
Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
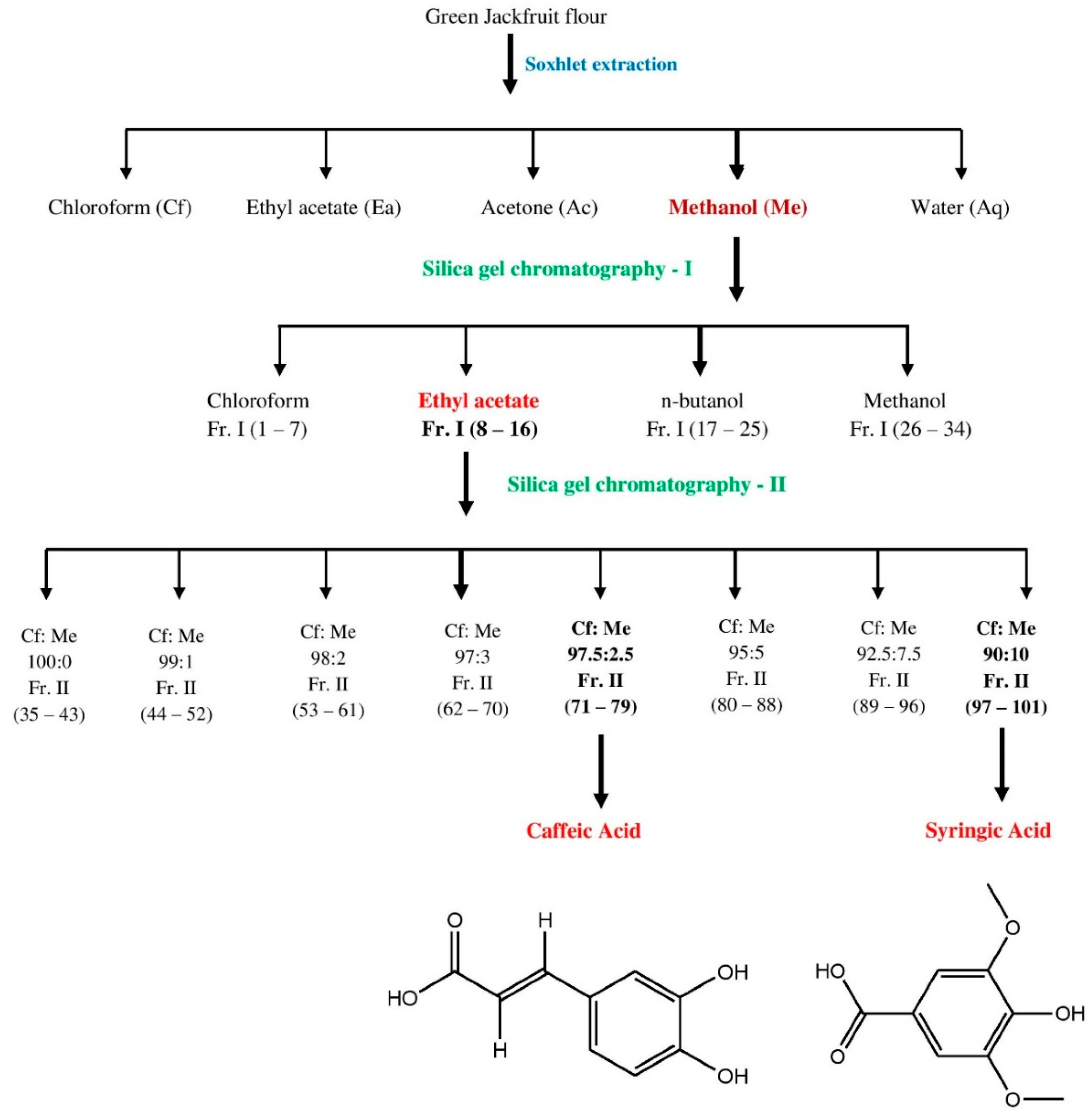
2.1. Extraction
2.2. Analysis of Phenolic Compounds and Ascorbic Acid by HPLC
2.3. In Vitro Enzymes Inhibition Assays
2.4. HSA Glycation Inhibition Assay at Multiple Stages
2.5. Antioxidant Activity
2.6. Isolation and Identification of Bioactive Compounds from MJ
2.7. Spectral Measurements
2.8. α-Glucosidase, α-Amylase and Aldose Reductase Inhibition Kinetics
2.9. Molecular Docking Simulation
2.10. Molecular Dynamics Simulation
2.11. Druglikeness and Pharmacokinetics Analysis
2.12. Binding Free Energy Calculations
2.13. Statistical Analysis
3. Results
3.1. In Vitro Inhibition of Carbohydrate Hydrolysing Enzymes
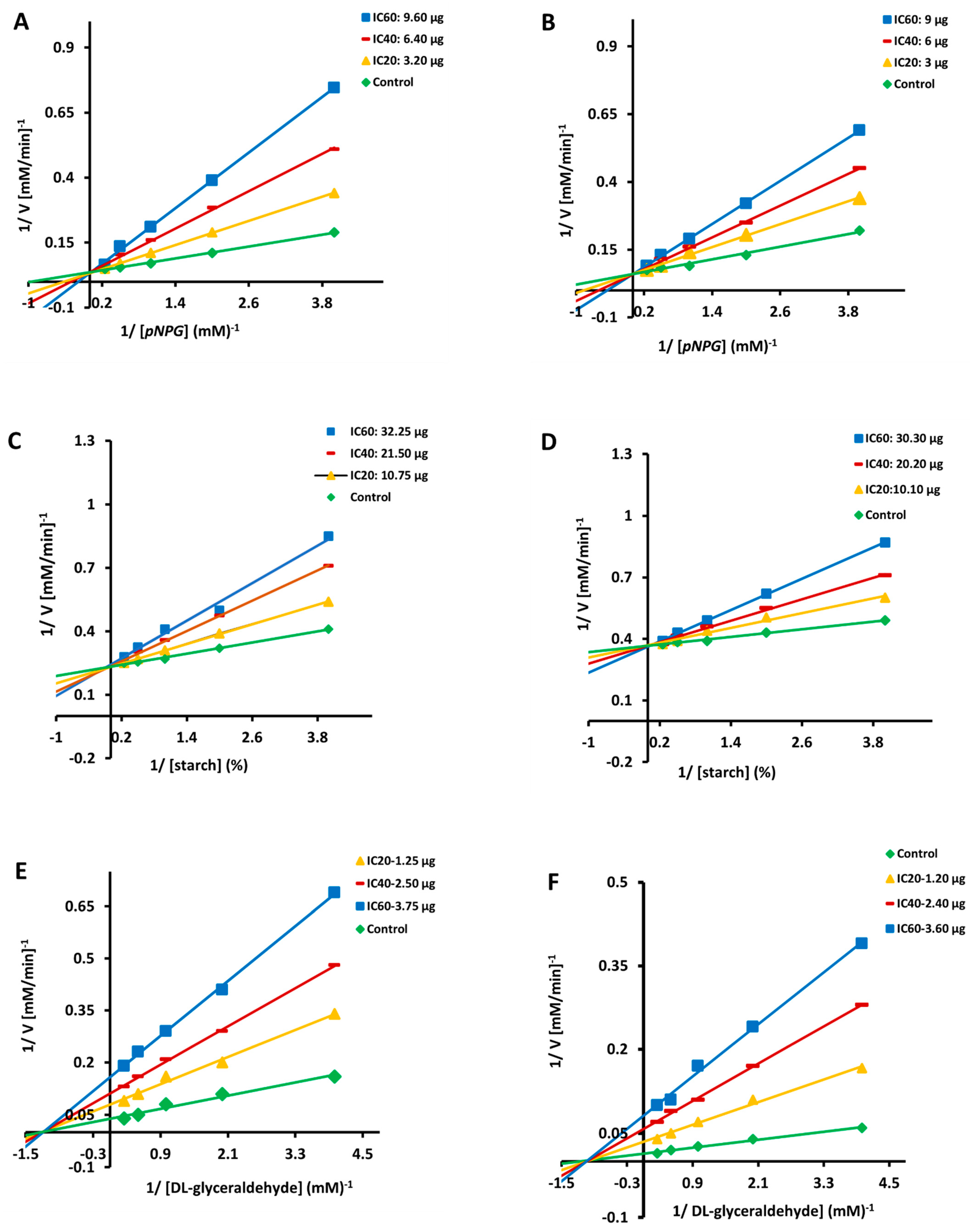
3.2. Kinetics of α-Glucosidase and α-Amylase Inhibition
3.3. In Vitro Inhibition of Aldose Reductase
3.4. CA and SA Inhibition of Aldose Reductase: A Kinetic Study
3.5. Antioxidant Ability, TPC and TFC
3.6. Identification of Various Phenolic Acids, Flavonoids and Ascorbic Acid in MJ by HPLC
3.7. In Vitro Inhibition of HSA Glycation at Multiple Stages
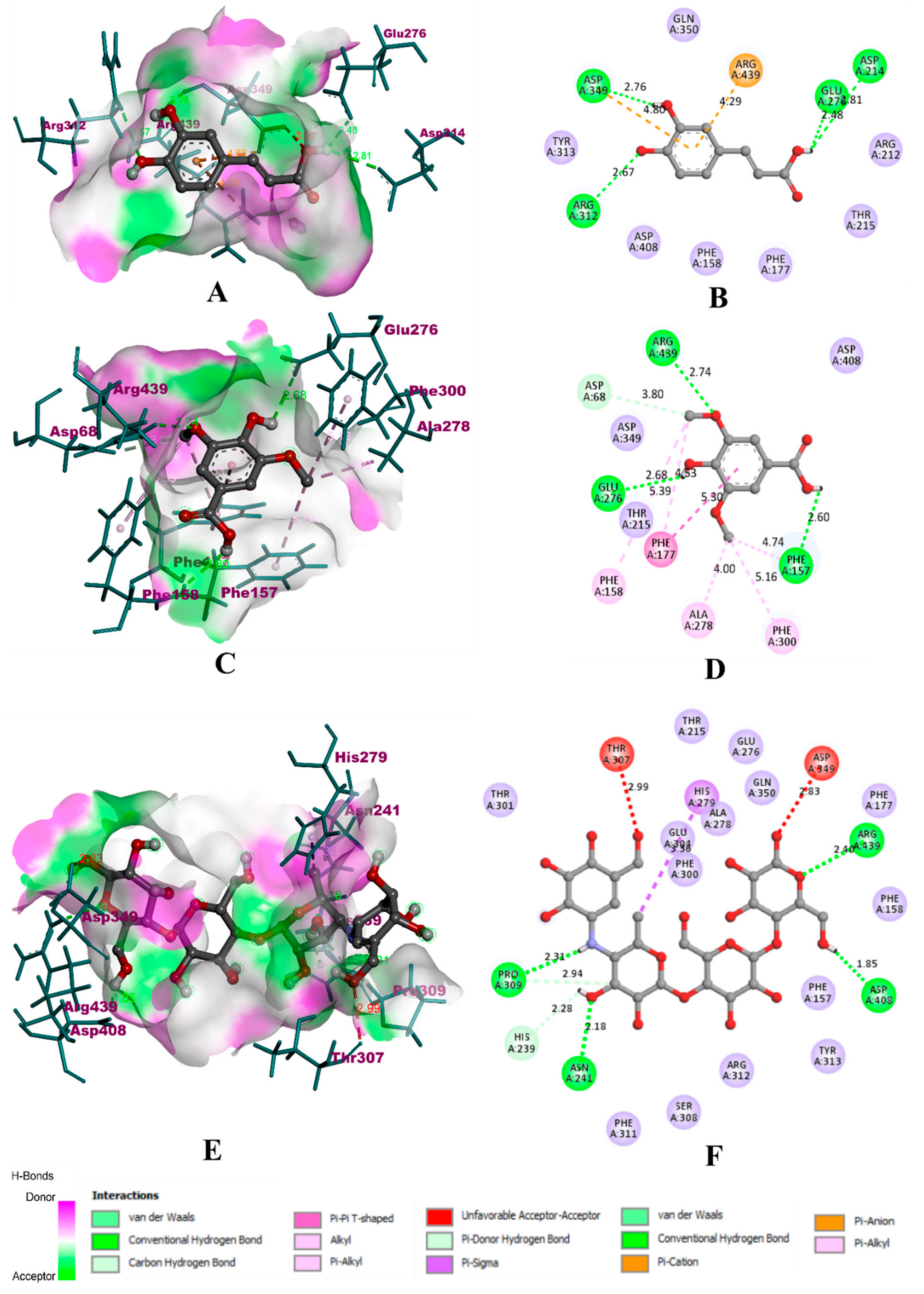
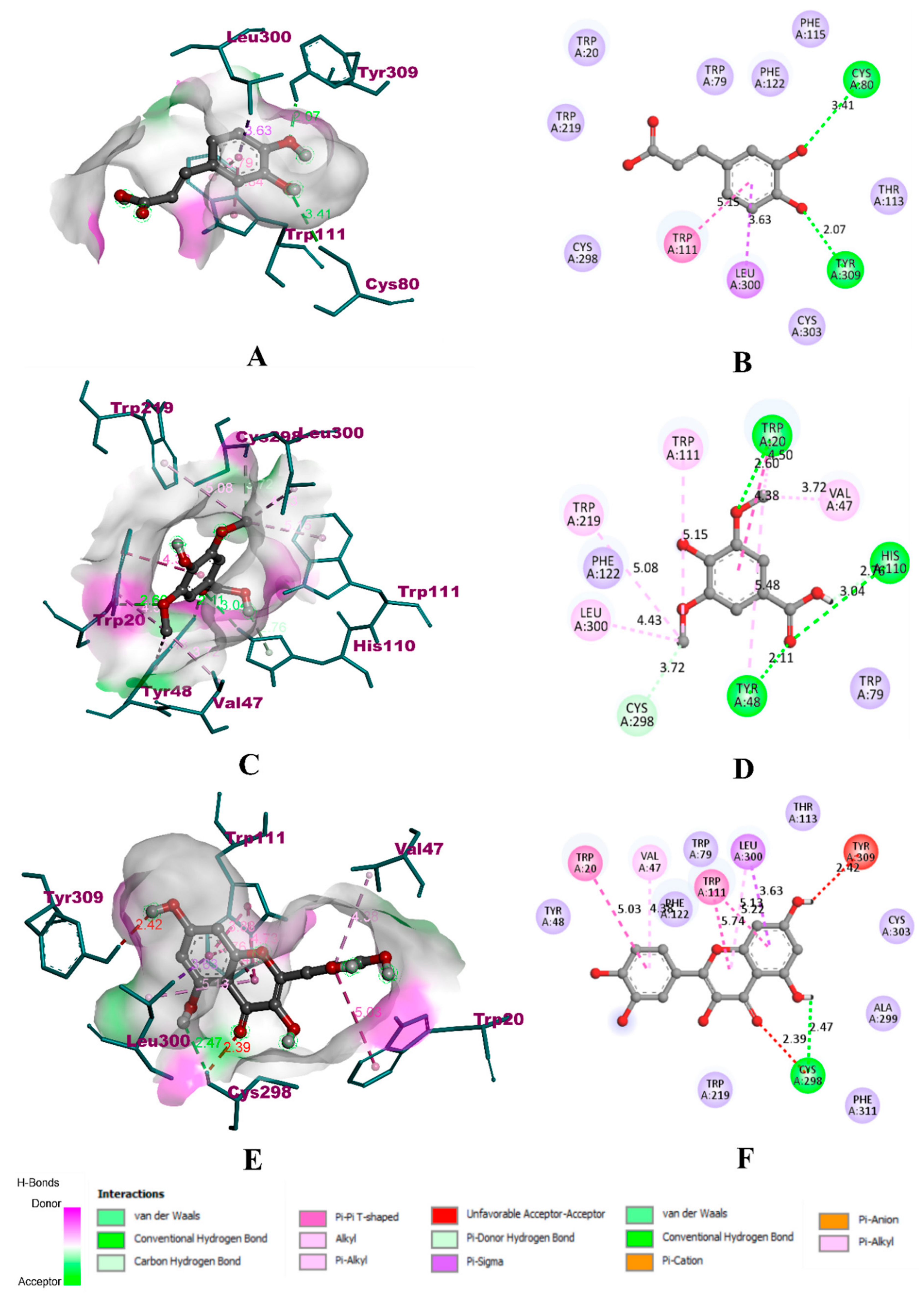
3.8. Molecular Docking Simulation
3.9. Molecular Dynamics Simulation
3.10. Druglikeness and Pharmacokinetics Analysis
3.11. Binding Free Energy Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Diabetes Federation (IDF). IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- Spinola, V.; Llorent-Martínez, E.J.; Castilho, P.C. Inhibition of α-amylase; α-glucosidase and pancreatic lipase by phenolic compounds of Rumex maderensis (Madeira sorrel). Influence of simulated gastrointestinal digestion on hyperglycaemia-related damage linked with aldose reductase activity and protein glycation. LWT 2020, 118, 108727. [Google Scholar]
- Hossain, U.; Das, A.K.; Ghosh, S.; Sil, P.C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. [Google Scholar] [CrossRef]
- Sharma, P.; Hajam, Y.A.; Kumar, R.; Rai, S. Complementary and alternative medicine for the treatment of diabetes and assoiated complications: A review on therapeutic role of polyphenols. Phytomed. Plus 2022, 2, 100188. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, Z.C.; Xie, X.; Wang, H.; Wang, H.; Wang, Z.X.; Sha, X.M.; Lu, Y. Jackfruit (Artocarpus heterophyllus Lam.) peel: A better source of antioxidants and a-glucosidase inhibitors than pulp; flake and seed; and phytochemical profile by HPLC-QTOF-MS/MS. Food Chem. 2017, 234, 303–313. [Google Scholar] [CrossRef]
- Baliga, M.S.; Shivashankara, A.R.; Haniadka, R.; Dsouza, J.; Bhat, H.P. Phytochemistry; nutritional and pharmacological properties of Artocarpus heterophyllus Lam. (jackfruit): A review. Food Res. Int. 2011, 44, 1800–1811. [Google Scholar] [CrossRef]
- Swami, S.B.; Thakor, N.J.; Haldankar, P.M.; Kalse, S.B. Jackfruit and its many functional components as related to human health: A review. Compr. Rev. Food Sci. Food Saf. 2012, 11, 565–576. [Google Scholar] [CrossRef]
- Ong, B.T.; Nazimah, S.A.; Osman, A.; Quek, S.Y.; Voon, Y.Y.; Hashim, D.M.; Chew, P.M.; Kong, Y.W. Chemical and flavour changes in jackfruit (Artocarpus heterophyllus Lam.) cultivar J3 during ripening. Post Biol. Technol. 2006, 40, 279–286. [Google Scholar] [CrossRef]
- Harbone, J.B. Phytochemical Methods, 1st ed.; Chapman and Hall Ltd.: London, UK, 1973; pp. 49–188. [Google Scholar]
- Ordon-Ez, A.A.; Gomez, J.D.; Vattuone, M.A.; Isla, M.I. Antioxidant activities of Sechiumedule (Jacq.) Swart extracts. Food Chem. 2006, 97, 452–458. [Google Scholar] [CrossRef]
- Shuxia, C.; Xiaoqing, S.; Siqiong, C.; Panpan, L.; Junna, D.; Yanxia, C.; Huanwen, M. Evaluation of garlic cultivars for polyphenolic content and antioxidant properties. PLoS ONE 2013, 8, e79730. [Google Scholar]
- Seal, T. Quantitative HPLC analysis of phenolic acids; flavonoids and ascorbic acid in four different solvent extracts of two wild edible leaves; Sonchus arvensis and Oenanthe linearis of North-Eastern region in India. J. Appl. Pharm. Sci. 2016, 6, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Nagendra, P.M.N. Inhibitory effect of banana (Musa sp. var. Nanjangud rasa bale) flower extract and its constituents umbelliferone and lupeol on α-glucosidase; aldose reductase and glycation at multiple stages. S. Afr. J. Bot. 2014, 95, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Nurhanani, R.; Rasyidah, R.; Sarni, M.J.; Azlina, A.A. Radical scavenging and reducing properties of extracts of cashew shoots (Anacardium occidentale). Food Chem. 2008, 111, 38–44. [Google Scholar]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNAdependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Steric hindrance by 2 amino acid residues determines the substrate specificity of isomaltase from Saccharomyces cerevisiae. J. Biosci. Bioeng. 2011, 112, 545–550. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Prot. Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acid Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 7, 1–5. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-Glucosidase; α-Amylase Inhibition; Kinetics and Docking Studies of Novel (2-Chloro-6-(trifluoromethyl) benzyloxy) arylidene) Based Rhodanine and Rhodanine Acetic Acid Derivatives. Chem. Select. 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Patil, S.M.; Maruthi, K.R.; Bajpe, N.S.; Vyshali, V.M.; Sushmitha, S.; Chagalamari, A.; Ramith, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 7, 932–939. [Google Scholar]
- Kaushik, G.; Satya, S.; Khandelwal, R.K.; Naik, S.N. Commonly consumed Indian plant food materials in the management of diabetes mellitus. Diabetes Metab. Syndr. Clin. Res. Rev. 2010, 4, 21–40. [Google Scholar]
- Joseph, J. Nutritional;glycemic and ecological assessment of green jackfruit for diabetes in Kerala. Int. J. Diabetes 2019, 1, 14–18. [Google Scholar]
- Rao, A.G.; Naik, K.S.; Unnikrishnan, A.G.; Joseph, J. Efficacy of green jackfruit flour as a medical nutrition therapy replacing rice or wheat in patients with type 2 diabetes mellitus: A randomized; double-blind; placebo-controlled study. Nutr. Diabetes 2021, 11, 1–6. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Zameer, F.; Nagendra Prasad, M.N. Investigation of antihyperglycaemic activity of banana (Musa sp. var. Nanjangud rasa bale) pseudostem in normal and diabetic rats. J. Sci. Food Agric. 2015, 95, 165–173. [Google Scholar] [CrossRef]
- Espindola, K.M.; Ferreira, R.G.; Narvaez, L.E.; Silva Rosario, A.C.; da Silva, A.H.; Silva, A.G.; Vieira, A.P.; Monteiro, M.C. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Srinivasulu, C.; Ramgopal, M.; Ramanjaneyulu, G.; Anuradha, C.M.; Kumar, C.S. Syringic acid (SA)—A review of its occurrence; biosynthesis; pharmacological and industrial importance. Biomed. Pharmacother. 2018, 108, 547–557. [Google Scholar] [CrossRef]
- Martin, A.E.; Montgomery, P.A. Acarbose: An α-glucosidase inhibitor. Am. J. Health Syst. Pharm. 1996, 53, 2277–2290. [Google Scholar] [CrossRef]
- Aleixandre, A.; Gil, J.V.; Sineiro, J.; Rosell, C.M. Understanding phenolic acids inhibition of α-amylase and α-glucosidase and influence of reaction conditions. Food Chem. 2022, 372, 131231. [Google Scholar] [CrossRef]
- Cao, J.; Yan, S.; Xiao, Y.; Han, L.; Sun, L.; Wang, M. Number of galloyl moiety and intramolecular bonds in galloyl-based polyphenols affect their interaction with alpha-glucosidase. Food Chem. 2022, 367, 129846. [Google Scholar] [CrossRef]
- Sun, L.; Warren, F.J.; Gidley, M.J. Natural products for glycaemic control: Polyphenols as inhibitors of alpha-amylase. Trend Food Sci. Technol. 2019, 91, 262–273. [Google Scholar] [CrossRef]
- Islam, M.N.; Ishita, I.J.; Jung, H.A.; Choi, J.S. Vicenin 2 isolated from Artemisia capillaris exhibited potent anti-glycation properties. Food Chem. Toxicol. 2014, 69, 55–62. [Google Scholar] [CrossRef]
- Chethan, S.; Dharmesh, S.M.; Malleshi, N.G. Inhibition of aldose reductase from cataracted eye lenses by finger millet (Eleusine coracana) polyphenols. Bioorg. Med. Chem. 2008, 16, 10085–10090. [Google Scholar] [CrossRef]
- Veeresham, C.; Rama Rao, A.; Asres, K. Aldose reductase inhibitors of plant origin. Phytother. Res. 2014, 28, 317–333. [Google Scholar] [CrossRef]
- Goh, S.Y.; Cooper, M.E. The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metabol. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Nivas, D.; Sonar, B.A.; Shaikh, S.S.; Patil, U.H.; Gaikwad, D.K.; Chavan, N.S.; Sabale, A.B.; Chavan, P.D. Screening of some coastal plant resources for their antioxidant potential; total polyphenol and flavonoid content. Pharmacogn. J. 2010, 2, 151–156. [Google Scholar] [CrossRef]
- Choi, C.W.; Kim, S.C.; Hwang, S.S.; Choi, B.K.; Ahn, H.J.; Lee, M.Y.; Park, S.H.; Kim, S.K. Antioxidant activity and free radical scavenging capacity between Korean medicinal plants and flavonoids by assay-guided comparison. Plant Sci. 2002, 163, 1161–1168. [Google Scholar] [CrossRef]
- Perry, G.; Raina, A.K.; Nunomura, A.; Wataya, T.; Sayre, L.M.; Smith, M.A. How important is oxidative damage? Lessons from Alzheimer’s disease. Free Rad. Biol. Med. 2000, 28, 831–834. [Google Scholar] [CrossRef]
- Wang, H.; Gan, D.; Zhang, X.; Pan, Y. Antioxidant capacity of the extracts from pulp of Osmanthus fragrans and its components. LWT-Food Sci. Technol. 2010, 43, 319–325. [Google Scholar] [CrossRef]
- Natic, M.M.; Dabic, D.C.; Papetti, A.; Aksic, M.M.; Ognjanov, V.; Ljubojevic, M.; Tesic, Z.L. Analysis and characterisation of phytochemicals in mulberry (Morus alba L.) fruits grown in Vojvodina, North Serbia. Food Chem. 2015, 171, 128–136. [Google Scholar] [CrossRef]
- Metrouh-Amir, H.; Duarte, C.M.; Maiza, F. Solvent effect on total phenolic contents, antioxidant, and antibacterial activities of Matricaria pubescens. Ind. Crops Prod. 2015, 67, 249–256. [Google Scholar] [CrossRef]
- Liu, Y.; Zhan, L.; Xu, C.; Jiang, H.; Zhu, C.; Sun, L.; Sun, C.; Li, X. α-Glucosidase inhibitors from Chinese bayberry (Morella rubra Sieb. et Zucc.) fruit: Molecular docking and interaction mechanism of flavonols with different B-ring hydroxylations. RSC Adv. 2020, 10, 29347–29361. [Google Scholar] [CrossRef]
- Narayanan, C.; Dias, C.L. Hydrophobic interactions and hydrogen bonds in β-sheet formation. J. Chem. Phys. 2013, 139, 09B640_1. [Google Scholar] [CrossRef] [Green Version]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holla, B.S.; Poojary, V.; et al. In vitro and in silico studies of fluorinated 2; 3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2021, 12, e2100342. [Google Scholar] [CrossRef]
- Shahzad, D.; Saeed, A.; Larik, F.A.; Channar, P.A.; Abbas, Q.; Alajmi, M.F.; Arshad, M.I.; Erben, M.F.; Hassan, M.; Raza, H.; et al. Novel C-2 symmetric molecules as α-glucosidase and α-amylase inhibitors: Design; synthesis; kinetic evaluation; molecular docking and pharmacokinetics. Molecules 2019, 24, 1511. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Martin, K.A.; Hwa, J. Aldose reductase; oxidative stress; and diabetic mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [Green Version]
- Sebastian-Perez, V.; García-Rubia, A.; Seif el-Din, S.H.; Sabra, A.N.; El-Lakkany, N.M.; William, S.; Blundell, T.L.; Maes, L.; Martinez, A.; Campillo, N.E.; et al. Deciphering the enzymatic target of a new family of antischistosomal agents bearing a quinazoline scaffold using complementary computational tools. J. Enzym. Inhib. Med. Chem. 2020, 35, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Martinez-Mayorga, K. The interplay between molecular modeling and chemoinformatics to characterize protein–ligand and protein–protein interactions landscapes for drug discovery. Adv. Protein Chem. Struct. Biol. 2014, 96, 1–37. [Google Scholar]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (−)-Kusunokinin as a Potential Aldose Reductase Inhibitor: Equivalency Observed via AKR1B1 Dynamics Simulation. ACS Omega 2020, 6, 606–614. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.N.; Dong, J.; Deng, Y.H.; Zhu, M.F.; Wen, M.; Yao, Z.J.; Lu, A.P.; Wang, J.B.; Cao, D.S. ADME properties evaluation in drug discovery: Prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef]
- McCarren, P.; Springer, C.; Whitehead, L. An investigation into pharmaceutically relevant mutagenicity data and the influence on Ames predictive potential. J. Cheminform. 2011, 3, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Morris, K.F.; Billiot, E.J.; Billiot, F.H.; Gladis, A.A.; Lipkowitz, K.B.; Southerland, W.M.; Fang, Y. A molecular dynamics simulation study of the association of 1,1′-binaphthyl-2,2′-diyl hydrogenphosphate enantiomers with a chiral molecular micelle. Chem. Phys. 2014, 439, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Batiha, G.E.; Beshbishy, A.M.; Ikram, M.; Mulla, Z.S.; El-Hack, M.E.; Taha, A.E.; Algammal, A.M.; Elewa, Y.H. The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: Quercetin. Foods 2020, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Machin, L.; Monzote, L.; Sharifi-Rad, J.; Ezzat, S.M.; Salem, M.A.; Merghany, R.M.; El Mahdy, N.M.; Kılıç, C.S.; Sytar, O.; et al. Therapeutic potential of quercetin: New insights and perspectives for human health. Acs Omega 2020, 5, 11849–118472. [Google Scholar] [CrossRef]
- Maalik, A.; Khan, F.A.; Mumtaz, A.; Mehmood, A.; Azhar, S.; Atif, M.; Karim, S.; Altaf, Y.; Tariq, I. Pharmacological applications of quercetin and its derivatives: A short review. Trop. J. Pharm. Res. 2014, 13, 1561–1566. [Google Scholar] [CrossRef]
- Utesch, D.; Feige, K.; Dasenbrock, J.; Broschard, T.H.; Harwood, M.; Danielewska-Nikiel, B.; Lines, T.C. Evaluation of the potential in vivo genotoxicity of quercetin. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 654, 38–44. [Google Scholar] [CrossRef]
- Pérez-Pastén, R.; Martínez-Galero, E.; Chamorro-Cevallos, G. Quercetin and naringenin reduce abnormal development of mouse embryos produced by hydroxyurea. J. Pharm. Pharmacol. 2010, 62, 1003–1009. [Google Scholar] [CrossRef]
- Vanhees, K.; de Bock, L.; Godschalk, R.W.; van Schooten, F.J.; van Waalwijk van Doorn-Khosrovani, S.B. Prenatal exposure to flavonoids: Implication for cancer risk. Toxic. Sci. 2011, 120, 59–67. [Google Scholar] [CrossRef]
- Harwood, M.; Danielewska-Nikiel, B.; Borzelleca, J.F.; Flamm, G.W.; Williams, G.M.; Lines, T.C. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem. Toxicol. 2007, 45, 2179–2205. [Google Scholar] [CrossRef]
- Wang, Y.H.; Chao, P.D.; Hsiu, S.L.; Wen, K.C.; Hou, Y.C. Lethal quercetin-digoxin interaction in pigs. Life Sci. 2004, 74, 1191–1197. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Name | Ret. Time | Area | Height | Concentration (µg/mg) in MJ |
|---|---|---|---|---|---|
| 1 | Ascorbic acid | 4.222 | 72,576 | 8335 | 6.828 |
| 2 | Gallic acid | 5.708 | 65,639 | 11,867 | 6.176 |
| 3 | Methyl gallate | 12.675 | 60,233 | 5868 | 5.667 |
| 4 | Caffeic acid | 15.060 | 22,368 | 2486 | 2.104 |
| 5 | Syringic acid | 15.451 | 23,443 | 2626 | 2.206 |
| 6 | Ferulic acid | 22.084 | 11,293 | 905 | 1.062 |
| 7 | Quercetin | 29.964 | 6706 | 787 | 0.631 |
| 8 | Kaempferol | 34.914 | 54,923 | 5186 | 5.167 |
| Enzymes | IC50 x,y (µg/mL) | ||||
|---|---|---|---|---|---|
| MJ | EaFr. | Caffeic Acid | Syringic Acid | Acarbose/* Quercetin | |
| α-Amylase | 28.00 ± 0.03 d | 27.80 ± 0.06 c | 26.90 ± 0.05 b | 25.25 ± 1.00 a | 28.50 ± 0.05 d |
| α-Glucosidase | 10.00 ± 0.14 b | 09.55 ± 0.87 b | 8.00 ± 0.40 a | 7.50 ± 1.05 a | 11.00 ± 0.11 c |
| Aldose reductase | 3.75 ± 0.75 b,c | 3.60 ± 0.00 b | 3.10 ± 0.33 a | 3.00 ± 0.00 a | 4.10 ± 0.22 c,* |
| Enzymes | Compound | Treatment | Mode of Inhibition x | Km (µM) | Vmax (103(µM/min)−1 | Ki (mg) y,z |
|---|---|---|---|---|---|---|
| α-Amylase | CA | Control | Competitive | 0.79 | 25.25 | 1.03 ± 0.15 |
| IC20 10.75 µg | 0.91 | 26.15 | ||||
| IC40 21.50 µg | 1.82 | 25.75 | ||||
| IC60 32.25 µg | 3.02 | 26.10 | ||||
| SA | Control | Competitive | 0.94 | 50.55 | 1.25 ± 0.04 | |
| IC20 10.10 µg | 1.55 | 51.10 | ||||
| IC40 20.20 µg | 2.88 | 51.11 | ||||
| IC60 30.30 µg | 3.99 | 50.95 | ||||
| α-Glucosidase | CA | Control | Competitive | 1.01 | 40.05 | 0.52 ± 0.02 |
| IC20 3.20 µg | 2.44 | 39.98 | ||||
| IC40 6.40 µg | 3.33 | 40.40 | ||||
| IC60 9.60 µg | 5.01 | 40.37 | ||||
| SA | Control | Competitive | 1.22 | 34.00 | 0.96 ± 0.22 | |
| IC20 3.0 µg | 3.58 | 33.55 | ||||
| IC40 6.0 µg | 5.54 | 33.43 | ||||
| IC60 9.0 µg | 8.80 | 33.79 | ||||
| Aldose reductase | CA | Control | Non-competitive | 0.79 | 27.71 | 1.11 ± 0.36 |
| IC20 1.25 µg | 0.81 | 13.35 | ||||
| IC40 2.50 µg | 0.81 | 10.15 | ||||
| IC60 3.75 µg | 0.80 | 5.05 | ||||
| SA | Control | Non-competitive | 0.81 | 78.60 | 1.64 ± 0.65 | |
| IC20 1.20 µg | 0.91 | 34.47 | ||||
| IC40 2.40 µg | 0.92 | 20.05 | ||||
| IC60 3.60 µg | 0.92 | 11.50 |
| Sample | TPC (mg GAE/g) | TFC (mg QE/g) | EC50 x,y (μg/mL) | ||
|---|---|---|---|---|---|
| Radical Scavenging Activities | |||||
| DPPH | ABTS | Superoxide | |||
| MJ | 252.07 ± 0.15 b | 601.05 ± 0.24 b | 24.30 ± 0.82 c | 20.80 ± 1.32 c | 44.50 ± 2.40 c |
| EaFr. | 153.75 ± 0.36 a | 365.04 ± 2.00 a | 24.02 ± 1.87 c | 20.01 ± 0.33 c | 44.06 ± 1.78 c |
| Caffeic acid | - | 18.50 ± 0.08 b | 12.44 ± 1.60 b | 30.13 ± 2.05 b | |
| Syringic acid | - | 16.00 ± 0.13 a | 11.40 ± 2.04 a | 28.00 ± 1.19 a | |
| BHA | - | - | 40.25 ± 0.30 d | 31.00 ± 0.55 d | 64.75 ± 0.13 d |
| A. | Fructosamine (mmol/mg Protein) | ||||||
| Week | HSA | HSA/Fructose | MJ | Caffeic Acid | Syringic Acid | Aminoguanidine | |
| 1 | - | 42.15 ± 1.06 e | 27.15 ± 0.31 c | 26.82 ± 0.14 a | 26.30 ± 1.21 b | 28.50 ± 1.16 d | |
| 2 | - | 54.08 ± 0.18 e | 28.00 ± 1.10 c | 27.06 ± 0.13 a | 26.99 ± 1.90 b | 30.00 ± 1.00 d | |
| 3 | - | 60.50 ± 1.06 e | 28.80 ± 1.72 c | 28.02 ± 1.04 a | 26.41 ± 1.18 b | 31.75 ± 0.97 d | |
| B. | Protein carbonyl Content (nmol/mg protein) | ||||||
| 1 | 0.50 ± 0.20 a | 2.06 ± 2.12 f | 0.57 ± 0.12 d | 0.49 ± 2.17 b | 0.49 ± 1.06 c | 0.64 ± 0.54 e | |
| 2 | 0.52 ± 0.14 a | 4.20 ± 0.00 f | 0.58 ± 0.02 d | 0.57 ± 1.25 b | 0.54 ± 0.62 c | 0.66 ± 1.05 e | |
| 3 | 0.55 ± 0.27 a | 6.66 ± 0.76 f | 0.58 ±1.44 d | 0.57 ± 2.22 b | 0.54 ± 1.32 c | 0.70 ± 1.11 e | |
| C. | Thiols Group (nmol/mg protein) | ||||||
| 1 | 2.20 ± 1.16 d | 1.64 ± 0.14 c | 0.76 ± 0.57 b | 0.69 ± 0.55 a | 0.64 ± 0.62 a | 0.95 ± 1.46 b | |
| 2 | 2.45 ± 1.32 d | 1.95 ± 0.31 c | 0.79 ± 0.48 b | 0.79 ± 0.05 a | 0.76 ± 0.53 a | 1.04 ± 1.00 b | |
| 3 | 2.99 ± 0.42 d | 2.04 ± 1.04 c | 0.81 ± 0.98 b | 0.80 ± 0.88 a | 0.78 ± 0.34 a | 1.17 ± 0.89 b | |
| Compound | α-Glucosidase | α-Amylase | HAR | ||||||
|---|---|---|---|---|---|---|---|---|---|
| BA | NB | HB | BA | NB | HB | BA | NB | HB | |
| Ascorbic acid | −7.6 | 4 | 0 | −6.8 | 5 | 1 | −4.2 | 7 | 0 |
| Gallic acid | −5.3 | 7 | 2 | −5.2 | 7 | 2 | −6.3 | 6 | 2 |
| Methyl gallate | −4.5 | 2 | 1 | −6.1 | 5 | 0 | −7.1 | 9 | 2 |
| Caffeic acid | −8.2 | 6 | 4 | −8.1 | 6 | 3 | −7.4 | 4 | 2 |
| Syringic acid | −11.4 | 10 | 4 | −12.5 | 11 | 3 | −12.9 | 10 | 4 |
| Ferulic acid | −5.2 | 5 | 1 | −6.7 | 7 | 1 | −8.9 | 8 | 3 |
| Quercetin | −6.2 | 7 | 2 | −7.1 | 6 | 3 | −10.3 | 7 | 1 |
| Kaempferol | −4.7 | 2 | 0 | −8.1 | 6 | 2 | −9.1 | 9 | 3 |
| Acarbose | −10.2 | 7 | 6 | −6.2 | 2 | 2 | - | - | - |
| Categories | Parameters | Caffeic Acid | Syringic Acid | Acarbose | Amino-Guanidine | Quercetin |
|---|---|---|---|---|---|---|
| Druglikeliness | Mol. Wt. | 180.04 | 198.05 | 645.25 | 74.06 | 302.04 |
| nHA | 4 | 5 | 19 | 4 | 7 | |
| nHD | 3 | 2 | 14 | 6 | 5 | |
| TPSA | 77.76 | 75.99 | 321.17 | 87.92 | 131.36 | |
| LogP | 1.43 | 1.212 | −4.37 | −2.376 | 2.155 | |
| Absorption | Caco-2 | −5.22 | −5.142 | −6.149 | −5.448 | −5.204 |
| MDCK | 1.1 | 1.1 | 0.00089 | 0.001687 | 8.0 | |
| Distribution | VD | 0.37 | 0.259 | 0.071 | 0.918 | 0.579 |
| BBB | 0.119 | 0.457 | 0.385 | 0.361 | 0.008 | |
| Metabolism | CYP1A2 | 0.048 | 0.032 | 0.0 | 0.029 | 0.943 |
| CYPC19 | 0.069 | 0.025 | 0.002 | 0.025 | 0.053 | |
| CYP2C9 | 0.036 | 0.028 | 0.0 | 0.011 | 0.598 | |
| CYP2D6 | 0.014 | 0.012 | 0.0 | 0.011 | 0.411 | |
| CYP3A4 | 0.043 | 0.016 | 0.0 | 0.006 | 0.348 | |
| Excretion | Clearance | 10.973 | 7.208 | 0.373 | 5.857 | 8.284 |
| Toxicity | hERG | 0.018 | 0.034 | 0.04 | 0.0051 | 0.099 |
| AMES | 0.183 | 0.009 | 0.0099 | 0.875 | 0.657 |
| Protein–Ligand Complexes | Types of Binding Free Energies | |||||
|---|---|---|---|---|---|---|
| Values and Standard Deviations | Van der Waal’s Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy | Binding Energy | |
| α-Glucosidase- caffeic acid | Values (KJ/mol) | −76.593 | −28.312 | 56.039 | −7.771 | −56.637 |
| Standard deviation (KJ/mol) | +/−86.888 | +/−37.350 | +/−63.731 | +/−8.772 | +/−72.011 | |
| α-Glucosidase- syringic acid | Values (KJ/mol) | −170.549 | −17.803 | 50.133 | −13.855 | −152.074 |
| Standard deviation (KJ/mol) | +/−85.257 | +/−10.149 | +/−31.329 | +/−7.492 | +/−78.335 | |
| α-Glucosidase- acarbose | Values (KJ/mol) | −131.001 | −6.710 | 58.293 | −10.168 | −89.586 |
| Standard deviation (KJ/mol) | +/−177.536 | +/−9.451 | +/−61.431 | +/−13.817 | +/−158.089 | |
| α-Amylase- caffeic acid | Values (KJ/mol) | −29.394 | −0.791 | 4.329 | −2.644 | −26.918 |
| Standard deviation (KJ/mol) | +/−66.039 | +/−4.572 | +/−34.555 | +/−6.106 | +/−57.630 | |
| α-Amylase- syringic acid | Values (KJ/mol) | −109.781 | −33.898 | 52.824 | −9.420 | −100.275 |
| Standard deviation (KJ/mol) | +/−102.373 | +/−34.252 | +/−86.454 | +/−8.557 | +/−83.569 | |
| α-Amylase- acarbose | Values (KJ/mol) | −122.109 | −30.198 | 42.314 | −10.521 | −90.275 |
| Standard deviation (KJ/mol) | +/−102.373 | +/−24.152 | +/−56.245 | +/−7.522 | +/−63.569 | |
| Human aldose reductase-caffeic acid | Values (KJ/mol) | −84.938 | −21.601 | 37.285 | −7.192 | −76.445 |
| Standard deviation (KJ/mol) | +/−62.813 | +/−25.688 | +/−32.088 | +/−5.183 | +/−62.792 | |
| Human aldose reductase-syringic acid | Values (KJ/mol) | −149.669 | −6.992 | 79.945 | −12.899 | −109.615 |
| Standard deviation (KJ/mol) | +/−101.479 | +/−11.374 | +/−50.793 | +/−7.329 | +/−73.901 | |
| Human aldose reductase-quercetin | Values (KJ/mol) | −159.669 | −30.870 | 82.920 | −13.796 | −100.299 |
| Standard deviation (KJ/mol) | +/−100.389 | +/−26.576 | +/−72.468 | +/−11.937 | +/−98.464 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations. Molecules 2022, 27, 1888. https://doi.org/10.3390/molecules27061888
Maradesha T, Patil SM, Al-Mutairi KA, Ramu R, Madhunapantula SV, Alqadi T. Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations. Molecules. 2022; 27(6):1888. https://doi.org/10.3390/molecules27061888
Chicago/Turabian StyleMaradesha, Tejaswini, Shashank M. Patil, Khalid Awadh Al-Mutairi, Ramith Ramu, SubbaRao V. Madhunapantula, and Taha Alqadi. 2022. "Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations" Molecules 27, no. 6: 1888. https://doi.org/10.3390/molecules27061888
APA StyleMaradesha, T., Patil, S. M., Al-Mutairi, K. A., Ramu, R., Madhunapantula, S. V., & Alqadi, T. (2022). Inhibitory Effect of Polyphenols from the Whole Green Jackfruit Flour against α-Glucosidase, α-Amylase, Aldose Reductase and Glycation at Multiple Stages and Their Interaction: Inhibition Kinetics and Molecular Simulations. Molecules, 27(6), 1888. https://doi.org/10.3390/molecules27061888