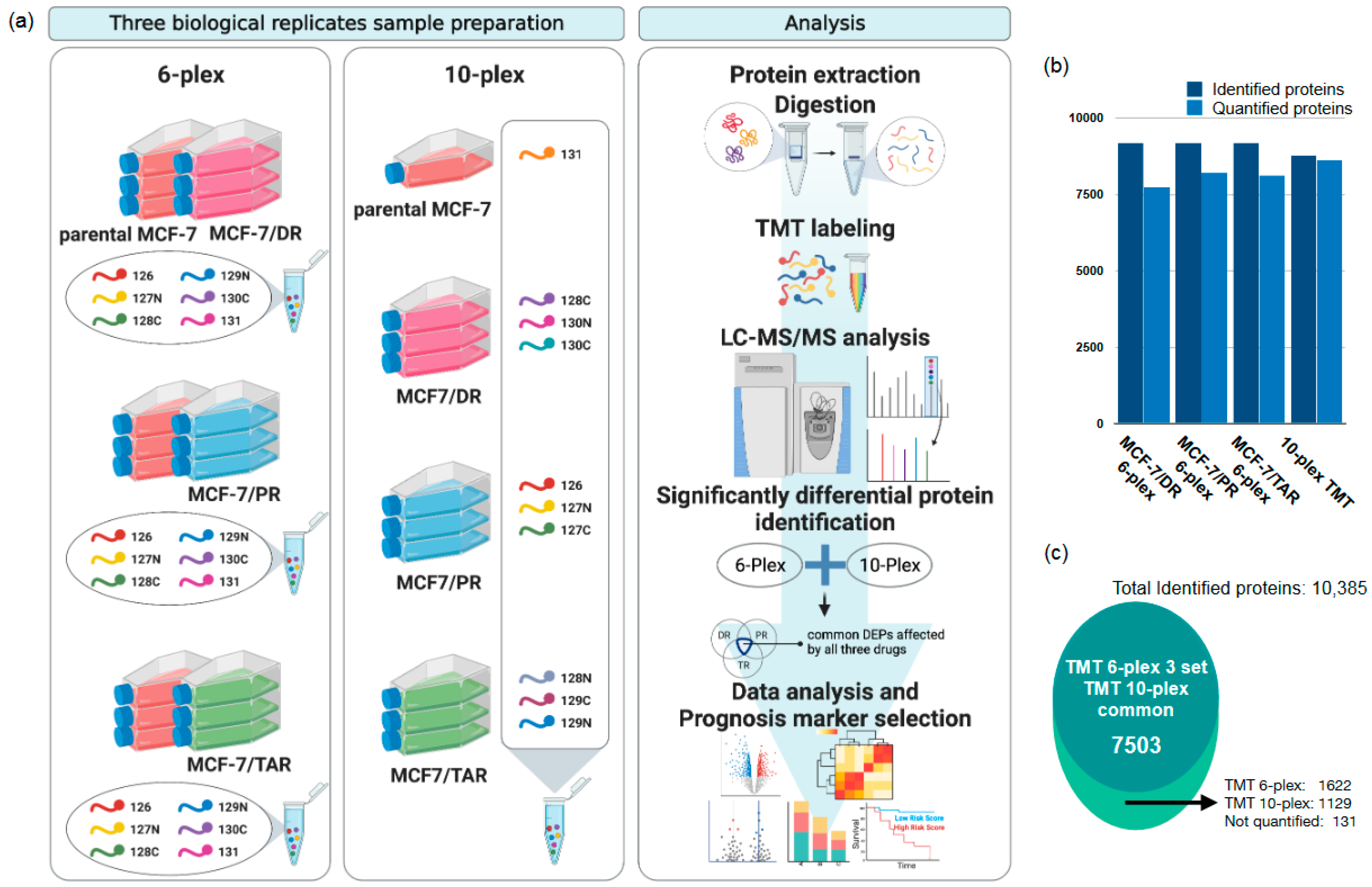
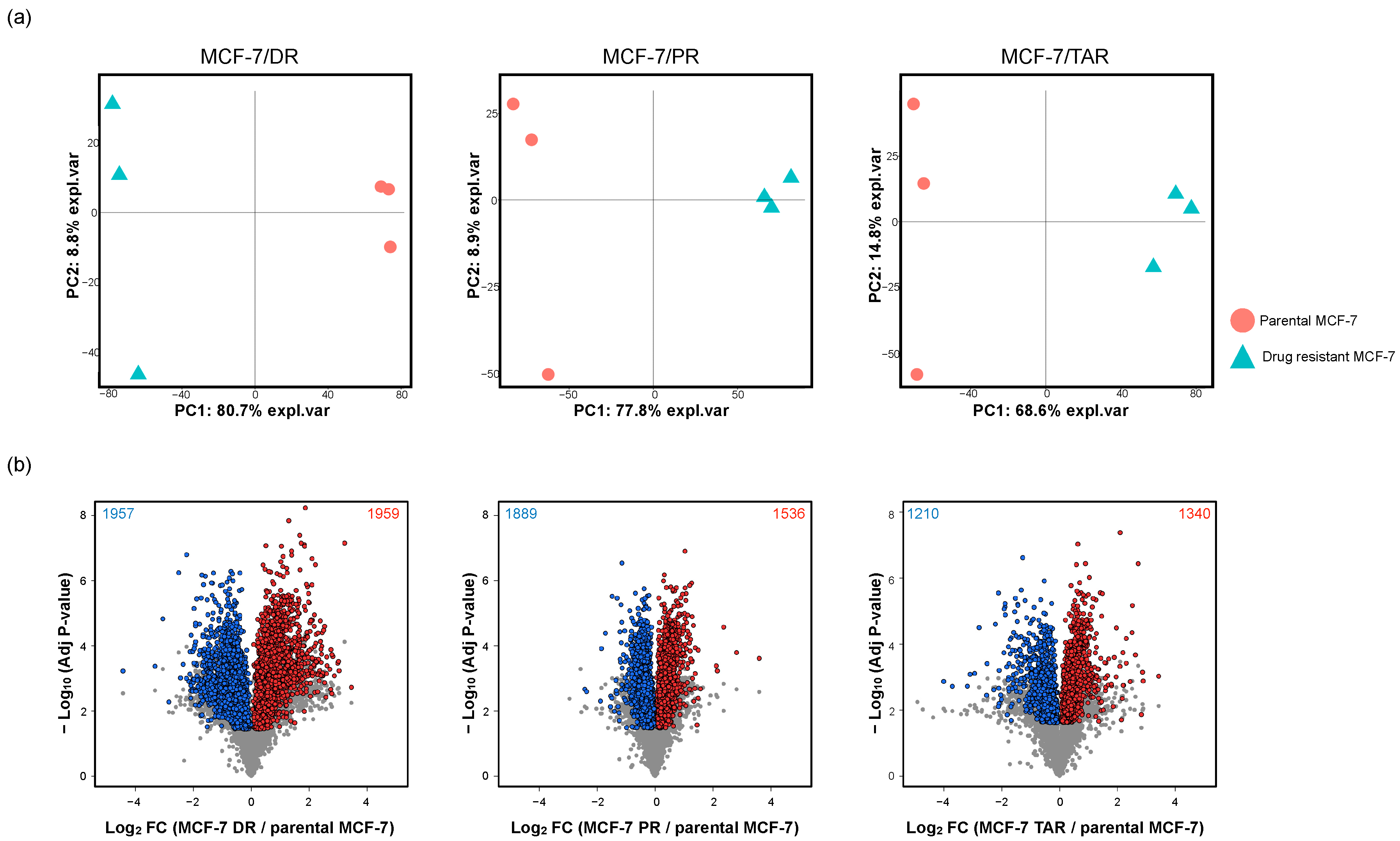
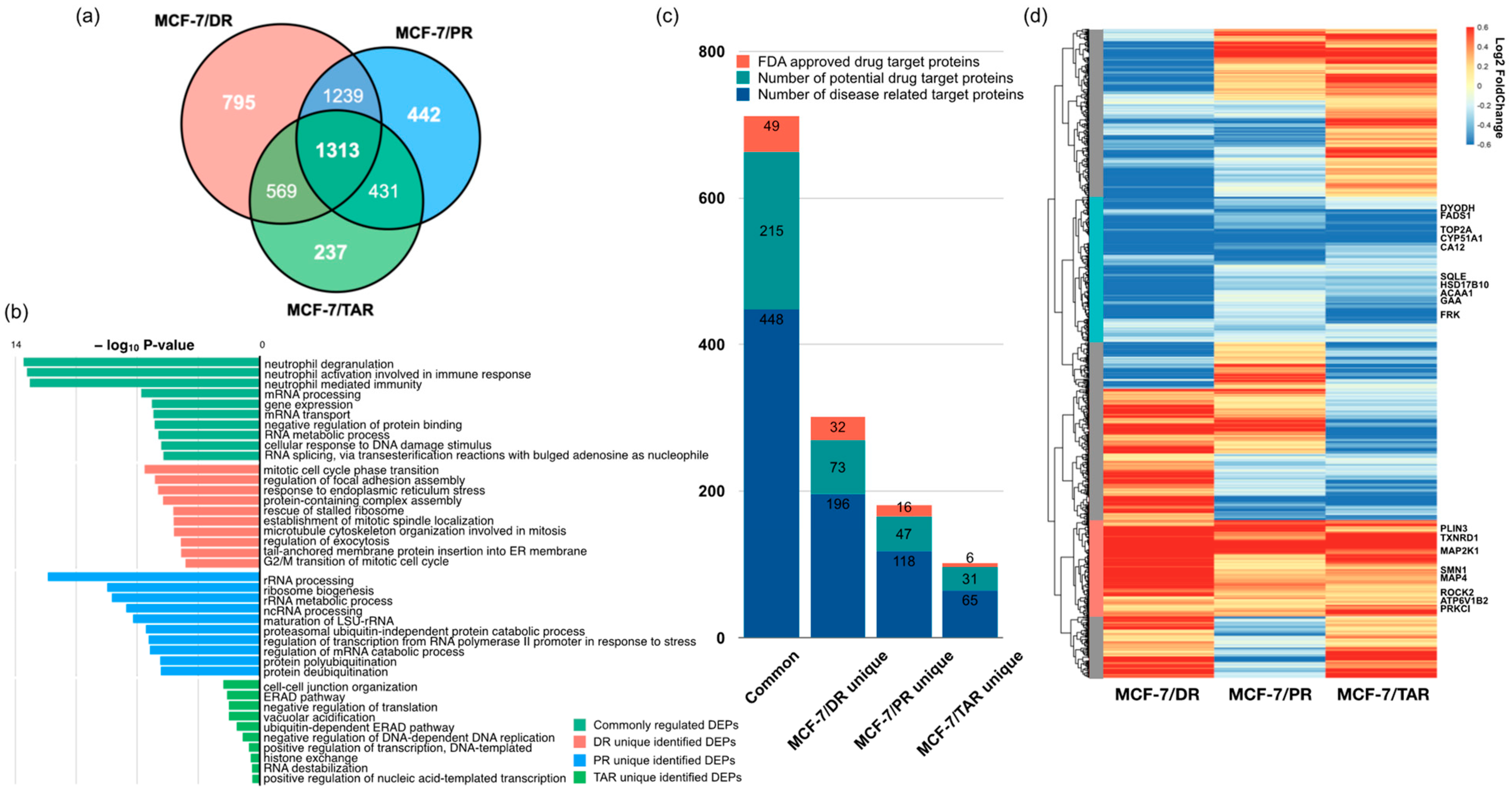
2.1. Comparative Proteomic Analysis of Drug-Resistant Breast Cancer Cell Lines
2.2. Identification of Protein Expression in Individual Drug-Resistant Cells
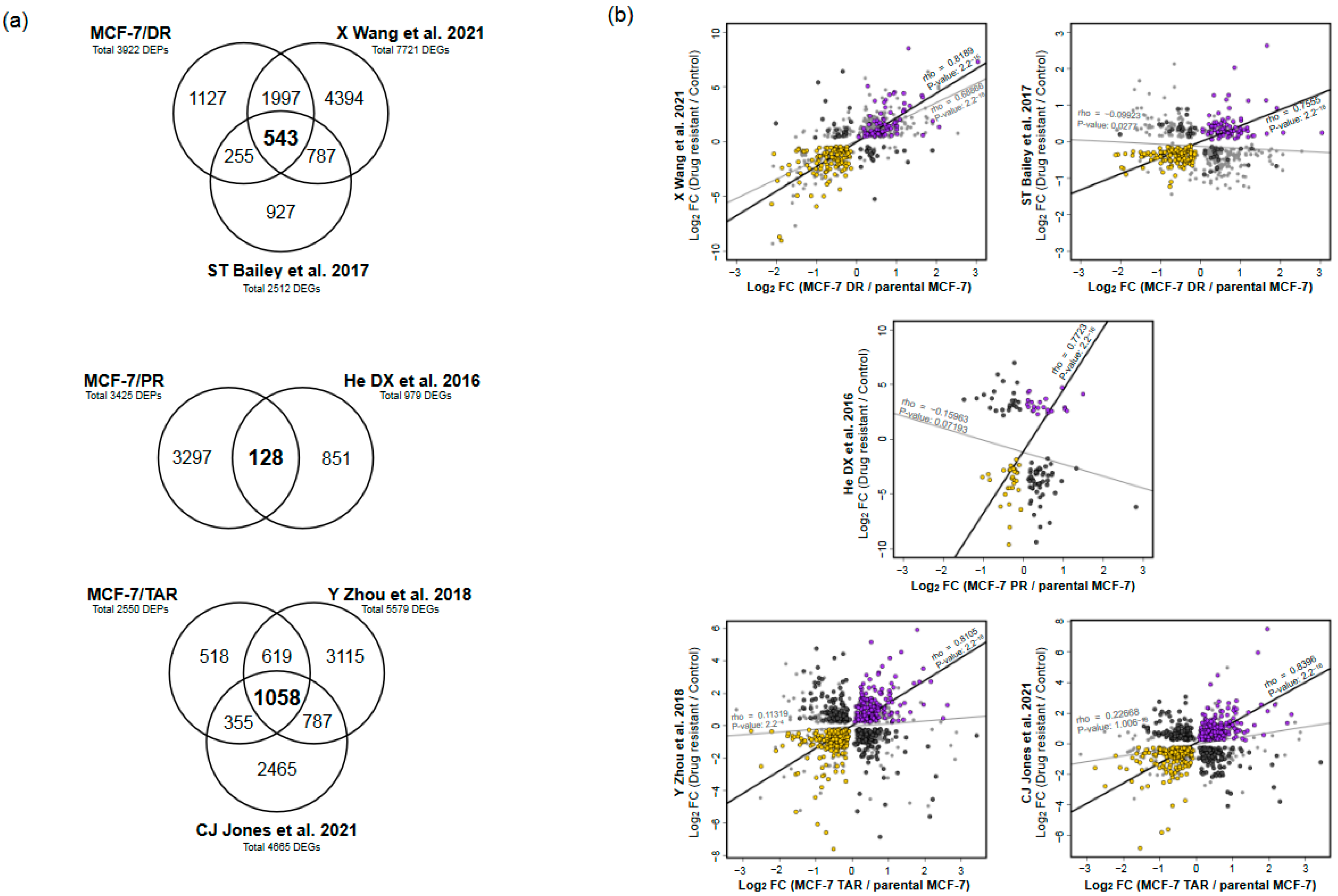
2.3. Integrative Analysis of Proteome and Transcriptome from Drug-Resistant Cells
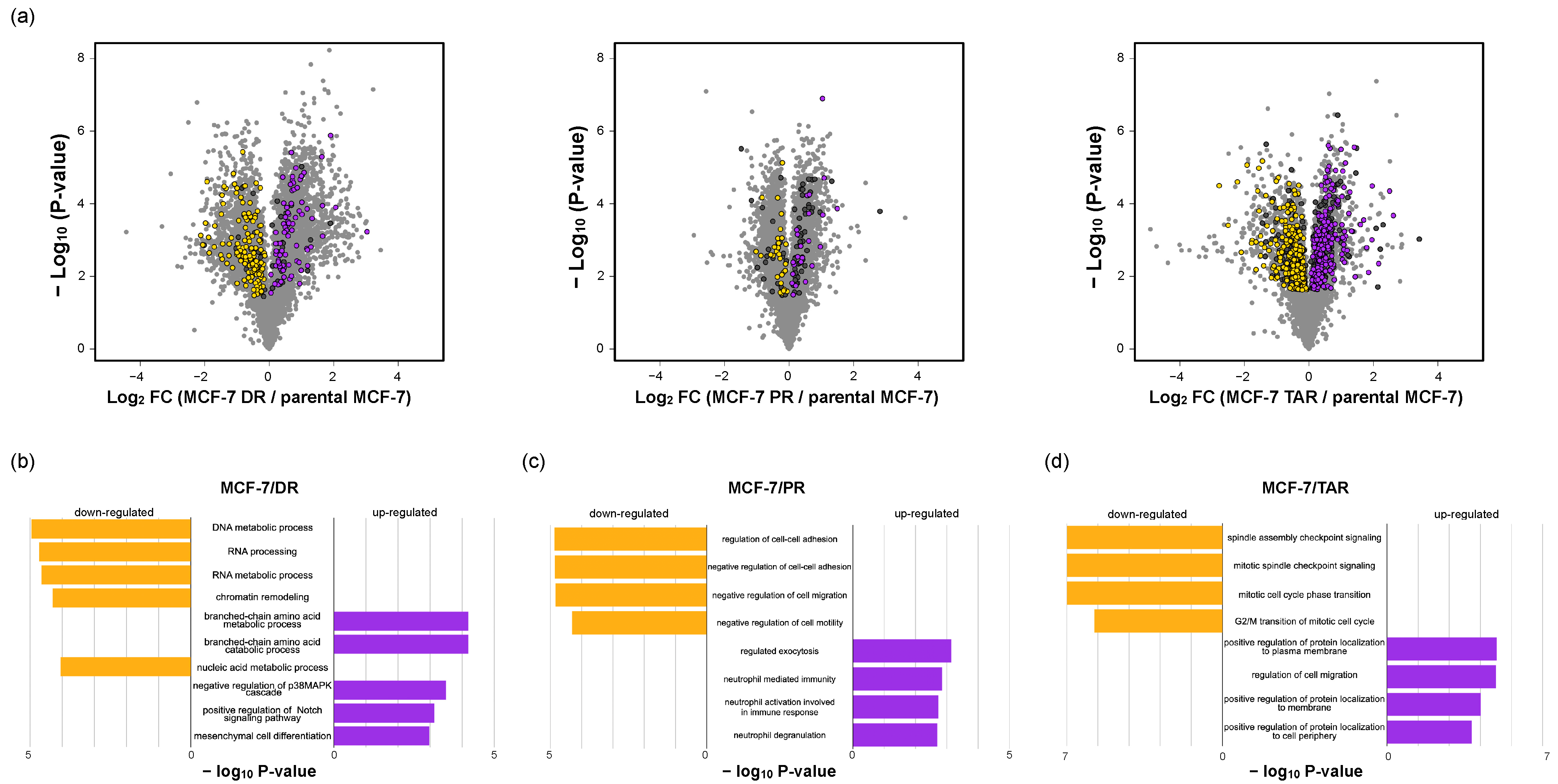
2.4. Functional Analysis of Correlated Expression of Genes between Proteome and Transcriptome in Resistant Cells
2.5. Analysis of Three Types of Drug-Resistant-Cell-Expressed Proteins and Commonly Regulated Proteins
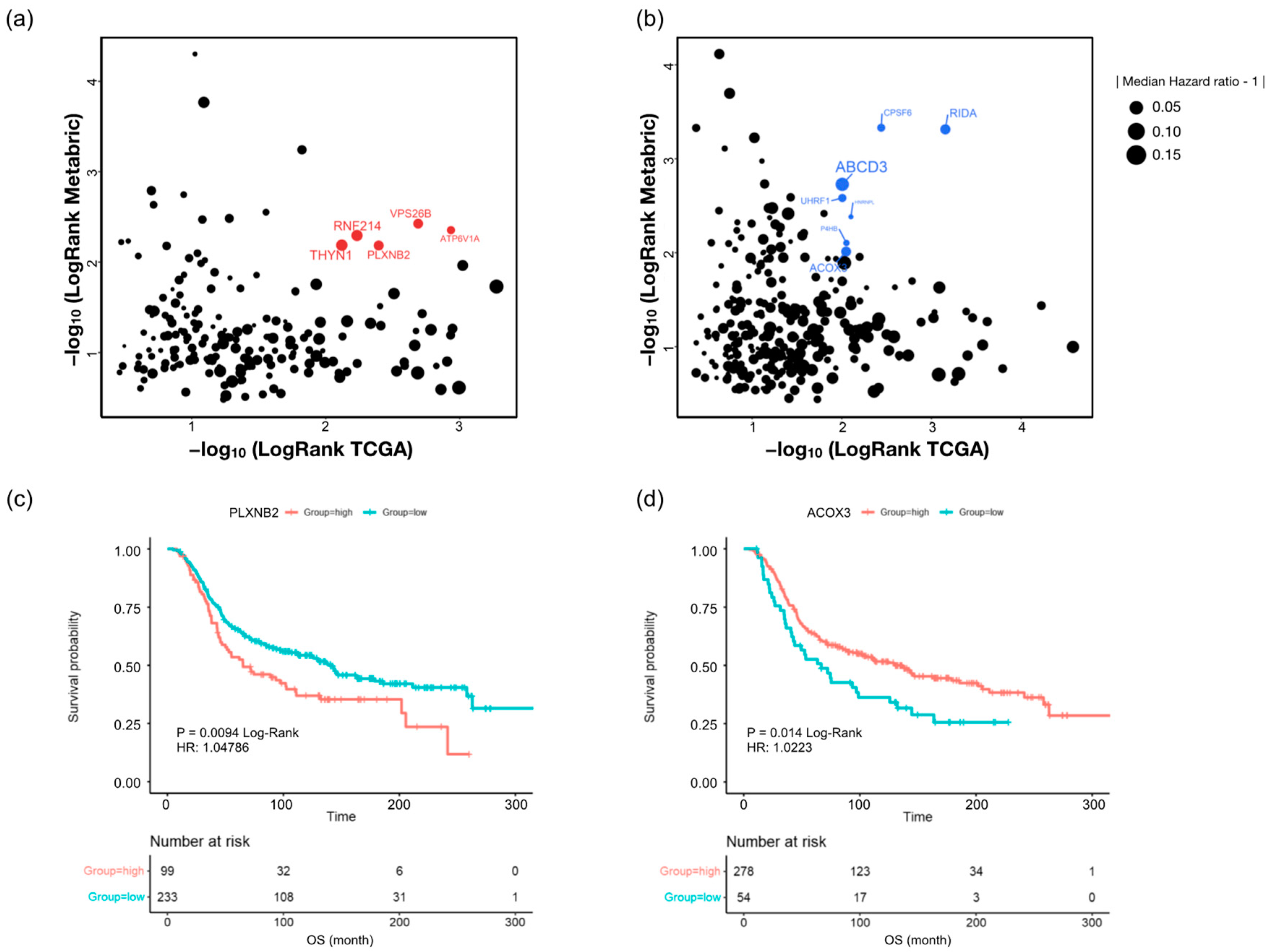
2.6. Discovery of Candidate Anti-Cancer-Drug-Resistant Marker with TCGA and Metabric Survival Data
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Lysis and Protein Digestion
4.3. TMT Labeling
4.4. High-pH Peptide Fractionation
4.5. Mass Spectrometry and Proteomic Data Analysis
4.6. Transcriptome Data Analysis
4.7. Statistical Analysis
4.8. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sager, C.B. A preferred future through informed choice. Dent. Clin. N. Am. 1988, 32, 17–27. [Google Scholar] [PubMed]
- Luond, F.; Tiede, S.; Christofori, G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br. J. Cancer 2021, 125, 164–175. [Google Scholar] [CrossRef]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Wen, S.H.; Su, S.C.; Liou, B.H.; Lin, C.H.; Lee, K.R. Sulbactam-enhanced cytotoxicity of doxorubicin in breast cancer cells. Cancer Cell Int. 2018, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Ghebeh, H.; Al-Sayed, A.; Eiada, R.; Cabangon, L.; Ajarim, D.; Suleman, K.; Tulbah, A.; Al-Tweigeri, T. Weekly Paclitaxel given concurrently with Durvalumab has a favorable safety profile in triple-negative metastatic breast cancer. Sci. Rep. 2021, 11, 19154. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Binkhorst, L.; van Gelder, T.; Mathijssen, R.H. Individualization of tamoxifen treatment for breast carcinoma. Clin. Pharmacol. Ther. 2012, 92, 431–433. [Google Scholar] [CrossRef]
- Zaal, E.A.; Berkers, C.R. The Influence of Metabolism on Drug Response in Cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tian, Y.; Leong, W.S.; Song, H.; Yang, W.; Wang, M.; Wang, X.; Kong, J.; Shan, B.; Song, Z. Efficient and tumor-specific knockdown of MTDH gene attenuates paclitaxel resistance of breast cancer cells both in vivo and in vitro. Breast Cancer Res. 2018, 20, 113. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, N.P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Cox, J.; Weinman, S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepat. Oncol. 2016, 3, 57–59. [Google Scholar] [CrossRef]
- Wei, Y.; Guo, Y.; Zhou, J.; Dai, K.; Xu, Q.; Jin, X. Nicotinamide Overcomes Doxorubicin Resistance of Breast Cancer Cells through Deregulating SIRT1/Akt Pathway. Anticancer. Agents Med. Chem. 2019, 19, 687–696. [Google Scholar] [CrossRef]
- Cervellera, M.; Raschella, G.; Santilli, G.; Tanno, B.; Ventura, A.; Mancini, C.; Sevignani, C.; Calabretta, B.; Sala, A. Direct transactivation of the anti-apoptotic gene apolipoprotein J (clusterin) by B-MYB. J. Biol. Chem. 2000, 275, 21055–21060. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.K.; Hermawan, A.; Ljepoja, B.; Frohlich, T.; Arnold, G.J.; Wagner, E.; Roidl, A. A proteomic analysis of chemoresistance development via sequential treatment with doxorubicin reveals novel players in MCF7 breast cancer cells. Int. J. Mol. Med. 2018, 42, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wei, S.; Ma, Y.; Lu, J.; Niu, G.; Xue, Y.; Chen, X.; Yang, F. Quantitative proteomics analysis identifies mitochondria as therapeutic targets of multidrug-resistance in ovarian cancer. Theranostics 2014, 4, 1164–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci. Rep. 2015, 5, 9301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yan, J.; Shen, B.; Wei, G. Integrated Chromatin Accessibility and Transcriptome Landscapes of Doxorubicin-Resistant Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 708066. [Google Scholar] [CrossRef]
- He, D.X.; Gu, F.; Gao, F.; Hao, J.J.; Gong, D.; Gu, X.T.; Mao, A.Q.; Jin, J.; Fu, L.; Ma, X. Genome-wide profiles of methylation, microRNAs, and gene expression in chemoresistant breast cancer. Sci. Rep. 2016, 6, 24706. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gerrard, D.L.; Wang, J.; Li, T.; Yang, Y.; Fritz, A.J.; Rajendran, M.; Fu, X.; Stein, G.; Schiff, R.; et al. Temporal dynamic reorganization of 3D chromatin architecture in hormone-induced breast cancer and endocrine resistance. Nat. Commun. 2019, 10, 1522. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.J.; Subramaniam, M.; Emch, M.J.; Bruinsma, E.S.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. Development and Characterization of Novel Endoxifen-Resistant Breast Cancer Cell Lines Highlight Numerous Differences from Tamoxifen-Resistant Models. Mol. Cancer Res. 2021, 19, 1026–1039. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Mayo, A.; Keren, L.; Alon, U. Central dogma rates and the trade-off between precision and economy in gene expression. Nat. Commun. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 2000, 67, 704–713, reprinted in Biotechnol. Bioeng. 1969, XI, 1101–1110. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; McAndrew, N.P.; Bardia, A.; Press, M.F.; Pegram, M.; Crown, J.P.; Fasching, P.A.; Ejlertsen, B.; Yang, E.H.; Glaspy, J.A.; et al. A careful reassessment of anthracycline use in curable breast cancer. NPJ Breast Cancer 2021, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.H.; Carter, S.K. Adriamycin: A new anticancer drug with significant clinical activity. Ann. Intern. Med. 1974, 80, 249–259. [Google Scholar] [CrossRef]
- Marinello, P.C.; Panis, C.; Silva, T.N.X.; Binato, R.; Abdelhay, E.; Rodrigues, J.A.; Mencalha, A.L.; Lopes, N.M.D.; Luiz, R.C.; Cecchini, R.; et al. Metformin prevention of doxorubicin resistance in MCF-7 and MDA-MB-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci. Rep. 2019, 9, 5864. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Biswas, D.; Duffley, L.; Pulinilkunnil, T. Role of branched-chain amino acid-catabolizing enzymes in intertissue signaling, metabolic remodeling, and energy homeostasis. FASEB J. 2019, 33, 8711–8731. [Google Scholar] [CrossRef]
- Biswas, D.; Slade, L.; Duffley, L.; Mueller, N.; Dao, K.T.; Mercer, A.; Pakkiriswami, S.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. Inhibiting BCKDK in triple negative breast cancer suppresses protein translation, impairs mitochondrial function, and potentiates doxorubicin cytotoxicity. Cell Death Discov. 2021, 7, 241. [Google Scholar] [CrossRef]
- Tan, W.; Yu, H.G.; Luo, H.S. Inhibition of the p38 MAPK pathway sensitizes human gastric cells to doxorubicin treatment in vitro and in vivo. Mol. Med. Rep. 2014, 10, 3275–3281. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, K.; Jing, J.; Wang, C.; Hu, Y.; Shi, Y.; Li, E.; Geng, Q. Recombinant Dual-target MDM2/MDMX Inhibitor Reverses Doxorubicin Resistance through Activation of the TAB1/TAK1/p38 MAPK Pathway in Wild-type p53 Multidrug-resistant Breast Cancer Cells. J. Cancer 2020, 11, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Brennan, K. Notch Signalling in Breast Development and Cancer. Front. Cell Dev. Biol. 2021, 9, 692173. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Chen, C.; Yang, Y.; Wang, C.; Yang, T.; Yang, X.; Liu, S.C. Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int J. Clin. Exp. Pathol 2015, 8, 4378–4387. [Google Scholar] [PubMed]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017, 32, 731–747.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, M.; Yaoi, T.; Itoh, K.; Kato, D.; Terauchi, K.; Shimada, J.; Fushiki, S. Drug resistance to paclitaxel is not only associated with ABCB1 mRNA expression but also with drug accumulation in intracellular compartments in human lung cancer cell lines. Int J. Oncol. 2012, 40, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Monzo, M.; Rosell, R.; Sanchez, J.J.; Lee, J.S.; O’Brate, A.; Gonzalez-Larriba, J.L.; Alberola, V.; Lorenzo, J.C.; Nunez, L.; Ro, J.Y.; et al. Paclitaxel resistance in non-small-cell lung cancer associated with beta-tubulin gene mutations. J. Clin. Oncol. 1999, 17, 1786–1793. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [Green Version]
- Giannakakou, P.; Sackett, D.L.; Kang, Y.K.; Zhan, Z.; Buters, J.T.; Fojo, T.; Poruchynsky, M.S. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J. Biol. Chem. 1997, 272, 17118–17125. [Google Scholar] [CrossRef] [Green Version]
- Aoki, D.; Oda, Y.; Hattori, S.; Taguchi, K.; Ohishi, Y.; Basaki, Y.; Oie, S.; Suzuki, N.; Kono, S.; Tsuneyoshi, M.; et al. Overexpression of class III beta-tubulin predicts good response to taxane-based chemotherapy in ovarian clear cell adenocarcinoma. Clin. Cancer Res. 2009, 15, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Benelli, R.; Albini, A.; Noonan, D. Neutrophils and angiogenesis: Potential initiators of the angiogenic cascade. Chem Immunol. Allergy 2003, 83, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, E.; Forni, G.; Musiani, P. Neutrophils in the antitumoral immune response. Chem. Immunol. Allergy 2003, 83, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Whalen, G.F. Solid tumours and wounds: Transformed cells misunderstood as injured tissue? Lancet 1990, 336, 1489–1492. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Goel, P.; Prajapati, D.R.; Wang, C.; Singh, R.K. Breast Cancer Cell-Neutrophil Interactions Enhance Neutrophil Survival and Pro-Tumorigenic Activities. Cancers 2020, 12, 2884. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Loutherback, K.; Lambert, G.; Estevez-Salmeron, L.; Tlsty, T.D.; Austin, R.H.; Sturm, J.C. Cell motility and drug gradients in the emergence of resistance to chemotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 16103–16108. [Google Scholar] [CrossRef] [Green Version]
- Mezencev, R.; Matyunina, L.V.; Jabbari, N.; McDonald, J.F. Snail-induced epithelial-to-mesenchymal transition of MCF-7 breast cancer cells: Systems analysis of molecular changes and their effect on radiation and drug sensitivity. BMC Cancer 2016, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of epithelial-mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef] [Green Version]
- Selvaggio, G.; Canato, S.; Pawar, A.; Monteiro, P.T.; Guerreiro, P.S.; Bras, M.M.; Janody, F.; Chaouiya, C. Hybrid Epithelial-Mesenchymal Phenotypes Are Controlled by Microenvironmental Factors. Cancer Res. 2020, 80, 2407–2420. [Google Scholar] [CrossRef] [Green Version]
- De Las Rivas, J.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef]
- Jordan, V.C. The role of tamoxifen in the treatment and prevention of breast cancer. Curr. Probl. Cancer 1992, 16, 129–176. [Google Scholar] [CrossRef] [PubMed]
- Olawuyi, O.; Tidman, M. Evaluation of Docetaxel vs. Tamoxifen in Combined Therapies Based on Overall Survival Rate (OSR) Endpoint among Female Breast Cancer Patients. Adv. Cancer Prev. 2017, 2, 122. [Google Scholar] [CrossRef] [Green Version]
- Vassilomanolakis, M.; Koumakis, G.; Barbounis, V.; Demiri, M.; Panopoulos, C.; Chrissohoou, M.; Apostolikas, N.; Efremidis, A.P. First-line chemotherapy with docetaxel and cisplatin in metastatic breast cancer. Breast 2005, 14, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Deblock, M.; Esterni, B.; Tarpin, C.; Bertucci, F.; Gilabert, M.; Charafe-Jauffret, E.; Jacquemier, J.; Houvenaeghel, G.; Extra, J.M.; et al. Docetaxel first-line therapy in HER2-negative advanced breast cancer: A cohort study in patients with prospectively determined HER2 status. Anticancer Drugs 2009, 20, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Schoene, K.; Franz, J.T.; Masuch, G. The effect of ozone on pollen development in Lolium perenne L. Environ. Pollut. 2004, 131, 347–354. [Google Scholar] [CrossRef]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Tanic, N.; Milovanovic, Z.; Tanic, N.; Dzodic, R.; Juranic, Z.; Susnjar, S.; Plesinac-Karapandzic, V.; Tatic, S.; Dramicanin, T.; Davidovic, R.; et al. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol. Ther. 2012, 13, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; Van Dam, P.; Pauwels, P.; Dirix, L.Y.; Van Laere, S.J. The interaction between ER and NFkappaB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14, 212. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell Endocrinol. 2020, 506, 110762. [Google Scholar] [CrossRef]
- Nair, B.C.; Vadlamudi, R.K. Regulation of hormonal therapy resistance by cell cycle machinery. Gene Ther. Mol. Biol. 2008, 12, 395. [Google Scholar] [PubMed]
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Brief. Bioinform. 2020, 21, 1937–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Goncalves, K.A.; Li, S.; Kishikawa, H.; Sun, G.; Yang, H.; Vanli, N.; Wu, Y.; Jiang, Y.; Hu, M.G.; et al. Plexin-B2 Mediates Physiologic and Pathologic Functions of Angiogenin. Cell 2017, 171, 849–864.e25. [Google Scholar] [CrossRef] [Green Version]
- Fett, J.W.; Strydom, D.J.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 1985, 24, 5480–5486. [Google Scholar] [CrossRef] [PubMed]
- Conrotto, P.; Corso, S.; Gamberini, S.; Comoglio, P.M.; Giordano, S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene 2004, 23, 5131–5137. [Google Scholar] [CrossRef] [Green Version]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, H.; Kong, F.; Lan, J.; Yi, S.; Jia, W.; Zheng, S.; Guo, Y.; Zhan, X. Comprehensive Analysis of Alteration Landscape and Its Clinical Significance of Mitochondrial Energy Metabolism Pathway-Related Genes in Lung Cancers. Oxid. Med. Cell. Longev. 2021, 2021, 9259297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Sun, S.; Wang, K.; Qian, J.; Cui, Z.; Tao, T.; Zhou, J. ACOX2 is a prognostic marker and impedes the progression of hepatocellular carcinoma via PPARalpha pathway. Cell Death Dis. 2021, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, S.S.; Kristensen, V.N.; Seiler, M.; Kumar, S.; Alnaes, G.I.; Ming, Y.; Kerrigan, J.; Naume, B.; Sachidanandam, R.; Bhanot, G.; et al. Expression of an estrogen-regulated variant transcript of the peroxisomal branched chain fatty acid oxidase ACOX2 in breast carcinomas. BMC Cancer 2015, 15, 524. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Jin, J.; Woo, J.; Min, H.; Kim, Y. Proteomic analysis of mouse astrocytes and their secretome by a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics 2014, 14, 1604–1609. [Google Scholar] [CrossRef]
- Woo, J.; Han, D.; Park, J.; Kim, S.J.; Kim, Y. In-depth characterization of the secretome of mouse CNS cell lines by LC-MS/MS without prefractionation. Proteomics 2015, 15, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Yoon, J.H.; Long, N.P.; Koo, G.B.; Noh, H.J.; Oh, S.J.; Lee, S.B.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; et al. Spheroid-Induced Epithelial-Mesenchymal Transition Provokes Global Alterations of Breast Cancer Lipidome: A Multi-Layered Omics Analysis. Front. Oncol. 2019, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.; Dan, K.; Kim, S.I.; Park, S.H.; Han, D.; Kim, Y.H. In-depth proteomic profiling captures subtype-specific features of craniopharyngiomas. Sci. Rep. 2021, 11, 21206. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | MCF-7/DR | MCF-7/PR | MCF-7/TAR | |||
|---|---|---|---|---|---|---|
| Log2 FC | Adj p-Value | Log2 FC | Adj p-Value | Log2 FC | Adj p-Value | |
| ATP6V1B2 | 0.2987 | 0.0007 | 0.1741 | 0.0167 | 0.4997 | 0.0034 |
| MAP2K1 | 0.8756 | 0.0015 | 0.5689 | 0.0076 | 0.4647 | 0.0125 |
| MAP4 | 0.7893 | 0.0005 | 0.2916 | 0.0218 | 0.3371 | 0.0141 |
| PLIN3 | 0.8894 | 0.0007 | 0.6368 | 0.0057 | 0.2839 | 0.0189 |
| PRKCI | 0.1833 | 0.0195 | 0.2564 | 0.0060 | 0.6548 | 0.0060 |
| ROCK2 | 0.2936 | 0.0131 | 0.2128 | 0.0198 | 0.1766 | 0.0322 |
| SMN1 | 0.6062 | 0.0016 | 0.2697 | 0.0241 | 0.3360 | 0.0226 |
| TXNRD1 | 0.8700 | 0.0007 | 0.3042 | 0.0137 | 0.6149 | 0.0121 |
| FADS1 | −0.2534 | 0.0226 | −0.5072 | 0.0069 | −0.8856 | 0.0137 |
| FRK | −0.6422 | 0.0006 | −0.2322 | 0.0043 | −0.9870 | 0.0043 |
| GAA | −0.6750 | 0.0010 | −0.1442 | 0.0440 | −0.3608 | 0.0133 |
| HSD17B10 | −0.6633 | 0.0054 | −0.2812 | 0.0143 | −0.3145 | 0.0308 |
| CYP51A1 | −1.0314 | 0.0072 | −0.7026 | 0.0014 | −0.6652 | 0.0209 |
| DHODH | −0.9274 | 0.0051 | −0.4792 | 0.0015 | −0.1451 | 0.0329 |
| CA12 | −1.5826 | 0.0025 | −0.7594 | 0.0020 | −0.7171 | 0.0051 |
| ACAA1 | −1.3387 | 0.0002 | −0.3037 | 0.0039 | −0.3683 | 0.0282 |
| Protein Name | MCF-7/DR | MCF-7/PR | MCF-7/TAR | |||
|---|---|---|---|---|---|---|
| Log2 FC | Adj p-Value | Log2 FC | Adj p-Value | Log2 FC | Adj p-Value | |
| ATP6V1A | 0.3835 | 0.0084 | 0.3850 | 0.0082 | 0.4489 | 0.0181 |
| VPS26B | 0.7674 | 0.0013 | 0.6040 | 0.0014 | 0.4039 | 0.0040 |
| PLXNB2 | 0.6396 | 0.0017 | 0.3654 | 0.0017 | 0.6469 | 0.0055 |
| RNF214 | 1.2672 | 0.0005 | 0.1830 | 0.0081 | 0.3928 | 0.0114 |
| THYN1 | 0.6490 | 0.0006 | 0.2734 | 0.0438 | 0.2793 | 0.0043 |
| RIDA | −0.5185 | 0.0025 | −0.2993 | 0.0059 | −0.1191 | 0.0104 |
| CPSF6 | −0.4050 | 0.0085 | −0.1964 | 0.0028 | −0.2366 | 0.0139 |
| ABCD3 | −0.9008 | 0.0040 | −0.2900 | 0.0055 | −0.8820 | 0.0104 |
| UHRF1 | −0.1969 | 0.0021 | −0.1794 | 0.0328 | −0.4596 | 0.0088 |
| HNRNPL | −0.2741 | 0.0220 | −0.1738 | 0.0414 | −0.2100 | 0.0353 |
| P4HB | −1.0898 | 0.0050 | −0.3169 | 0.0336 | −0.3561 | 0.0023 |
| ACOX3 | −1.5011 | 0.0069 | −0.2912 | 0.0193 | −0.5299 | 0.0098 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cha, H.K.; Cheon, S.; Kim, H.; Lee, K.-M.; Ryu, H.S.; Han, D. Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis. Molecules 2022, 27, 1762. https://doi.org/10.3390/molecules27061762
Cha HK, Cheon S, Kim H, Lee K-M, Ryu HS, Han D. Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis. Molecules. 2022; 27(6):1762. https://doi.org/10.3390/molecules27061762
Chicago/Turabian StyleCha, Hyo Kyeong, Seongmin Cheon, Hyeyoon Kim, Kyung-Min Lee, Han Suk Ryu, and Dohyun Han. 2022. "Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis" Molecules 27, no. 6: 1762. https://doi.org/10.3390/molecules27061762