
Preparation and Synthetic Applications of Five-to-Seven-Membered Cyclic α-Diazo Monocarbonyl Compounds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cyclic α-Diazo Ketoness
2.1. Methods of Synthesis of Cyclic α-Diazo Ketones
2.2. The Wolff Rearrangement of Cyclic α-Diazo Ketones
2.3. Decomposition of Cyclic α-Diazo Ketones Followed by β-Migration
2.4. Decomposition of Cyclic α-Diazo Ketones in the Presence of Substrates Containing n-Nucleophiles
2.5. Reactions of Cyclic α-Diazo Ketones Not Involving Nitrogen Elimination
2.6. Other Reactions of Cyclic α-Diazo Ketones
2.7. 6-Diazo Cyclohex-2-Enones as Precursors to Phenols
3. α-Diazo Lactones
3.1. Methods of Synthesis
3.2. Decomposition of Cyclic α-Diazo Lactones Followed by β-Migration
3.3. Decomposition of α-Diazo Lactones in the Presence of Substrates Containing n-Nucleophiles
3.4. Other Reactions of α-Diazo Lactones
4. α-Diazo Lactams
4.1. Methods of Synthesis of α-Diazo Lactams
4.2. Reactions of α-Diazo Lactams
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, T.; McKervey, M.A. Organic Synthesis with a-Diazo Carbonyl Compounds. Chem. Rev. 1994, 94, 1091–1160. [Google Scholar] [CrossRef]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.D.; Grieco, P.A. Intramolecular Reactions of Diazocarbonyl Compounds. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1979; pp. 361–475. ISBN 9780471264187. [Google Scholar]
- Liu, L.; Zhang, J. Gold-Catalyzed Transformations of α-Diazocarbonyl Compounds: Selectivity and Diversity. Chem. Soc. Rev. 2016, 45, 506–516. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.; Dmitrenko, O.; Fox, J.M. Rh-Catalyzed Intermolecular Reactions of Cyclic α-Diazocarbonyl Compounds with Selectivity over Tertiary C–H Bond Migration. J. Am. Chem. Soc. 2012, 134, 11035–11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, R.; Shen, Y.; Liang, Z.; Hua, Y.; Zhang, Y. Total Synthesis of Aplydactone by a Conformationally Controlled C−H Functionalization. Angew. Chem. Int. Ed. 2017, 56, 8187–8190. [Google Scholar] [CrossRef]
- Mascitti, V.; Corey, E.J. Total Synthesis of (±)-Pentacycloanammoxic Acid. J. Am. Chem. Soc. 2004, 126, 15664–15665. [Google Scholar] [CrossRef]
- Wolff, S.; Venepalli, B.R.; George, C.F.; Agosta, W.C. Some Thermal and Photochemical Reactions of [4.4.4.5] Fenestranes. J. Am. Chem. Soc. 1988, 110, 6785–6790. [Google Scholar] [CrossRef]
- Fu, L.Q.; Guo, X.S.; Liu, X.; He, H.L.; Wang, Y.L.; Yang, Y.S. Synthesis and Antibacterial Activity of C-2(S)-Substituted Pleuromutilin Derivatives. Chin. Chem. Lett. 2010, 21, 507–510. [Google Scholar] [CrossRef]
- Thommen, C.; Jana, C.K.; Neuburger, M.; Gademann, K. Syntheses of Taiwaniaquinone F and Taiwaniaquinol A via an Unusual Remote C–H Functionalization. Org. Lett. 2013, 15, 1390–1393. [Google Scholar] [CrossRef]
- Line, N.J.; Witherspoon, B.P.; Hancock, E.N.; Brown, M.K. Synthesis of Ent-[3]-Ladderanol: Development and Application of Intramolecular Chirality Transfer [2+2] Cycloadditions of Allenic Ketones and Alkenes. J. Am. Chem. Soc. 2017, 139, 14392–14395. [Google Scholar] [CrossRef]
- Hu, P.; Snyder, S.A. Enantiospecific Total Synthesis of the Highly Strained (−)-Presilphiperfolan-8-Ol via a Pd-Catalyzed Tandem Cyclization. J. Am. Chem. Soc. 2017, 139, 5007–5010. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Li, R.; Luo, Y.; Li, J.; Zhou, S.; Li, Y.; Hu, J.; Li, A. Divergent Total Synthesis of Taiwaniaquinones A and F and Taiwaniaquinols B and D. Org. Lett. 2013, 15, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Corey, E.J. A Method for the Catalytic Enantioselective Synthesis of 6-Silylated 2-Cyclohexenones. Tetrahedron Lett. 2006, 47, 2319–2321. [Google Scholar] [CrossRef]
- Liffert, R.; Linden, A.; Gademann, K. Total Synthesis of the Sesquiterpenoid Periconianone A Based on a Postulated Biogenesis. J. Am. Chem. Soc. 2017, 139, 16096–16099. [Google Scholar] [CrossRef] [Green Version]
- Shen, R.; Corey, E.J. Studies of the Stereochemistry of [2+2]-Photocycloaddition Reactions of 2-Cyclohexenones with Olefins. Org. Lett. 2007, 9, 1057–1059. [Google Scholar] [CrossRef]
- Dar’in, D.; Kantin, G.; Krasavin, M. Practical Application of the Aqueous ‘Sulfonyl-Azide-Free’ (SAFE) Diazo Transfer Protocol to Less α-C–H Acidic Ketones and Esters. Synthesis 2019, 51, 4284–4290. [Google Scholar] [CrossRef]
- Norbeck, D.W.; Kramer, J.B. Synthesis of (-)-Oxetanocin. J. Am. Chem. Soc. 1988, 110, 7217–7218. [Google Scholar] [CrossRef]
- Meier, R.; Trauner, D. A Synthesis of (±)-Aplydactone. Angew. Chem. Int. Ed. 2016, 55, 11251–11255. [Google Scholar] [CrossRef]
- Zhang, L.; Koreeda, M. Total Synthesis of (+)-Acanthodoral by the Use of a Pd-Catalyzed Metal-Ene Reaction and a Nonreductive 5- exo -Acyl Radical Cyclization. Org. Lett. 2004, 6, 537–540. [Google Scholar] [CrossRef]
- Wang, B.; Xie, Y.; Yang, Q.; Zhang, G.; Gu, Z. Total Synthesis of Aquatolide: Wolff Ring Contraction and Late-Stage Nozaki–Hiyama–Kishi Medium-Ring Formation. Org. Lett. 2016, 18, 5388–5391. [Google Scholar] [CrossRef]
- Ando, W.; Hanyu, Y.; Kumamota, Y.; Takata, T. Synthesis of Diisopropylidenethiirane (Thiiranoradialene) and Its Reactions. Tetrahedron 1986, 42, 1989–1994. [Google Scholar] [CrossRef]
- Freeman, D.B.; Holubec, A.A.; Weiss, M.W.; Dixon, J.A.; Kakefuda, A.; Ohtsuka, M.; Inoue, M.; Vaswani, R.G.; Ohki, H.; Doan, B.D.; et al. Welwitindolinone C Synthetic Studies. Construction of the Welwitindolinone Carbon Skeleton via a Transannular Nitrone Cycloaddition. Tetrahedron 2010, 66, 6647–6655. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lam, S.M.; Ip, I.; Wong, W.; Chiu, P. Rearrangements of α-Diazo-β-Hydroxyketones for the Synthesis of Bicyclo[m.n.1]. Alkanones Org. Lett. 2017, 19, 4464–4467. [Google Scholar] [CrossRef]
- Padwa, A.; Hornbuckle, S.F.; Zhang, Z.; Zhi, L. Synthesis of 1,3-Diketones Using.Alpha.-Diazo Ketones and Aldehydes in the Presence of Tin(II) Chloride. J. Org. Chem. 1990, 55, 5297–5299. [Google Scholar] [CrossRef]
- Tsvetkov, N.P.; Bayir, A.; Schneider, S.; Brewer, M. A Ring Fragmentation Approach to Medium-Sized Cyclic 2-Alkynones. Org. Lett. 2012, 14, 264–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhdanova, O.V.; Korneev, S.M.; Nikolaev, V.A. Chemistry of Diazocarbonyl Compounds: XVIII. Synthesis and Spectral Parameters of 1,3-Dialkyl- 3-Hydroxy-2-Diazoketones. Russ. J. Org. Chem. 2004, 40, 316–328. [Google Scholar] [CrossRef]
- Popik, V.V. The Role of Molecular Geometry in the Wolff Rearrangement of α-Diazocarbonyl Compounds—Conformational Control or Structural Constraints? Can. J. Chem. 2005, 83, 1382–1390. [Google Scholar] [CrossRef]
- Sudrik, S.G.; Chavan, S.P.; Chandrakumar, K.R.S.; Pal, S.; Date, S.K.; Chavan, S.P.; Sonawane, H.R. Microwave Specific Wolff Rearrangement of α-Diazoketones and Its Relevance to the Nonthermal and Thermal Effect. J. Org. Chem. 2002, 67, 1574–1579. [Google Scholar] [CrossRef]
- McLachlan, M.M.W.; O’Connor, P.D.; Fairweather, K.A.; Willis, A.C.; Mander, L.N. Total Synthesis of the Galbulimima Alkaloid (±)-GB 13. Aust. J. Chem. 2010, 63, 742. [Google Scholar] [CrossRef]
- Conti, P.; Kozikowski, A.P. New Synthesis of 2-Aminobicyclo[2.1.1]Hexane-2,5-Dicarboxylic Acid-I (ABHxD-I), a Potent Metabotropic Receptor Agonist. Tetrahedron Lett. 2000, 41, 4053–4056. [Google Scholar] [CrossRef]
- Uyehara, T.; Takehara, N.; Ueno, M.; Sato, T. Rearrangement Approaches to Cyclic Skeletons. IX. Stereoselective Total Synthesis of (±)-Camphorenone Based on a Ring-Contraction of Bicyclo[3.2.1]Oct-6-En-2-One. Reliable One-Step Diazo Transfer Followed by a Wolff Rearrangement. Bull. Chem. Soc. Jpn. 1995, 68, 2687–2694. [Google Scholar] [CrossRef]
- Wheeler, T.N.; Meinwald, J. Formation and photochemical Wolff rearrangement of cyclic a-diazo ketones: D-norandrost-5-en-3b-ol-16-carboxylic acids. Org. Synth. 1972, 52, 53. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Krishnamurthy, N.; Nunn, D.S.; McPhail, A.T. Synthesis, Structure, and Properties of a 2-(Trimethylsilyl)Cyclobutenocyclooctatetraene. J. Am. Chem. Soc. 1991, 113, 4910–4917. [Google Scholar] [CrossRef]
- Tsuji, T.; Nishida, S.; Okuyama, M.; Osawa, E. Photochemical Generation of Bicyclo[4.2.2]Decapentaene from [4.2.2]Propellatetraene. Experimental and Theoretical Study of the.Pi.-Bond-Shift Isomers of Bicyclo[4.2.2]Decapentaene, [4]Paracyclophane-1,3-Diene, and 1,6-Ethenocycloocta-1,3,5,7-Tetraene. J. Am. Chem. Soc. 1995, 117, 9804–9813. [Google Scholar] [CrossRef]
- Liu, C.-X. Impact of a New Quinolone, DU-6859a, and Two Oral Carbapenems, CS-834 and L-084, on the Rat and Mouse Caecal Microflora. J. Antimicrob. Chemother. 2000, 46, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, H.W.; Arnold, M.J. Photolysis of 4-Diazopyrrolidine-2,3-Diones. A New Synthetic Route to Mono- and Bicyclic.Beta.-Lactams. J. Org. Chem. 1983, 48, 3365–3367. [Google Scholar] [CrossRef]
- Rodina, L.L.; Malashikhin, S.A.; Galkina, O.S.; Nikolaev, V.A. Photochemical Reactions of Regioisomeric 2,2-Dimethyl-5,5-Diphenyl- and 5,5-Dimethyl-2,2-Diphenyl-Substituted Diazo Ketones of a Tetrahydrofuran Series. Helv. Chim. Acta 2009, 92, 1990–1998. [Google Scholar] [CrossRef]
- Rodina, L.L.; Galkina, O.S.; Supurgibekov, M.B.; Grigor’ev, Y.M.; Utsal’, V.A. Photolysis of Regioisomeric α,α-Diphenyl-Substituted Diazotetrahydrofuranones: Primary and Secondary Photochemical Processes. Russ. J. Org. Chem. 2010, 46, 1542–1545. [Google Scholar] [CrossRef]
- Rao, P.; Hu, J.; Xuan, J.; Ding, H. Total Synthesis of (−)-Pavidolide B: A Ring Contraction Strategy. J. Org. Chem. 2019, 84, 9385–9392. [Google Scholar] [CrossRef]
- Zefirova, O.N.; Baranova, T.Y.; Ivanova, A.A.; Ivanov, A.A.; Zefirov, N.S. Application of the Bridgehead Fragments for the Design of Conformationally Restricted Melatonin Analogues. Bioorg. Chem. 2011, 39, 67–72. [Google Scholar] [CrossRef]
- Mander, L.N.; McLachlan, M.M. The Total Synthesis of the Galbulimima Alkaloid GB 13. J. Am. Chem. Soc. 2003, 125, 2400–2401. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Beck, J.C.; Lacker, C.R.; Wu, L.; Reisman, S.E. Evolution of a Strategy for the Enantioselective Total Synthesis of (+)-Psiguadial B. J. Org. Chem. 2018, 83, 6066–6085. [Google Scholar] [CrossRef]
- Wollenweber, M.; Etzkorn, M.; Reinbold, J.; Wahl, F.; Voss, T.; Melder, J.-P.; Grund, C.; Pinkos, R.; Hunkler, D.; Keller, M.; et al. [2.2.2.2]/[2.1.1.1]Pagodanes and [1.1.1.1]/[2.2.1.1]/[2.2.2.2]Isopagodanes: Syntheses, Structures, Reactivities−Benzo/Ene- and Benzo/Benzo-Photocycloadditions. Eur. J. Org. Chem. 2000, 2000, 3855–3886. [Google Scholar] [CrossRef]
- Kawai, H.; Suzuki, T.; Ohkita, M.; Tsuji, T. Kinetic Stabilization of the [1.1]Paracyclophane System: Isolation and X-Ray Structural Analysis of a [1.1]Paracyclophane Derivative and Its Interconversion with the Transannular Adduct. Chemistry 2000, 6, 4177–4187. [Google Scholar] [CrossRef]
- Belov, V.N.; Wurm, C.A.; Boyarskiy, V.P.; Jakobs, S.; Hell, S.W. Rhodamines NN: A Novel Class of Caged Fluorescent Dyes. Angew. Chem. Int. Ed. 2010, 49, 3520–3523. [Google Scholar] [CrossRef] [PubMed]
- Belov, V.N.; Mitronova, G.Y.; Bossi, M.L.; Boyarskiy, V.P.; Hebisch, E.; Geisler, C.; Kolmakov, K.; Wurm, C.A.; Willig, K.I.; Hell, S.W. Masked Rhodamine Dyes of Five Principal Colors Revealed by Photolysis of a 2-Diazo-1-Indanone Caging Group: Synthesis, Photophysics, and Light Microscopy Applications. Chem. Eur. J. 2014, 20, 13162–13173. [Google Scholar] [CrossRef] [Green Version]
- Halabi, E.A.; Püntener, S.; Rivera-Fuentes, P. A Simple Probe for Super-Resolution Imaging of the Endoplasmic Reticulum in Living Cells. Helv. Chim. Acta 2018, 101, e1800165. [Google Scholar] [CrossRef] [Green Version]
- Halabi, E.A.; Thiel, Z.; Trapp, N.; Pinotsi, D.; Rivera-Fuentes, P. A Photoactivatable Probe for Super-Resolution Imaging of Enzymatic Activity in Live Cells. J. Am. Chem. Soc. 2017, 139, 13200–13207. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Liu, S.; Li, M.-M.; Li, Y.; Lan, Y.; Lu, L.-Q.; Xiao, W.-J. Enantioselective Trapping of Pd-Containing 1,5-Dipoles by Photogenerated Ketenes: Access to 7-Membered Lactones Bearing Chiral Quaternary Stereocenters. J. Am. Chem. Soc. 2019, 141, 133–137. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, F.; Du, Z.; Ma, B. An Efficient Synthesis of 6-Oxa-Spiro[3.4]Octan-1-One Derivates Through 3-Diazochroman-4-One and Alkene. Adv. Synth. Catal. 2018, 360, 911–916. [Google Scholar] [CrossRef]
- Diethelm, S.; Schindler, C.S.; Carreira, E.M. Synthesis of Microcin SF608 through Nucleophilic Opening of an Oxabicyclo[2.2.1]Heptane. Org. Lett. 2010, 12, 3950–3953. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, K.; Kaliappan, K. A One-Pot Deprotection and Intramolecular Oxa-Michael Addition to Access Angular Trioxatriquinanes. Synlett 2011, 2011, 2580–2584. [Google Scholar] [CrossRef]
- Medvedev, J.; Semenok, D.; Azarova, X.; Rodina, L.; Nikolaev, V. A New Powerful Approach to Multi-Substituted 3(2H)-Furanones via Brønsted Acid-Catalyzed Reactions of 4-Diazodihydrofuran-3-Ones. Synthesis 2016, 48, 4525–4532. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, K.; Hori, K.; Hirai, T.; Takebayashi, M.; Ibata, T. The Acid-Catalyzed Decomposition of α-Diazo β-Hydroxy Ketones. Bull. Chem. Soc. Jpn. 1981, 54, 2142–2147. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.M.; Wong, W.; Chiu, P. An Approach to the Welwistatin Core via a Diazoketone Rearrangement–Ring Expansion Strategy. Org. Lett. 2017, 19, 4468–4471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, J.; Zheng, H.; Li, Y.; Yang, K.; Cheng, B.; Jin, X.; Yao, X.; Zhai, H. Silver(I)-Catalyzed Ring-Contractive Rearrangement: A New Entry to 5-Alkylidene-2-Cyclopentenones. Org. Lett. 2014, 16, 6378–6381. [Google Scholar] [CrossRef]
- De Kraker, J.-W.; Franssen, M.C.R.; Joerink, M.; De Groot, A.; Bouwmeester, H.J. Biosynthesis of Costunolide, Dihydrocostunolide, and Leucodin. Demonstration of Cytochrome P450-Catalyzed Formation of the Lactone Ring Present in Sesquiterpene Lactones of Chicory. Plant Physiol. 2002, 129, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Furuta, K.; Maeda, M.; Hirata, Y.; Shibata, S.; Kiuchi, K.; Suzuki, M. Synthesis of Neuroprotective Cyclopentenone Prostaglandin Analogs: Suppression of Manganese-Induced Apoptosis of PC12 Cells. Bioorg. Med. Chem. Lett. 2007, 17, 5487–5491. [Google Scholar] [CrossRef]
- Bayir, A.; Draghici, C.; Brewer, M. Preparation of Tethered Aldehyde Ynoates and Ynones by Ring Fragmentation of Cyclic γ-Oxy-β-Hydroxy-α-Diazo Carbonyls. J. Org. Chem. 2010, 75, 296–302. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Sadeghpour, B.M.; Marinozzi, M.; Snyder, J.P.; Williamson, B.L.; Kuethe, J.T.; Padwa, A. The Reaction of α-Diazo-β-Hydroxy Esters with Boron Trifluoride Etherate: Generation and Rearrangement of Destabilized Vinyl Cations. A Detailed Experimental and Theoretical Study. J. Am. Chem. Soc. 1996, 118, 1–12. [Google Scholar] [CrossRef]
- Shinada, T.; Kawakami, T.; Sakai, H.; Takada, I.; Ohfune, Y. An Efficient Synthesis of α-Acyloxyketone by Cu(Acac)2-Catalyzed Insertion Reaction of α-Diazoketone to Carboxylic Acid. Tetrahedron Lett. 1998, 39, 3757–3760. [Google Scholar] [CrossRef]
- Denmark, S.E.; Cullen, L.R. Development of a Phase-Transfer-Catalyzed, [2,3]-Wittig Rearrangement. J. Org. Chem. 2015, 80, 11818–11848. [Google Scholar] [CrossRef] [PubMed]
- Mateos, J.L.; Chao, O.; Flores, R.H. Some Reactions of 16-Diazo-Androstan-3β-Ol-17-One. Tetrahedron 1963, 19, 1051–1056. [Google Scholar] [CrossRef]
- Thomas, A.A.; Hunt, K.W.; Newhouse, B.; Watts, R.J.; Liu, X.; Vigers, G.; Smith, D.; Rhodes, S.P.; Brown, K.D.; Otten, J.N.; et al. 8-Tetrahydropyran-2-Yl Chromans: Highly Selective Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitors. J. Med. Chem. 2014, 57, 10112–10129. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujisawa, H.; Mukaiyama, T. Lewis Base-Catalyzed [2,3]-Wittig Rearrangement of Silyl Enolates Generated from α-Allyloxy Carbonyl Compounds. Bull. Chem. Soc. Jpn. 2006, 79, 1275–1287. [Google Scholar] [CrossRef]
- Yuan, W.; Eriksson, L.; Szabó, K.J. Rhodium-Catalyzed Geminal Oxyfluorination and Oxytrifluoro-Methylation of Diazocarbonyl Compounds. Angew. Chem. Int. Ed. 2016, 55, 8410–8415. [Google Scholar] [CrossRef]
- Lübcke, M.; Yuan, W.; Szabó, K.J. Trifluoromethylthiolation-Based Bifunctionalization of Diazocarbonyl Compounds by Rhodium Catalysis. Org. Lett. 2017, 19, 4548–4551. [Google Scholar] [CrossRef]
- Rosenfeld, M.J.; Shankar, B.K.R.; Shechter, H. Rhodium(II) Acetate-Catalyzed Reactions of 2-Diazo-1,3-Indandione and 2-Diazo-1-Indanone with Various Substrates. J. Org. Chem. 1988, 53, 2699–2705. [Google Scholar] [CrossRef]
- Anthony McKervey, M.; Ratananukul, P. Regiospecific Synthesis of α-(Phenylthio)Ketones via Rhodium(II) Acetate Catalysed Addition of Thiophenol to α-Diazoketones. Tetrahedron Lett. 1982, 23, 2509–2512. [Google Scholar] [CrossRef]
- Bagheri, V.; Doyle, M.P.; Taunton, J.; Claxton, E.E. A New and General Synthesis of.Alpha.-Silyl Carbonyl Compounds by Silicon-Hydrogen Insertion from Transition Metal-Catalyzed Reactions of Diazo Esters and Diazo Ketones. J. Org. Chem. 1988, 53, 6158–6160. [Google Scholar] [CrossRef]
- Carroll, F.I.; Blough, B.E.; Abraham, P.; Mills, A.C.; Holleman, J.A.; Wolckenhauer, S.A.; Decker, A.M.; Landavazo, A.; McElroy, K.T.; Navarro, H.A.; et al. Synthesis and Biological Evaluation of Bupropion Analogues as Potential Pharmacotherapies for Cocaine Addiction. J. Med. Chem. 2009, 52, 6768–6781. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cheng, P.; Jiang, L.; Yang, J.-L.; Zu, L. Bio-Inspired Fragmentations: Rapid Assembly of Indolones, 2-Quinolinones, and (−)-Goniomitine. Angew. Chem. Int. Ed. 2017, 56, 2754–2757. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Shan, X.; Zu, L. Divergent Coupling of 2-Carbonyl-Anilines and Diazo-Cyclopentanones: Asymmetric Total Synthesis of (+)-Leucomidine A. Org. Lett. 2018, 20, 6498–6501. [Google Scholar] [CrossRef] [PubMed]
- McKervey, M.A.; Ratananukul, P. Regiospecific Synthesis of α-(Phenylthio)Cycloalkenones and of α-Phenyl-α-(Phenylthio) Ketones via aα-Addition of Phenylsulphenyl Chloride to a-Diazoketones. Tetrahedron Lett. 1983, 24, 117–120. [Google Scholar] [CrossRef]
- Cronin, J.P.; Dilworth, B.M.; McKervey, M.A. Organic Synthesis with α-Chlorosulphides. Convenient Routes to Phenylthioacetals from α-Diazoketones and Alkyl Phenyl Sulphides via α-Chlorosulphides. Tetrahedron Lett. 1986, 27, 757–760. [Google Scholar] [CrossRef]
- Usuki, Y.; Iwaoka, M.; Tomoda, S. A New Synthesis of α-Fluoro-α,β-Unsaturated Ketones and Esters Based on Organoselenium Methodology. J. Chem. Soc. Chem. Commun. 1992, 1148–1150. [Google Scholar] [CrossRef]
- Buckley, D.J.; Kulkowit, S.; McKervey, A. Reactions of α-Diazoketones with Phenylselenenyl Chloride. A New Synthesis of α-Chloro- and α-Phenylselenenyl-Aβ-Unsaturated Ketones. J. Chem. Soc. Chem. Commun. 1980, 506–507. [Google Scholar] [CrossRef]
- Rodina, L.L.; Galiullina, S.V.; Matyushina, N.V.; Nikolaev, V.A. Staudinger Reaction in the Series of 4-Diazotetrahydrofuran-3-Ones: Effect of Substituents in the Diazo and Phosphine Components. Russ. J. Org. Chem. 2007, 43, 1882–1885. [Google Scholar] [CrossRef]
- Vuluga, D.; Legros, J.; Crousse, B.; Bonnet-Delpon, D. Synthesis of Pyrazoles through Catalyst-Free Cycloaddition of Diazo Compounds to Alkynes. Green Chem. 2009, 11, 156–159. [Google Scholar] [CrossRef]
- Jiang, N.; Li, C.-J. Novel 1,3-Dipolar Cycloaddition of Diazocarbonyl Compounds to Alkynes Catalyzed by InCl3 in Water. Chem. Commun. 2004, 4, 394–395. [Google Scholar] [CrossRef]
- Elzinga, J.; Hogeveen, H.; Schudde, E.P. Synthesis and Chemistry of a Bicyclobutane-Bridged.Alpha.-Diazoketone. J. Org. Chem. 1980, 45, 4337–4348. [Google Scholar] [CrossRef]
- Bolster, J.M.; Kellogg, R.M. Synthesis and Chemistry of 2,2,5,5-Tetramethylthiolane-3,4-Dione. A Route to Bicyclo[2.1.0]Pentyl-1-Sulfonium Intermediates. J. Org. Chem. 1982, 47, 4429–4439. [Google Scholar] [CrossRef]
- Franck-Neumann, M.; Dietrich-Buchecker, C. Spontaneous Transpositions from Pyrazolenines into Pyrazoles. Competitive [1,5] Migrations of an Acyl or Carbalkoxy Group to the Carbon and Nitrogen. Tetrahedron Lett. 1976, 24, 2069–2072. [Google Scholar] [CrossRef]
- Yagupolskii, Y.L.; Pavlenko, N.V.; Gerus, I.I.; Peng, S.; Nappa, M. 1,1,1,4,4,4-Hexafluorobut-2-yne (HFB) as a Versatile Dipolarophile for [3+2] Cycloaddition with 1,3-Dipoles toward Azoles with Adjacent CF3 Groups. ChemistrySelect 2019, 4, 4604–4610. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shechter, H. Rearrangement, Extrusion, and Polymerization Reactions upon Addition of Acetylenes to 3-Diazooxindole and Six-Membered Ring α-Diazo Ketones. Tetrahedron Lett. 1973, 14, 1417–1420. [Google Scholar] [CrossRef]
- Cheng, B.; Bao, B.; Zu, B.; Duan, X.; Duan, S.; Li, Y.; Zhai, H. Synthesis of Spiro-3 H-Indazoles via 1,3-Dipolar Cycloaddition of Arynes with 6-Diazocyclohex-2-En-1-One Derivatives and Fused-2 H-Indazoles by Subsequent Rearrangement. RSC Adv. 2017, 7, 54087–54090. [Google Scholar] [CrossRef] [Green Version]
- Bauta, W.; Dodd, J.; Bullington, J.; Gauthier, D.; Leo, G.; McDonnell, P. Stereoselectivity in the Rhodium(II) Acetate Catalysed Cyclopropanations of 2-Diazo-1-Indanone with Styrenes. Tetrahedron Lett. 2000, 41, 1491–1494. [Google Scholar] [CrossRef]
- Gong, J.; Zhao, Z.; Zhang, F.; Wu, S.; Yan, G.; Quan, Y.; Ma, B. One-Pot Novel Regioselective Cycloisomerization Synthesis of 2-Substituted or 3-Substituted 4H-Furo[3,2-c]Chromene through the Intermediate Cyclopropenes of 3-Diazochroman-4-One and Phenylacetylene. Org. Lett. 2014, 16, 5524–5527. [Google Scholar] [CrossRef]
- Peng, C.; Wang, Y.; Wang, J. Palladium-Catalyzed Cross-Coupling of α-Diazocarbonyl Compounds with Arylboronic Acids. J. Am. Chem. Soc. 2008, 130, 1566–1567. [Google Scholar] [CrossRef]
- Peng, C.; Yan, G.; Wang, Y.; Jiang, Y.; Zhang, Y.; Wang, J. Palladium-Catalyzed Coupling Reaction of α-Diazocarbonyl Compounds with Aromatic Boronic Acids or Halides. Synthesis 2010, 4154–4168. [Google Scholar] [CrossRef]
- Tsoi, Y.-T.; Zhou, Z.; Chan, A.S.C.; Yu, W.-Y. Palladium-Catalyzed Oxidative Cross-Coupling Reaction of Arylboronic Acids with Diazoesters for Stereoselective Synthesis of (E)-α,β-Diarylacrylates. Org. Lett. 2010, 12, 4506–4509. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Xia, Y.; Liu, Z.; Zhang, Y.; Wang, J. Palladium-Catalyzed Cross-Coupling Reaction of Diazo Compounds and Vinyl Boronic Acids: An Approach to 1,3-Diene Compounds. J. Org. Chem. 2014, 79, 7711–7717. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhang, W.; Yan, G.; Wang, J. Arylation and Vinylation of α-Diazocarbonyl Compounds with Boroxines. Org. Lett. 2009, 11, 1667–1670. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, J.; Li, Y.; Cheng, B.; Zhao, L.; Zhai, H. Facile Synthesis of 2-Arylphenols via Palladium-Catalyzed Cross-Coupling of Aryl Iodides with 6-Diazo-2-Cyclohexenones. Org. Lett. 2013, 15, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, K.; Zheng, H.; Ding, R.; Yin, F.; Wang, N.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. A One-Pot Synthesis of Dibenzofurans from 6-Diazo-2-Cyclohexenones. Org. Lett. 2015, 17, 5744–5747. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, G. Rhodium(III)-Catalyzed Cascade Redox-Neutral C-H Functionalization and Aromatization: Synthesis of Unsymmetrical Ortho-Biphenols. Adv. Synth. Catal. 2017, 359, 1643–1648. [Google Scholar] [CrossRef]
- Ding, D.; Lv, X.; Li, J.; Xu, G.; Ma, B.; Sun, J. Copper-Catalyzed N-H Insertion and Oxidative Aromatization Cascade: Facile Synthesis of 2-Arylaminophenols. Chem. Asian J. 2014, 9, 1539–1542. [Google Scholar] [CrossRef]
- Salomon, R.G.; Kochi, J.K. Copper(I) Catalysis in Cyclopropanations with Diazo Compounds. Role of Olefin Coordination. J. Am. Chem. Soc. 1973, 95, 3300–3310. [Google Scholar] [CrossRef]
- Ding, D.; Lv, X.; Li, J.; Qiu, L.; Xu, G.; Sun, J. A Pd-Catalyzed Cascade Reaction of N–H Insertion and Oxidative Dehydrogenative Aromatization: A New Entry to 2-Amino-Phenols. Org. Biomol. Chem. 2014, 12, 4084–4088. [Google Scholar] [CrossRef]
- Hostetler, E.D.; Jonson, S.D.; Welch, M.J.; Katzenellenbogen, J.A. Synthesis of 2-[18F]Fluoroestradiol, a Potential Diagnostic Imaging Agent for Breast Cancer: Strategies to Achieve Nucleophilic Substitution of an Electron-Rich Aromatic Ring with [8F]F−. J. Org. Chem. 1999, 64, 178–185. [Google Scholar] [CrossRef]
- Yasui, N.; Mayne, C.G.; Katzenellenbogen, J.A. Preparation of o-Fluorophenols from Nonaromatic Precursors: Mechanistic Considerations for Adaptation to Fluorine-18 Radiolabeling. Org. Lett. 2015, 17, 5540–5543. [Google Scholar] [CrossRef] [Green Version]
- Fissekis, J.D.; Myles, A.; Brown, G.B. Synthesis of 5-Hydroxyalkylpyrimidines from Lactones 1,2. J. Org. Chem. 1964, 29, 2670–2673. [Google Scholar] [CrossRef]
- Schmitz, A.; Kraatz, U.; Korte, F. Über die Synthese und Reaktivität von α-Diazo-γ-butyrolacton. Chem. Ber. 1975, 108, 1010–1016. [Google Scholar] [CrossRef]
- Brown, R.C.D.; Bataille, C.J.R.; Bruton, G.; Hinks, J.D.; Swain, N.A. C−H Insertion Approach to the Synthesis of e Ndo, e Xo -Furofuranones: Synthesis of (±)-Asarinin, (±)-Epimagnolin A, and (±)-Fargesin. J. Org. Chem. 2001, 66, 6719–6728. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.D. Synthesis of Endo, Exo-Furofuranones Using a Highly Diastereoselective C–H Insertion Reaction. Chem. Commun. 1998, 17, 1895–1896. [Google Scholar] [CrossRef]
- Sattely, E.S.; Meek, S.J.; Malcolmson, S.J.; Schrock, R.R.; Hoveyda, A.H. Design and Stereoselective Preparation of a New Class of Chiral Olefin Metathesis Catalysts and Application to Enantioselective Synthesis of Quebrachamine: Catalyst Development Inspired by Natural Product Synthesis. J. Am. Chem. Soc. 2009, 131, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dileep Kumar, J.S.; Dupradeau, F.-Y.; Strouse, M.J.; Phelps, M.E.; Toyokuni, T. Electrophilic Azidation for Stereoselective Synthesis of 2-Azido-2-Deoxyaldono-1,5-Lactones. J. Org. Chem. 2001, 66, 3220–3223. [Google Scholar] [CrossRef]
- Brown, R.C.D.; Bataille, C.J.R.; Hinks, J.D. Total Synthesis of (±)-Epimagnolin A. Tetrahedron Lett. 2001, 42, 473–475. [Google Scholar] [CrossRef]
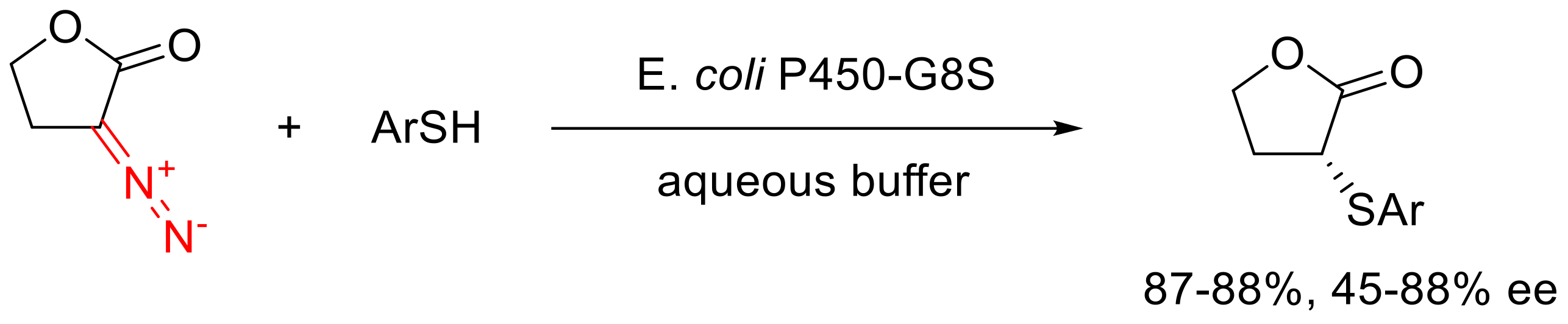
- Chen, K.; Huang, X.; Zhang, S.-Q.; Zhou, A.; Kan, S.B.J.; Hong, X.; Arnold, F. Engineered Cytochrome C-Catalyzed Lactone-Carbene B–H Insertion. Synlett 2019, 30, 378–382. [Google Scholar] [CrossRef] [Green Version]
- Skerry, P.S.; Swain, N.A.; Harrowven, D.C.; Smyth, D.; Bruton, G.; Brown, R.C.D. Intramolecular C–H Insertions Adjacent to Sulfur for the Diastereoselective Synthesis of Thienofuranones. Chem. Commun. 2004, 1772–1773. [Google Scholar] [CrossRef]
- Swain, N.A.; Brown, R.C.D.; Bruton, G. An Efficient Synthesis of Endo, Exo-Furofuranone Derivatives. Chem. Commun. 2002, 2042–2043. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, F.; Wu, C.; Zhang, Y.; Wang, J. Construction of All-Carbon Quaternary Centers through Cu-Catalyzed Sequential Carbene Migratory Insertion and Nucleophilic Substitution/Michael Addition. J. Org. Chem. 2015, 80, 8748–8757. [Google Scholar] [CrossRef]
- Shershnev, I.; Dar’in, D.; Chuprun, S.; Kantin, G.; Bakulina, O.; Krasavin, M. The Use of ∝-Diazo-γ-Butyrolactone in the Büchner-Curtius-Schlotterbeck Reaction of Cyclic Ketones: A Facile Entry into Spirocyclic Scaffolds. Tetrahedron Lett. 2019, 60, 1800–1802. [Google Scholar] [CrossRef]
- Nicolle, S.M.; Moody, C.J. Potassium N -Iodo p -Toluenesulfonamide (TsNIK, Iodamine-T): A New Reagent for the Oxidation of Hydrazones to Diazo Compounds. Chem. Eur. J. 2014, 20, 4420–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, E.L.; Raines, R.T. A Phosphine-Mediated Conversion of Azides into Diazo Compounds. Angew. Chem. Int. Ed. 2009, 48, 2359–2363. [Google Scholar] [CrossRef]
- Pinkerton, D.M.; Chow, S.; Eisa, N.H.; Kainth, K.; Vanden Berg, T.J.; Burns, J.M.; Guddat, L.W.; Savage, G.P.; Chadli, A.; Williams, C.M. Synthesis of the Seco-Limonoid BCD Ring System Identifies a Hsp90 Chaperon Machinery (P23) Inhibitor. Chem. Eur. J. 2019, 25, 1451–1455. [Google Scholar] [CrossRef]
- Bayir, A.; Brewer, M. Fragmentation of Bicyclic γ-Silyloxy-β-Hydroxy-α-Diazolactones as an Approach to Ynolides. J. Org. Chem. 2014, 79, 6037–6046. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, S.-Q.; Brandenberg, O.F.; Hong, X.; Arnold, F.H. Alternate Heme Ligation Steers Activity and Selectivity in Engineered Cytochrome P450-Catalyzed Carbene-Transfer Reactions. J. Am. Chem. Soc. 2018, 140, 16402–16407. [Google Scholar] [CrossRef] [Green Version]
- Solovyov, I.; Dar’in, D.; Krasavin, M. Convenient Approach to 2-Substituted (Thio)Morpholin-3-Ones from α-Diazoacetates via X-H Carbene Insertion—Lactamization Sequence: Convenient Approach to 2-Substituted (Thio)Morpholin-3-Ones from α-Diazoacetates via X-H Carbene Insertion—Lactamization Sequence. Eur. J. Org. Chem. 2019, 2019, 7432–7438. [Google Scholar] [CrossRef]
- Nicolle, S.M.; Lewis, W.; Hayes, C.J.; Moody, C.J. Stereoselective Synthesis of Functionalized Pyrrolidines by the Diverted N−H Insertion Reaction of Metallocarbenes with β-Aminoketone Derivatives. Angew. Chem. Int. Ed. 2016, 55, 3749–3753. [Google Scholar] [CrossRef]
- Nicolle, S.M.; Lewis, W.; Hayes, C.J.; Moody, C.J. Stereoselective Synthesis of Highly Substituted Tetrahydrofurans through Diverted Carbene O-H Insertion Reaction. Angew. Chem. Int. Ed. 2015, 54, 8485–8489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.K.; Chen, K.; Huang, X.; Wohlschlager, L.; Renata, H.; Arnold, F.H. Enzymatic Assembly of Carbon–Carbon Bonds via Iron-Catalysed Sp3 C–H Functionalization. Nature 2019, 565, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Swain, N.A.; Brown, R.C.D.; Bruton, G. A Versatile Stereoselective Synthesis of e Ndo, e Xo -Furofuranones: Application to the Enantioselective Synthesis of Furofuran Lignans. J. Org. Chem. 2004, 69, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, I.S.; Matlin, S.A.; Mete, A. The Synthesis and Chemistry of 3-Diazo-Piperidin-2-One. Tetrahedron 2002, 58, 3137–3143. [Google Scholar] [CrossRef]
- Sultanova, R.M.; Lobov, A.N.; Spirikhin, L.V. Synthesis of Dimethyl Esters of 7-Oxo-4,5,6,7-Tetrahydropyrazolo[1,5-c]Pyrimidine-2,3-Dicarboxylic Acid. Chem. Heterocycl. Compd. 2015, 51, 1048–1051. [Google Scholar] [CrossRef]
- Muthusamy, S.; Srinivasan, P. Regioselective Synthesis of 3-Heteroarylpiperidin-2-Ones and Diazacyclopenta[a]Phenalenone via Carbenoid Reactions. Tetrahedron 2009, 65, 1567–1573. [Google Scholar] [CrossRef]
- Zhukovsky, D.; Dar’in, D.; Kantin, G.; Krasavin, M. Synthetic Exploration of α-Diazo γ-Butyrolactams. Eur. J. Org. Chem. 2019, 2019, 2397–2400. [Google Scholar] [CrossRef]
- Eremeyeva, M.; Zhukovsky, D.; Dar’in, D.; Krasavin, M. Preparation and in Situ Use of Unstable N-Alkyl α-Diazo-γ-Butyrolactams in Rh II -Catalyzed X–H Insertion Reactions. Beilstein J. Org. Chem. 2020, 16, 607–610. [Google Scholar] [CrossRef]
- Zhukovsky, D.; Dar’in, D.; Krasavin, M. Tert-Butyl 3-Diazo-2-Oxopyrrolidine-1-Carboxylate—A New Reagent for Introduction of the 2-Oxopyrrolidin-3-Yl Motif via Rh II -Catalyzed X-H Insertion Reactions: Tert-Butyl 3-Diazo-2-Oxopyrrolidine-1-Carboxylate—A New Reagent for Introduction of the 2-Oxopyrrolidin-3-Yl Motif via RhII-Catalyzed X-H Insertion. Eur. J. Org. Chem. 2020, 2020, 3013–3018. [Google Scholar] [CrossRef]
- Kazantsev, A.; Zhukovsky, D.; Bubyrev, A.; Dar’in, D.; Krasavin, M. Fluorination of α-Diazo-γ-Lactams with the Olah Reagent. Tetrahedron Lett. 2021, 69, 152969. [Google Scholar] [CrossRef]
- Barkhatova, D.; Zhukovsky, D.; Dar’in, D.; Krasavin, M. Employing α-Diazocarbonyl Compound Chemistry in the Assembly of Medicinally Important Aryl(Alkyl)Thiolactam Scaffold: Employing α-Diazocarbonyl Compound Chemistry in the Assembly of Medicinally Important Aryl(Alkyl)Thiolactam Scaffold. Eur. J. Org. Chem. 2019, 2019, 5798–5800. [Google Scholar] [CrossRef]
- Muthusamy, S.; Gangadurai, C. Domino Reactions of Bis-Diazo Compounds: Rhodium(II) Acetate Catalyzed Diastereoselective Synthesis of Epoxy- and Epithio-Bridged Heterocycle-Fused Quinolizinone Analogues. Synthesis 2016, 48, 2213–2225. [Google Scholar] [CrossRef]
- Krasavin, M.; Zhukovsky, D.; Solovyev, I.; Barkhatova, D.; Dar’in, D.; Frank, D.; Martinelli, G.; Weizel, L.; Proschak, A.; Rotter, M.; et al. RhII -Catalyzed De-symmetrization of Ethane-1,2-dithiol and Propane-1,3-dithiol Yields Metallo-β-lactamase Inhibitors. ChemMedChem 2021, 16, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; Srinivasan, P. Facile Chemoselective Rhodium Carbenoid N–H Insertion Reactions: Synthesis of 3-Arylamino- or 3-Heteroarylpiperidin-2-Ones. Tetrahedron Lett. 2005, 46, 1063–1066. [Google Scholar] [CrossRef]
- Zhukovsky, D.; Dar’in, D.; Krasavin, M. Rh2(Esp)2 -Catalyzed Coupling of α-Diazo-γ-Butyrolactams with Aromatic Amines: Rh2 (Esp)2 -Catalyzed Coupling of α-Diazo-γ-Butyrolactams with Aromatic Amines. Eur. J. Org. Chem. 2019, 2019, 4377–4383. [Google Scholar] [CrossRef]
- Albrecht, D.; Vogt, F.; Bach, T. Diastereo- and Enantioselective Intramolecular [2+2] Photocycloaddition Reactions of 3-(Ω′-Alkenyl)- and 3-(Ω′-Alkenyloxy)-Substituted 5,6-Dihydro-1H-Pyridin-2-Ones. Chem. Eur. J. 2010, 16, 4284–4296. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.; Mete, A.; Wilson, L. Olefination of 3-Diazopiperidin-2-One. Tetrahedron Lett. 2003, 44, 6621–6624. [Google Scholar] [CrossRef]
- Bonderoff, S.A.; Padwa, A. Rhodium(II) Catalyzed Cyclopropanation/Cycloaddition Reactions of the Bis(Diazo)Piperidin-2-One System. Tetrahedron Lett. 2015, 56, 3127–3129. [Google Scholar] [CrossRef] [Green Version]
- Eremeyeva, M.; Zhukovsky, D.; Dar’in, D.; Krasavin, M. The Use of α-Diazo-γ-Butyrolactams in the Büchner–Curtius–Schlotterbeck Reaction of Cyclic Ketones Opens New Entry to Spirocyclic Pyrrolidones. Synlett 2020, 31, 982–986. [Google Scholar] [CrossRef]












































































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhukovsky, D.; Dar’in, D.; Bakulina, O.; Krasavin, M. Preparation and Synthetic Applications of Five-to-Seven-Membered Cyclic α-Diazo Monocarbonyl Compounds. Molecules 2022, 27, 2030. https://doi.org/10.3390/molecules27062030
Zhukovsky D, Dar’in D, Bakulina O, Krasavin M. Preparation and Synthetic Applications of Five-to-Seven-Membered Cyclic α-Diazo Monocarbonyl Compounds. Molecules. 2022; 27(6):2030. https://doi.org/10.3390/molecules27062030
Chicago/Turabian StyleZhukovsky, Daniil, Dmitry Dar’in, Olga Bakulina, and Mikhail Krasavin. 2022. "Preparation and Synthetic Applications of Five-to-Seven-Membered Cyclic α-Diazo Monocarbonyl Compounds" Molecules 27, no. 6: 2030. https://doi.org/10.3390/molecules27062030
APA StyleZhukovsky, D., Dar’in, D., Bakulina, O., & Krasavin, M. (2022). Preparation and Synthetic Applications of Five-to-Seven-Membered Cyclic α-Diazo Monocarbonyl Compounds. Molecules, 27(6), 2030. https://doi.org/10.3390/molecules27062030