
Copper-Free Halodediazoniation of Arenediazonium Tetrafluoroborates in Deep Eutectic Solvents-like Mixtures

 ,
,  ,
,  ,
, 

Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Abbott, A.P.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K. Deep Eutectic Solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Marcus, Y. Deep Eutectic Solvents; Springer International Publishing: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2020, 121, 1232–1285. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, S.; Tailor, Y.K.; Kumar, M. Deep eutectic solvents (DESs) as eco-friendly and sustainable solvent/catalyst systems in organic transformations. J. Mol. Liq. 2016, 215, 345–386. [Google Scholar] [CrossRef]
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramón, D.J. Deep Eutectic Solvents: The Organic Reaction Medium of the Century. Eur. J. Org. Chem. 2016, 2016, 612–632. [Google Scholar] [CrossRef] [Green Version]
- Hooshmand, S.E.; Afshari, R.; Ramón, D.J.; Varma, R.S. Deep eutectic solvents: Cutting-edge applications in cross-coupling reactions. Green Chem. 2020, 22, 3668–3692. [Google Scholar] [CrossRef]
- Kamble, S.S.; Shankarling, G.S. Room temperature diazotization and coupling reaction using a DES-ethanol system: A green approach towards the synthesis of monoazo pigments. Chem. Commun. 2019, 55, 5970–5973. [Google Scholar] [CrossRef]
- Lenne, Q.; Andrieux, V.; Levanen, G.; Bergamini, J.F.; Nicolas, P.; Paquin, L.; Lagrost, C.; Leroux, Y.R. Electrochemical grafting of aryl diazonium salts in deep eutectic solvents. Electrochim. Acta 2021, 369, 137672. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z. Synthesis of Diarylethynes from Aryldiazonium Salts by Using Calcium Carbide as an Alkyne Source in a Deep Eutectic Solvent. Synlett 2021, 32, 631–635. [Google Scholar] [CrossRef]
- Antenucci, A.; Bonomo, M.; Ghigo, G.; Gontrani, L.; Barolo, C.; Dughera, S. How do arenediazonium salts behave in deep eutectic solvents? A combined experimental and computational approach. J. Mol. Liq. 2021, 339, 116743. [Google Scholar] [CrossRef]
- Saunders, K.H.; Allen, R.L.M. Aromatic Diazo Compounds; Edward Arnold: London, UK, 1985. [Google Scholar]
- Zollinger, H. Diazochemistry: Aromatic and Heteroaromatic Compounds; Wiley-VCH: Weinheim, Germany, 1994. [Google Scholar]
- Schareina, T.; Bellen, M. Copper-Catalyzed Cyanations of Aryl Halides and Related Compounds. In Copper-Mediated Cross-Coupling Reactions; Evano, G., Blanchard, N., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 313–334. [Google Scholar]
- Sandmeyer, T. Ueber die Ersetzung der Amid-gruppe durch Chlor, Brom und Cyan in den aromatischen Substanzen. Ber. Dtsch. Chem. Ges. 1884, 17, 2650–2653. [Google Scholar] [CrossRef] [Green Version]
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A new, one-step, effective protocol for the iodination of aromatic and heterocyclic compounds via aprotic diazotization of amines. Synthesis 2007, 2007, 81–84. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Sigeev, A.S.; Peregudov, A.S.; Petrovskii, P.V. Catalytic Sandmeyer bromination. Synthesis 2007, 2007, 2534–2538. [Google Scholar] [CrossRef]
- Hubbard, A.; Okazaki, T.; Laali, K.K. Halo- and azidodediazoniation of arenediazonium tetrafluoroborates with trimethylsilyl halides and trimethylsilyl azide and Sandmeyer-type bromodediazoniation with Cu(I)Br in [BMIM][PF6] ionic liquid. J. Org. Chem. 2008, 73, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, V.D.; Semenischeva, N.I.; Krasnokutskaya, E.A.; Tretyakov, A.N.; Ho, Y.H.; Chi, K.W. Sulfonic acid based cation-exchange resin: A novel proton source for one-pot diazotization-iodination of aromatic amines in water. Synthesis 2008, 2008, 185–187. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef]
- Ayogu, J.I.; Onoabedje, E.A. Recent advances in transition metal-catalysed cross-coupling of (hetero)aryl halides and analogues under ligand-free conditions. Catal. Sci. Technol. 2019, 9, 5233–5255. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Cabrita, M.T.; Vale, C.; Rauter, A.P. Halogenated compounds from marine algae. Mar. Drugs 2010, 8, 2301–2317. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef]
- Mo, F.; Qiu, D.; Zhang, Y.; Wang, J. Renaissance of Sandmeyer-Type Reactions: Conversion of Aromatic C-N Bonds into C-X Bonds (X = B, Sn, P, or CF3). Acc. Chem. Res. 2018, 51, 496–506. [Google Scholar] [CrossRef]
- Mo, F.; Qiu, D.; Zhang, L.; Wang, J. Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds. Chem. Rev. 2021, 121, 5741–5829. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.J.; Fang, C.; Xiao, B.; Yi, J.; Xu, J.; Liu, Z.J.; Lu, X.; Liu, L.; Fu, Y. Copper-promoted sandmeyer trifluoromethylation reaction. J. Am. Chem. Soc. 2013, 135, 8436–8439. [Google Scholar] [CrossRef] [PubMed]
- Danoun, G.; Bayarmagnai, B.; Grünberg, M.F.; Matheis, C.; Risto, E.; Gooßen, L.J. Sandmeyer trifluoromethylation. Synthesis 2014, 46, 2283–2286. [Google Scholar] [CrossRef]
- Zhao, C.J.; Xue, D.; Jia, Z.H.; Wang, C.; Xiao, J. Methanol-promoted borylation of arylamines: A simple and green synthetic method to arylboronic acids and arylboronates. Synlett 2014, 25, 1577–1584. [Google Scholar] [CrossRef]
- Qi, X.; Li, H.P.; Peng, J.B.; Wu, X.F. Borylation of aryldiazonium salts at room temperature in an aqueous solution under catalyst-free conditions. Tetrahedron Lett. 2017, 58, 3851–3853. [Google Scholar] [CrossRef]
- Qiu, D.; Wang, S.; Tang, S.; Meng, H.; Jin, L.; Mo, F.; Zhang, Y.; Wang, J. Synthesis of trimethylstannyl arylboronate compounds by sandmeyer-type transformations and their applications in chemoselective cross-coupling reactions. J. Org. Chem. 2014, 79, 1979–1988. [Google Scholar] [CrossRef]
- Zhong, T.; Pang, M.K.; Chen, Z.D.; Zhang, B.; Weng, J.; Lu, G. Copper-free Sandmeyer-type Reaction for the Synthesis of Sulfonyl Fluorides. Org. Lett. 2020, 22, 3072–3078. [Google Scholar] [CrossRef]
- Li, Y.; Pu, J.; Jiang, X. A highly efficient Cu-catalyzed S-transfer reaction: From amine to sulfide. Org. Lett. 2014, 16, 2692–2695. [Google Scholar] [CrossRef]
- Matheis, C.; Bayarmagnai, B.; Jouvin, K.; Goossen, L.J. Convenient synthesis of pentafluoroethyl thioethers: Via catalytic Sandmeyer reaction with a stable fluoroalkylthiolation reagent. Org. Chem. Front. 2016, 3, 949–952. [Google Scholar] [CrossRef] [Green Version]
- Koziakov, D.; Majek, M.; Jacobi Von Wangelin, A. Metal-free radical thiolations mediated by very weak bases. Org. Biomol. Chem. 2016, 14, 11347–11352. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Qiu, D.; Mo, F.; Zhang, Y.; Wang, J. Metal-Free Aromatic Carbon-Phosphorus Bond Formation via a Sandmeyer-Type Reaction. J. Org. Chem. 2016, 81, 11603–11611. [Google Scholar] [CrossRef] [PubMed]
- Estruch-Blasco, M.; Felipe-Blanco, D.; Bosque, I.; Gonzalez-Gomez, J.C. Radical arylation of triphenyl phosphite catalyzed by salicylic acid: Mechanistic investigations and synthetic applications. J. Org. Chem. 2020, 85, 14473–14485. [Google Scholar] [CrossRef] [PubMed]
- Kutonova, K.V.; Trusova, M.E.; Postnikov, P.S.; Filimonov, V.D. The first example of the copper-free chloro- and hydrodediazoniation of aromatic amines using sodium nitrite, CCl4, and CHCl3. Russ. Chem. Bull. 2012, 61, 206–208. [Google Scholar] [CrossRef]
- Leas, D.A.; Dong, Y.; Vennerstrom, J.L.; Stack, D.E. One-Pot, Metal-Free Conversion of Anilines to Aryl Bromides and Iodides. Org. Lett. 2017, 19, 2518–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Batra, S. Direct Transformation of Arylamines to Aryl Halides via Sodium Nitrite and N-Halosuccinimide. Chem. Eur. J. 2018, 24, 14622–14626. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, B.; Liu, Z.; Kao, Y.; Dong, B.W.; Jiang, S.D.; Li, F.; Liu, G.; Yang, Y.; Mo, F. A general electrochemical strategy for the Sandmeyer reaction. Chem. Sci. 2018, 9, 8731–8737. [Google Scholar] [CrossRef] [Green Version]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwang, H.Y.; Kim, H.; Chi, K.W. Unusually stable, versatile, and pure arenediazonium tosylates: Their preparation, structures, and synthetic applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef]
- Chromá, R.; Vilková, M.; Shepa, I.; Makoś-Chełstowska, P.; Andruch, V. Investigation of tetrabutylammonium bromide-glycerol-based deep eutectic solvents and their mixtures with water by spectroscopic techniques. J. Mol. Liq. 2021, 330, 115617. [Google Scholar] [CrossRef]
- Claudy, P.; Commerçon, J.C.; Lètoffé, J.M. Quasi-static study of the glass transition of glycerol by DSC. Thermochim. Acta 1988, 128, 251–260. [Google Scholar] [CrossRef]
- Lane, L.B. Freezing Points of Glycerol and Its Aqueous Solutions. Ind. Eng. Chem. 1925, 17, 924. [Google Scholar] [CrossRef]
- Abbott, A.P.; D’Agostino, C.; Davis, S.J.; Gladden, L.F.; Mantle, M.D. Do group 1 metal salts form deep eutectic solvents? Phys. Chem. Chem. Phys. 2016, 18, 25528–25537. [Google Scholar] [CrossRef] [PubMed]
- Littke, A.F.; Fu, G.C. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry and Solution Manual, 2nd ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry, 6th ed.; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry—Part A; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Bergstrom, R.G.; Landells, R.G.M.; Wahl, G.H.; Zollinger, H. Dediazoniation of arenediazonium ions in homogeneous solution. 7. Intermediacy of the phenyl cation. J. Am. Chem. Soc. 1976, 98, 3301–3305. [Google Scholar] [CrossRef]
- Himeshima, Y.; Kobayashi, H.; Sonoda, T. A first example of generating aryl cations in the solvolysis of aryl triflates in trifluoroethanol. J. Am. Chem. Soc. 1985, 107, 5286–5288. [Google Scholar] [CrossRef]
- Martínez, A.G.; Cerero, S.d.; Barcina, J.O.; Jiménez, F.M.; Maroto, B.L. The mechanism of hydrolysis of aryldiazonium ions revisited: Marcus theory vs. canonical Variational transition state theory. Eur. J. Org. Chem. 2013, 2013, 6098–6107. [Google Scholar] [CrossRef]
- Wu, Z.; Glaser, R. Ab Initio Study of the SN1Ar and SN2Ar Reactions of Benzenediazonium Ion with Water. On the Conception of “Unimolecular Dediazoniation” in Solvolysis Reactions. J. Am. Chem. Soc. 2004, 126, 10632–10639. [Google Scholar] [CrossRef]
- Laidler, K.J.; King, M.C. The development of transition-state theory. J. Phys. Chem. 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Swain, C.G.; Sheats, J.E.; Gorenstein, D.G.; Harbison, K.G. Aromatic hydrogen isotope effects in reactions of benzenediazonium salts. J. Am. Chem. Soc. 1975, 97, 791–795. [Google Scholar] [CrossRef]
- Swain, C.G.; Rogers, R.J. Mechanism of formation of aryl fluorides from arenediazonium fluoborates. J. Am. Chem. Soc. 1975, 97, 799–800. [Google Scholar] [CrossRef]
- Roe, A. Preparation of Aromatic Fluorine Compounds from Diazonium Fluoborates. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 193–228. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. In Horizons Quantum Chem; Springer: Berlin/Heidelberg, Germany, 1980; pp. 5–15. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–224. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Foresman, J.; Frisch, A. Exploring Chemistry with Electronic Structure Methods; Gaussian Inc.: Pittsburgh, PA, USA, 1996; Available online: http://gaussian.com/expchem3/ (accessed on 4 June 2021).
- Ribeiro, R.F.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B. 2011, 115, 14556–14562. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, D.J.; Trucks, M.J.; Schlegel, G.W.; Scuseria, H.B.; Robb, G.E.; Cheeseman, M.A.; Scalmani, R.J.; Barone, V.G.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre-and post-processing program for molecular and electronic structures. J. Comput. Aided. Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Zarei, M.; Noroozizadeh, E.; Moosavi-Zare, A.R.; Zolfigol, M.A. Synthesis of Nitroolefins and Nitroarenes under Mild Conditions. J. Org. Chem. 2018, 83, 3645–3650. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, F.; Liu, C.; Zeng, H.; Yang, Z.; Wu, W.; Jiang, H. Copper-Catalyzed Unstrained C–C Single Bond Cleavage of Acyclic Oxime Acetates Using Air: An Internal Oxidant-Triggered Strategy toward Nitriles and Ketones. J Org. Chem. 2018, 83, 14713–14722. [Google Scholar] [CrossRef]
- Xin, H.-L.; Pang, B.; Choi, J.; Akkad, W.; Morimoto, H.; Ohshima, T. C–C Bond Cleavage of Unactivated 2-Acylimidazoles. J. Org. Chem. 2020, 85, 11592–11606. [Google Scholar] [CrossRef]
- Li, T.; Cui, X.; Sun, L.; Li, C. Economical and efficient aqueous reductions of high melting-point imines and nitroarenes to amines: Promotion effects of granular PTFE. RSC Adv. 2014, 4, 33599–33606. [Google Scholar] [CrossRef]
- Shao, H.; Foley, D.W.; Huang, S.; Abbas, A.Y.; Lam, F.; Gershkovich, P.; Bradshaw, T.D.; Pepper, C.; Fischer, P.M.; Wang, S. Structure-based design of highly selective 2,4,5-trisubstituted pyrimidine CDK9 inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2021, 214, 113244. [Google Scholar] [CrossRef] [PubMed]
- Lerch, U.; Moffatt, J.G. Carbodiimide-Sulfoxide Reactions. XIII. Reactions of Amines and Hydrazine Derivatives. J. Org. Chem. 1971, 36, 3861–3869. [Google Scholar]
- Baudet, H.P. The replaceability of the halogen atom in 1-chloro- and 1-bromo-2-cyano-4-nitrobenzene. Recl. Trav. Chim. Pays-Bas 1924, 43, 707–726. [Google Scholar] [CrossRef]
- Gianni, J.; Pirovano, V.; Abbiati, G. Silver triflate/p-TSA co-catalysed synthesis of 3-substituted isocoumarins from 2-alkynylbenzoates. Org. Biomol. Chem. 2018, 16, 3213–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, J.; Ren, J.; Zeng, B.-B. Oxidation of aromatic amines into nitroarenes with m-CPBA. Tetrahedron Lett. 2014, 55, 1581–1584. [Google Scholar] [CrossRef]
- Ramananda, D.; Uchil, J. 15Cl NQR study of charge-transfer complexes. J. Mol. Struct. 1994, 319, 193–196. [Google Scholar] [CrossRef]
- Wilshire, J.F.K. The preaparation of 2-Fluoro-5-nitrobenzonitrile and the proton magnetic resonance spectra of some compounds containing the N-(2-Cyano-4-nitrophenyl)group. Aust. J. Chem. 1967, 20, 1663–1670. [Google Scholar] [CrossRef]
- Sloan, N.; Luthra, S.K.; McRobbie, G.; Pimlott, S.L.; Sutherland, A. A one-pot radioiodination of aryl amines via stable diazonium salts: Preparation of 125I-imaging agents. Chem. Commun. 2017, 53, 11008–11011. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ansari, A.; Sashidhara, K.V. Base promoted peroxide systems for the efficient synthesis of nitroarenes and benzamides. Tetrahedron Lett. 2019, 60, 151076. [Google Scholar] [CrossRef]
- Fu, Z.; Li, Z.; Song, Y.; Yang, R.; Liu, Y.; Cai, H. Decarboxylative Halogenation and Cyanation of Electron-Deficient Aryl Carboxylic Acids via Cu Mediator as Well as Electron-Rich Ones through Pd Catalyst under Aerobic Conditions. J. Org. Chem. 2016, 81, 2794–2803. [Google Scholar] [CrossRef] [PubMed]

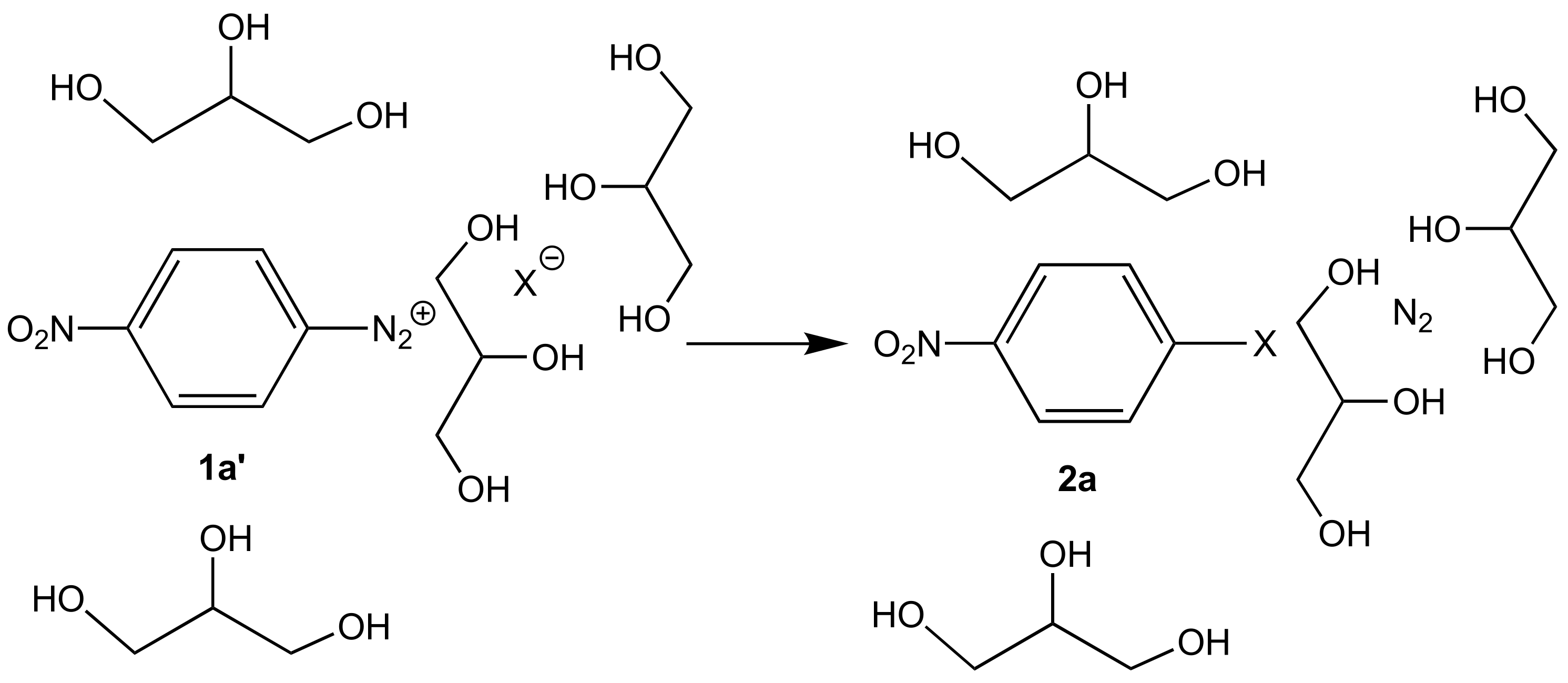
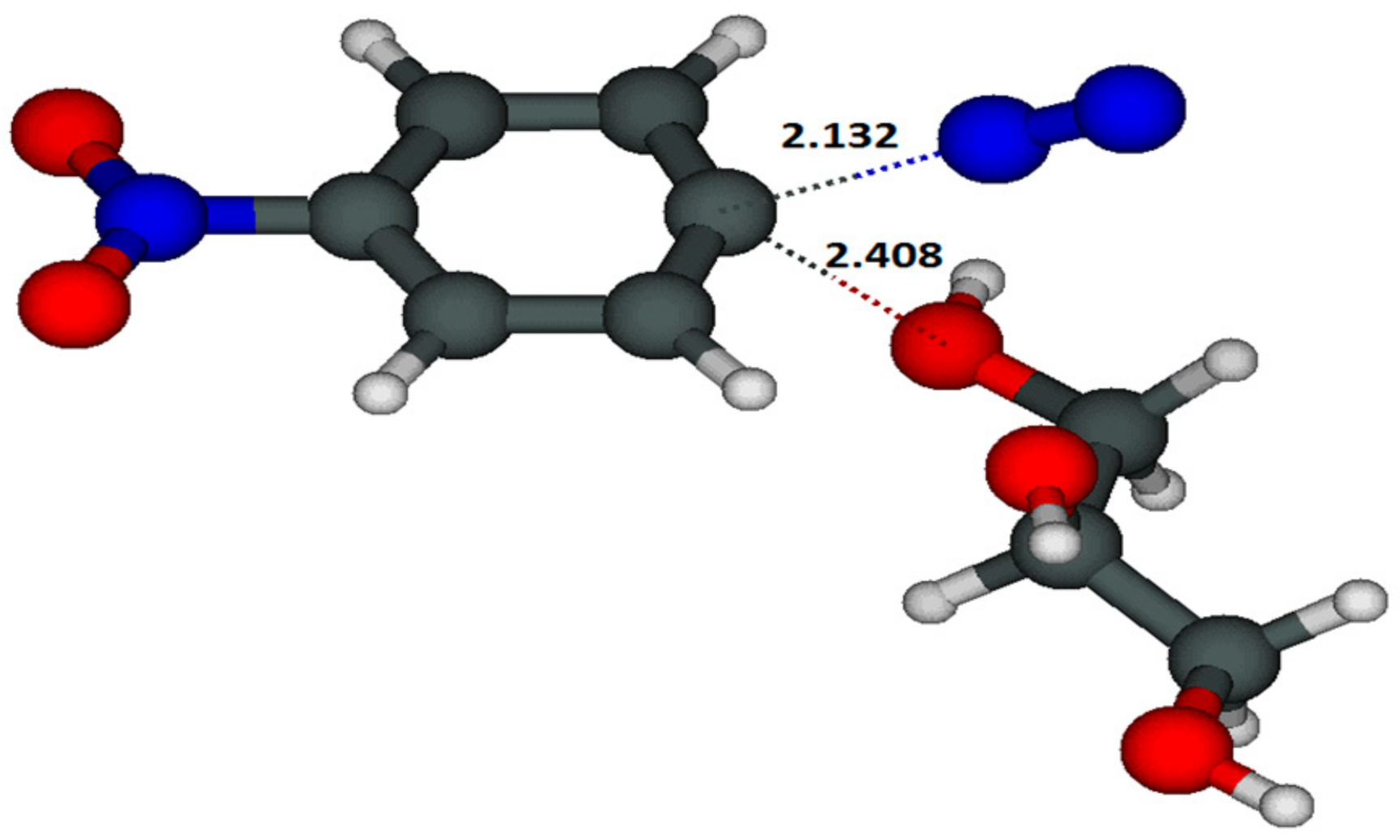

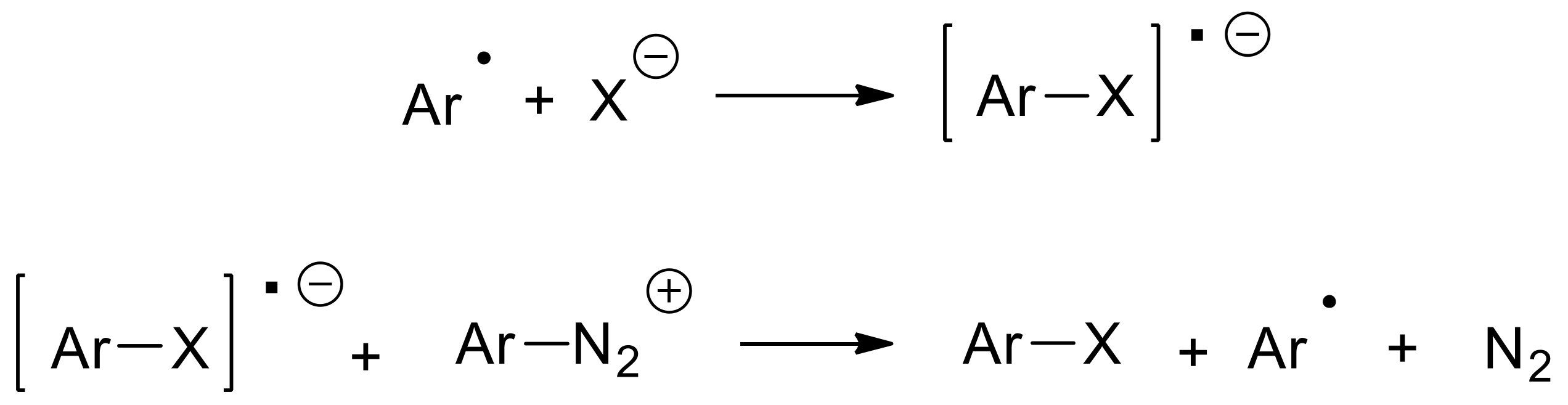

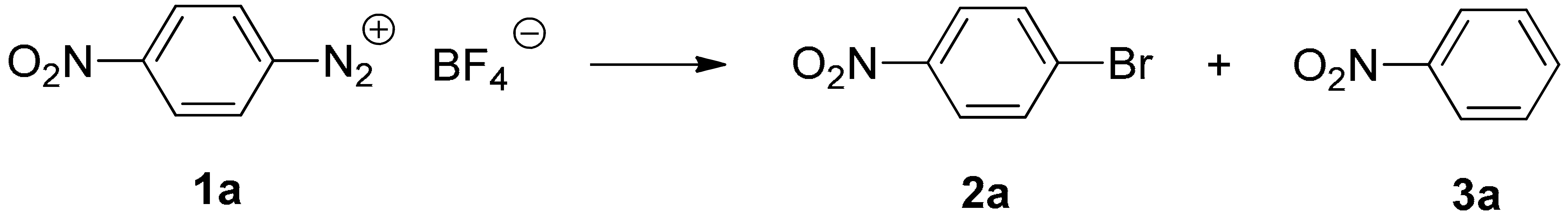
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | HBD | HBA | HBD/HBA Ratio | Melting Temperature/K |
|---|---|---|---|---|
| Gly/KBr 4:1 | Glycerol | KBr | 4:1 | 238 |
| Gly/KBr 6:1 | Glycerol | KBr | 6:1 | 225 |
| Gly/KBr 10:1 | Glycerol | KBr | 10:1 | 247 |
| Gly/KCl 6:1 | Glycerol | KCl | 6:1 | 239 |
| Gly/KI 6:1 | Glycerol | KI | 6:1 | 251 |
| Gly/TBABr 6:1 | Glycerol | TBABr | 6:1 | 245 |
| Gly/TBACl 6:1 | Glycerol | TBACl | 6:1 | 241 |
| Glycerol 1 | Glycerol | Glycerol | - | 291 1 |
| Glycerol 2 | Glycerol | Glycerol | - | 269 2 |
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Amount | Time (h) | Yields of 2a 1 (%) |
| 1 | Gly and KBr 2 | 2.5 mL | 24 | - 3 |
| 2 | Gly and KBr 4 | 2.5 mL | 24 | - 3 |
| 3 | EtOH and KBr 2 | 2.5 mL | 24 | - 3 |
| 4 | DMSO and KBr 2 | 2.5 mL | 24 | - 3 |
| 5 | MeCN and KBr 2 | 2.5 mL | 24 | - 3 |
| 6 | Gly/KBr 4:1 5 | 2.5 mL | 24 | Traces 3 |
| 7 | Gly/KBr 6:1 5 | 2.5 mL | 24 | Traces 3 |
| 8 | Gly/KBr 10:1 5 | 2.5 mL | 24 | Traces 3 |
| 9 | Gly/KBr 4:1 5 | 3.5 mL | 12 | 34 6 |
| 10 | Gly/KBr 6:1 5 | 3.5 mL | 12 | 32 6 |
| 11 | Gly/KBr 10:1 5 | 3.5 mL | 12 | 29 6 |
| 12 | Gly/KBr 6:1 5 | 5 mL | 4 | 72 6 |
| 13 | Gly/KBr 4:1 5 | 5 mL | 4 | 62 6 |
| 14 | Gly/KBr 6:1 5 | 10 mL | 4 | 72 6,7,8 |
| 15 | Gly/KBr 4:1 5 | 10 mL | 4 | 70 6 |
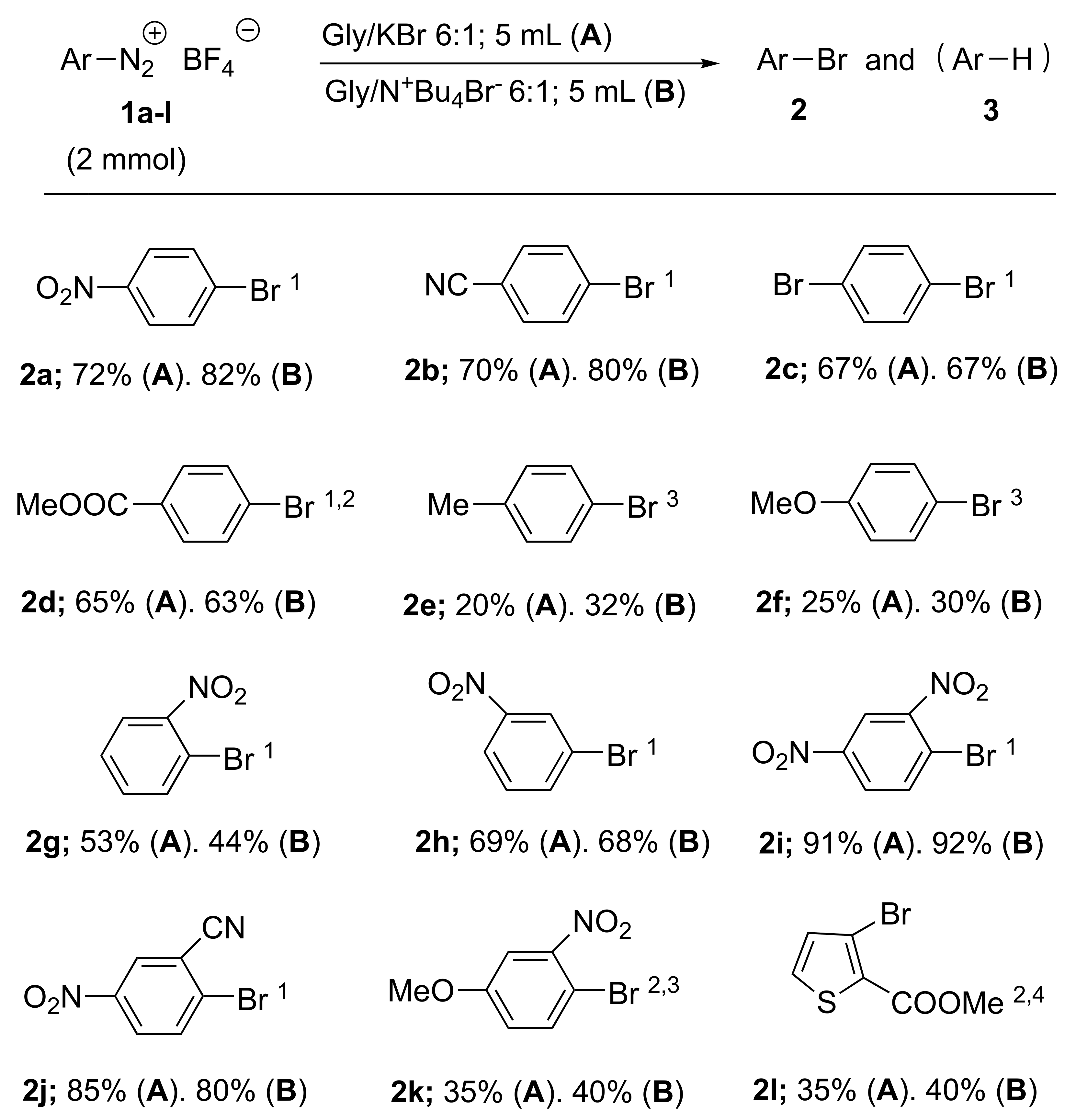
| Entry | Salt | Estimated Yield (%) of 2 or 4 | Estimated Yield (%) of 3 | Yields (%) of Isolated 2a | Method |
|---|---|---|---|---|---|
| 1 | 1a | 2a; 75 | 3a; 24 1 | 2a; 72 | A |
| 2 | 1a | 2a; 88 | 3a; 10 | 2a; 82 | B |
| 3 | 1b | 2b; 78 | 3b; 22 | 2b; 70 | A |
| 4 | 1b | 2b; 82 | 3b; 17 | 2b; 80 | B |
| 5 | 1c | 2c; 65 | 3c; 34 | 2c; 67 | A |
| 6 | 1c | 2c; 71 | 3c; 28 | 2c; 80 | B |
| 7 | 1d | 2d; 69 | 3d; 30 | 2d; 65 | A |
| 8 | 1d | 2d; 67 | 3d; 33 2 | 2d; 63 | B |
| 9 | 1e | 2e; 28 | 3e; 72 | 2e; 20 | A |
| 11 | 1f | 2f; 32 | 3f; 68 | 2f; 25 | A |
| 12 | 1f | 2f; 36 | 3f; 63 | 2f; 30 | B |
| 13 | 1g | 2g; 60 | 3g; 38 | 2g; 53 | A |
| 14 | 1g | 2g; 36 | 3g; 63 | 2g; 30 | B |
| 15 | 1h | 2h; 74 | 3h; 25 | 2h; 69 | A |
| 16 | 1h | 2h; 77 | 3h; 22 | 2h; 68 | B |
| 17 | 1i | 2i; 98 | 3i; traces | 2i; 91 | A |
| 18 | 1i | 2i; 98 | 3i; traces | 2i; 92 | B |
| 19 | 1j | 2j; 94 | 3j; traces | 2j; 85 | A |
| 20 | 1j | 2j; 93 | 3j; traces | 2j; 80 | B |
| 21 | 1k | 2k; 37 | 3k; 60 | 2k; 35 | A |
| 22 | 1k | 2k; 42 | 3k; 55 | 2k; 40 | B |
| 23 | 1l | 2l; 39 | 3l; 58 3 | 2l; 35 | A |
| 24 | 1l | 2l; 44 | 3l; 56 | 2l; 40 | B |
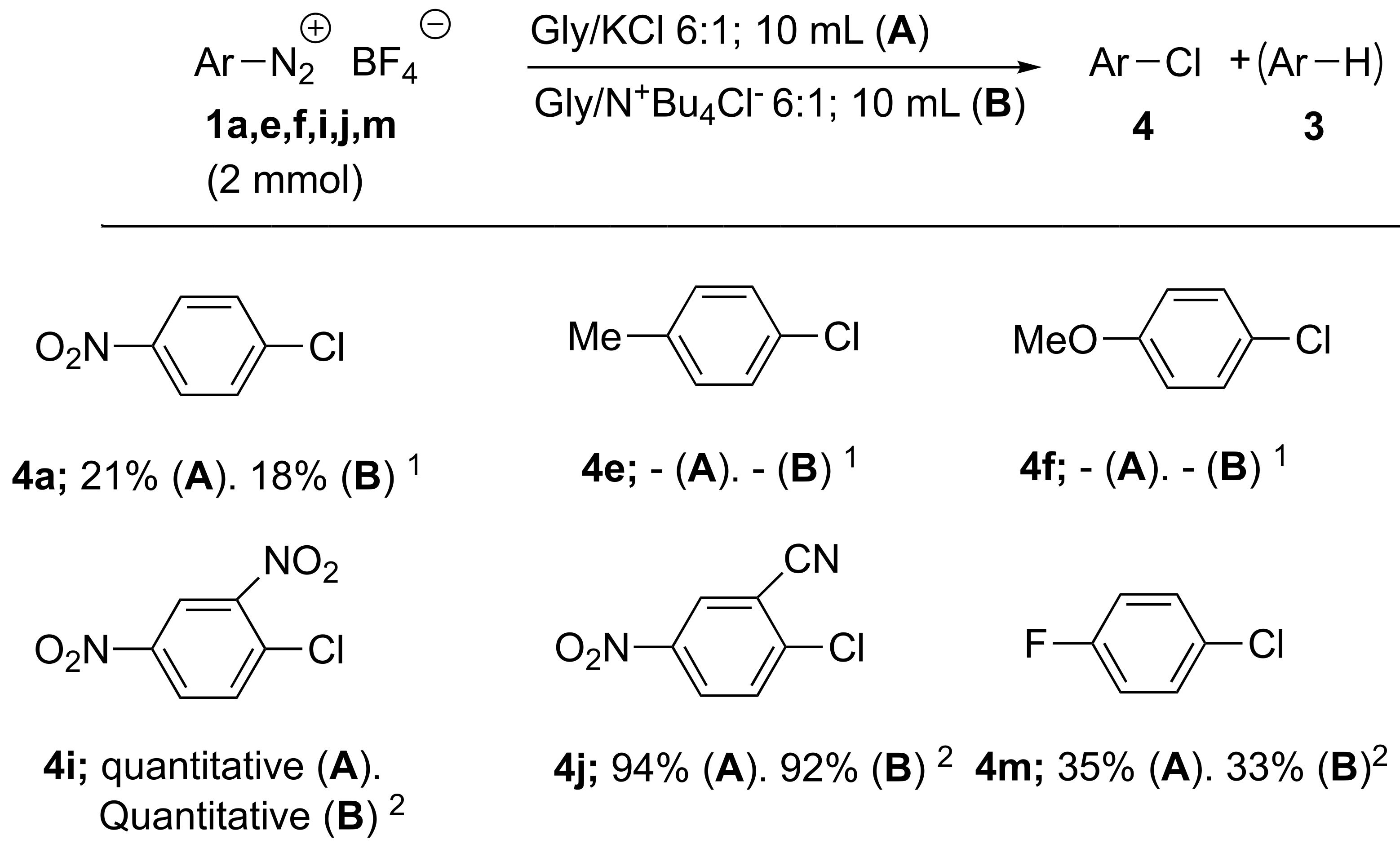
| 25 | 1a | 4a; 27 | 3a; 65 | 4a; 21 | A |
| 26 | 1a | 4a; 25 | 3a; 67 | 4a; 18 | B |
| 27 | 1e | 4e; - | 3e; 74 | 4e; - | A |
| 28 | 1e | 4e; - | 3e; 76 | 4e; - | B |
| 29 | 1f | 4f; - | 3f; 81 | 4f; - | A |
| 30 | 1f | 4f; - | 3f; 81 | 4f; - | B |
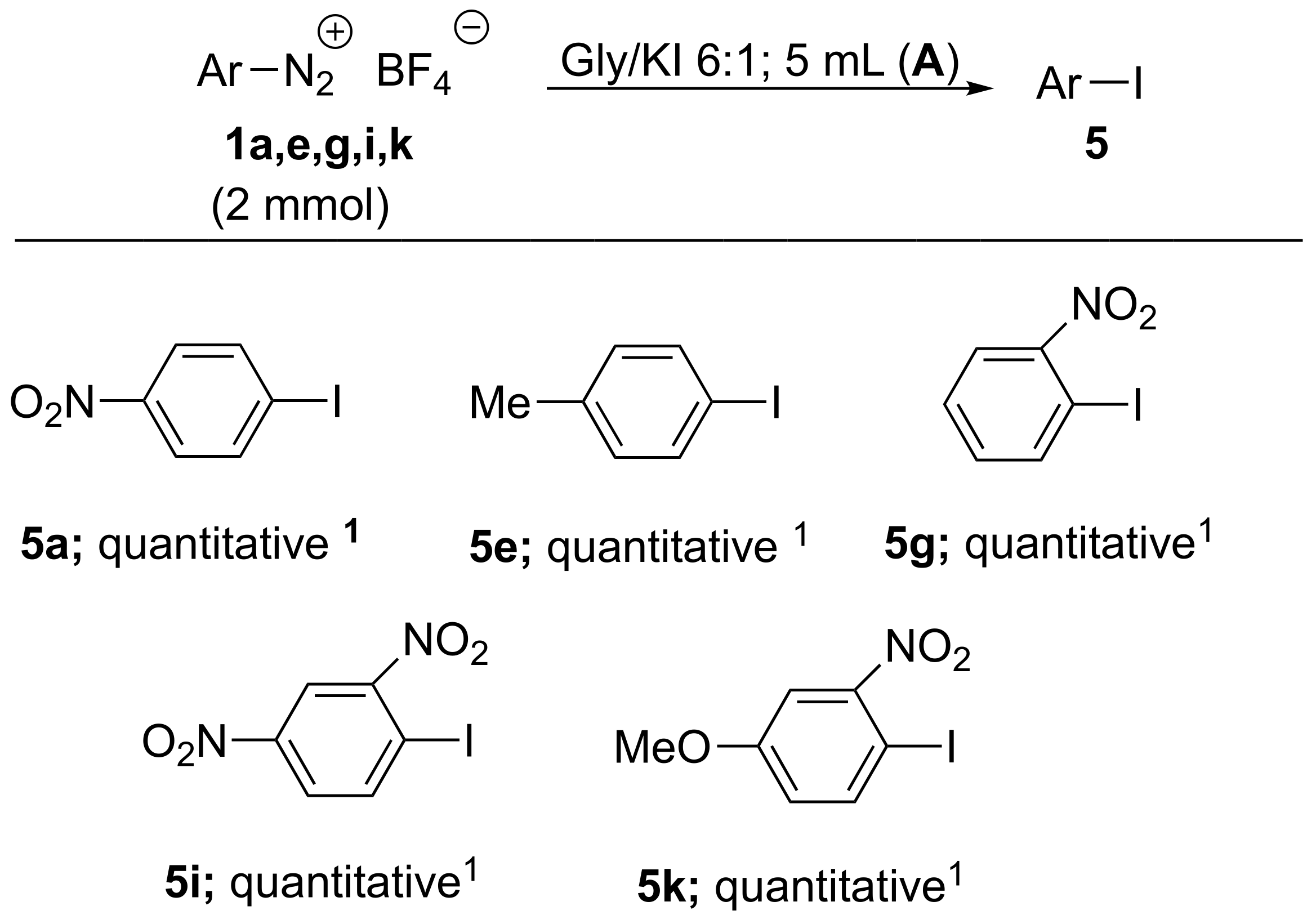
| 31 | 1i | 4i; 100 | 3i; - | 4i; 100 | A |
| 32 | 1i | 4i; 100 | 3i; - | 4i; 100 | B |
| 33 | 1j | 4j; 100 | 3j; - | 4j; 94 | A |
| 34 | 1j | 4j; 100 | 3j;- | 4j; 92 | B |
| 35 | 1m | 4m; 42 | 3m; 55 | 4m; 35 | A |
| 36 | 1m | 4m; 43 | 3m; 53 | 4m; 33 | B |
| Entry | Time (h) | Yield of 2a (%) 1,2 | Recovery of Solvent |
|---|---|---|---|
| 1 | 4 | 72 | 4.9 mL 3 |
| 2 | 4 | 70 | 4.9 mL 4 |
| 3 | 4 | 67 | 4.7 mL 5 |
| 4 | 6 | 70 | 4.6 mL 6 |
| 5 | 6 | 68 | 4.5 mL |
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Amount | Time (h) | Yields of 4i 1 (%) |
| 1 | Gly and KCl 2 | 2.5 mL | 24 | - 3 |
| 2 | Gly and KCl 4 | 2.5 mL | 24 | - 3 |
| 3 | EtOH and KCl 2 | 2.5 mL | 24 | - 3 |
| 4 | DMSO and KCl 2 | 2.5 mL | 24 | - 3 |
| 5 | MeCN and KCl 2 | 2.5 mL | 24 | - 3 |
| 6 | Gly/KCl 4:1 5 | 5 mL | 12 | 65 6,7 |
| 7 | Gly/KCl 6:1 5 | 5 mL | 12 | 63 6,7 |
| 8 | Gly/KCl 10:1 5 | 5 mL | 12 | 49 6.7 |
| 9 | Gly/KCl 6:1 5 | 5 mL | 4 | 87 6,7 |
| 10 | Gly/KCl 6:1 5 | 10 mL | 4 | 100 6 |
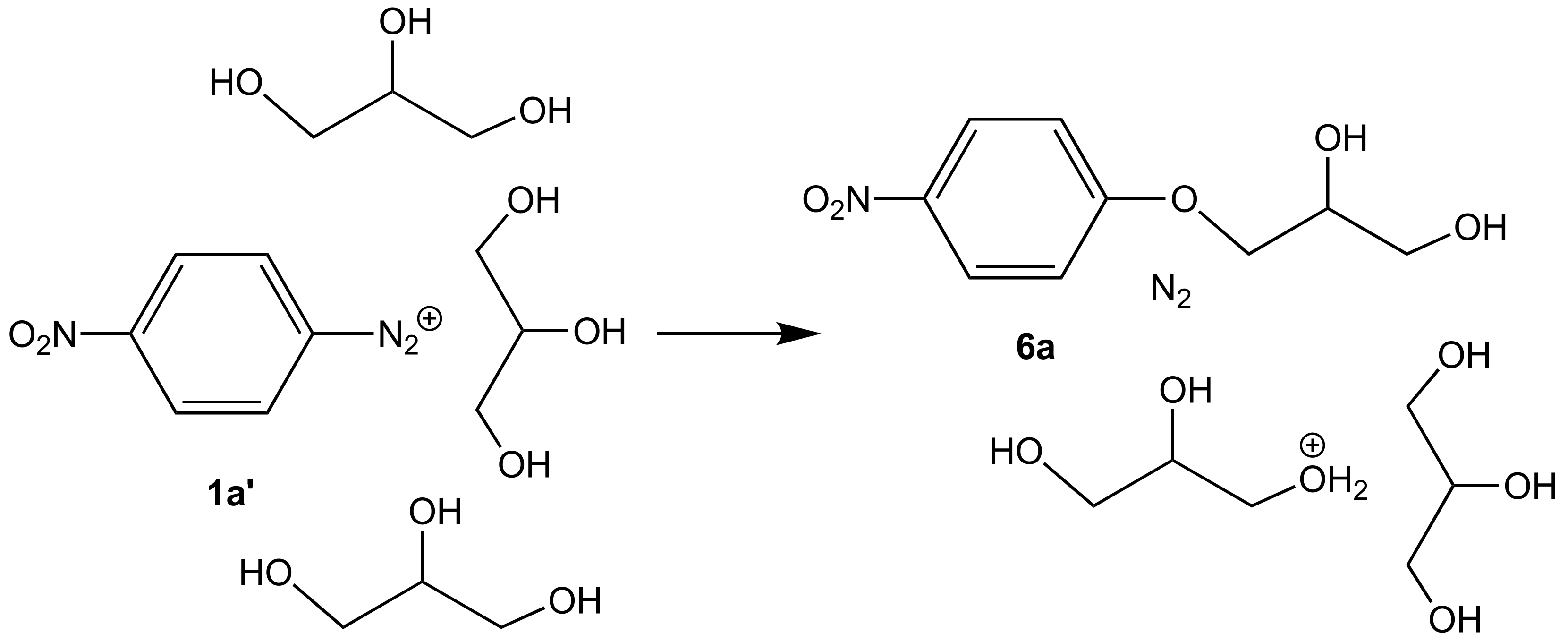
| 1a’ | 1f’ | |||
|---|---|---|---|---|
| K1 | τ | K1 | τ | |
| Fluorination | 2.3 × 101 | 0.002 s | 1.2 × 10−5 | 1.1 h |
| Chlorination | 1.2 × 10−1 | 0.4 s | 2.7 × 10−5 | 0.5 h |
| Bromination | 3.6 × 10−2 | 1.2 s | 1.0 × 10−5 | 1.1 h |
| Iodination | 4.6 × 10−1 | 0.1 s | - 2 | - 2 |
| Solvolysis | 4.1 × 10−6 | 2.8 days | 5.3 × 10−10 | 60 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghigo, G.; Bonomo, M.; Antenucci, A.; Reviglio, C.; Dughera, S. Copper-Free Halodediazoniation of Arenediazonium Tetrafluoroborates in Deep Eutectic Solvents-like Mixtures. Molecules 2022, 27, 1909. https://doi.org/10.3390/molecules27061909
Ghigo G, Bonomo M, Antenucci A, Reviglio C, Dughera S. Copper-Free Halodediazoniation of Arenediazonium Tetrafluoroborates in Deep Eutectic Solvents-like Mixtures. Molecules. 2022; 27(6):1909. https://doi.org/10.3390/molecules27061909
Chicago/Turabian StyleGhigo, Giovanni, Matteo Bonomo, Achille Antenucci, Chiara Reviglio, and Stefano Dughera. 2022. "Copper-Free Halodediazoniation of Arenediazonium Tetrafluoroborates in Deep Eutectic Solvents-like Mixtures" Molecules 27, no. 6: 1909. https://doi.org/10.3390/molecules27061909
APA StyleGhigo, G., Bonomo, M., Antenucci, A., Reviglio, C., & Dughera, S. (2022). Copper-Free Halodediazoniation of Arenediazonium Tetrafluoroborates in Deep Eutectic Solvents-like Mixtures. Molecules, 27(6), 1909. https://doi.org/10.3390/molecules27061909