Abstract
Significant efforts have been made in recent years to identify more environmentally benign and safe alternatives to side-chain protection and deprotection in solid-phase peptide synthesis (SPPS). Several protecting groups have been endorsed as suitable candidates, but finding a greener protecting group in SPPS has been challenging. Here, based on the 2-(o-nitrophenyl) propan-1-ol (Npp-OH) photolabile protecting group, a structural modification was carried out to synthesize a series of derivatives. Through experimental verification, we found that 3-(o-Nitrophenyl) butan-2-ol (Npb-OH) had a high photo-release rate, high tolerance to the key conditions of Fmoc-SPPS (20% piperidine DMF alkaline solution, and pure TFA acidic solution), and applicability as a carboxyl-protective group in aliphatic and aromatic carboxyl groups. Finally, Npb-OH was successfully applied to the synthesis of head–tail cyclic peptides and side-chain–tail cyclic peptides. Moreover, we found that Npb-OH could effectively resist diketopiperazines (DKP). The α-H of Npb-OH was found to be necessary for its photosensitivity in comparison to 3-(o-Nitrophenyl)but-3-en-2-ol (Npbe-OH) during photolysis-rate verification.
1. Introduction
With advances in biotechnology, many active peptides and proteins have been developed into drugs, been used in clinical practice, and played vital roles in maintaining the normal functions of the body due to their high specificity, low required dosages, clear functions, and low numbers of side effects [1]. However, linear peptides are unstable in the body, susceptible to degradation, and easily removed by the kidneys, leading to short half-lives. Therefore, the study of long-acting peptide drugs has become a hot topic. Studies have shown that a polypeptide’s cyclization can stabilize its dominant configuration, effectively improving its stability against proteases and, thereby, its pharmacokinetic properties and biological activity [1]. The self-cyclization of the amino acid side chain on a polypeptide includes head-to-tail, head-to-side, and side-to-tail processes [2]. Traditional synthetic cyclic peptide methods rely on three dimensions of orthogonal protecting groups to allow for the synthesis of the linear peptide, the selective deprotection of the reactive ends (N- or C-terminus or side chain), cyclization, and then the final cleavage from the solid support. A common third-dimensional protecting group is an allyl group, which is easy to synthesize, has high acid–base tolerance, and can be catalytically removed with Pd reagents. However, Pd reagents are expensive, and the endpoint of the reaction is difficult to monitor, limiting their further application. A recent review of the greening of photolabile protecting groups (PPGs) showed that the allyl group has advantages for the compound protection and deprotection processes in lowering the environmental burden. The greening of the SPPS process has also seen improvements in chain assembly, resin cleavage, side-chain deprotection, and peptide work-up [3,4]. Based on previous laboratory studies on PPGs [5], we decided to use them as the third-dimensional protecting groups for amino acid side chains to verify the possibility of their application in cyclic peptide synthesis.
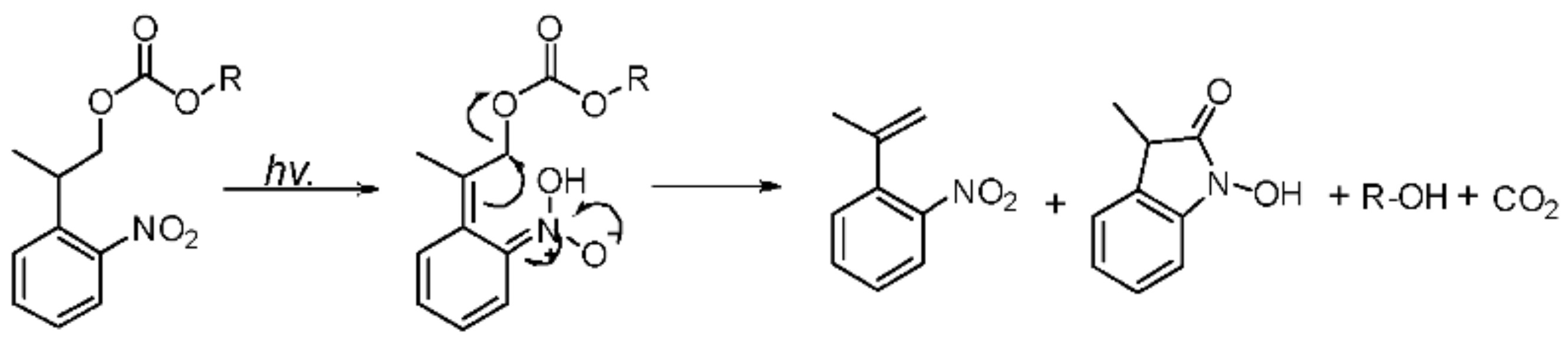
PPGs enjoyed a resurgence in interest after the publications of Kaplan [6] and Engels [7] in the late 1970s [8]. PPGs have an interesting feature: they do not require any reagent for their cleavage, only UV light. A fundamental role is played by PPGs in the process of greening. This category of protecting groups opens the possibility of dealing with sensitive molecules that are otherwise incompatible with acids or bases [9]. Moreover, the greening use of PPGs in nucleic acid [10], carbohydrate [11], and peptide [12,13,14,15] chemistry has been well-established [16]. In contrast to chemically cleavable protecting groups, PPGs use light as a traceless tool in the deprotection process [17], which not only simplifies the reaction step but also improves the purity and operability of the final product. Simultaneously, as the third-dimensional protective group, PPGs are convenient tools for the site-specific modification of peptides and the preparation of cyclic peptides, which also play significant roles in the structural modification of drugs. The most commonly used photolabile groups are currently o-nitrobenzyl derivatives, which have proven to be highly versatile and are used to protect a wide variety of functional groups [8,18]. Pfleiderer et al. [16] synthesized a novel type of PPG, 2-(o-nitrophenyl)-propyloxycarbonyl [19,20] (Nppoc), which was used to protect the hydroxyl groups in nucleosides and to conduct a series of nucleoside modifications. Unlike the conventional o-nitrobenzyl structural group, the photocyclization of Nppoc [19] (Scheme 1) to N-hydroxyindole ketone has been postulated to involve the initial formation of nitrostyrene products that could cyclize with the cleavage of the Cβ-O bond [21] because several of the side products are completely inactive, which is advantageous in the synthesis of cyclic peptides. Additionally, Steiner et al. [21] reported that, when the benzylic site was alkylated, the photolysis quantum yield was significantly higher.
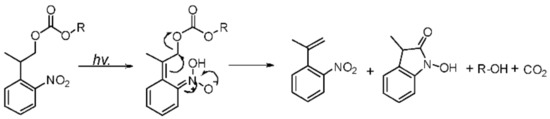
Scheme 1.
Photo-deprotection mechanism of Nppoc.
Recently, we used the 2-(o-nitrophenyl) propan-1-ol (Npp-OH) group—a rapid and effective PPG for carboxyl groups in standard solid-phase peptide synthesis (SPPS) that is tolerated in the reaction conditions used to cleave the t-Bu ester and the Fmoc group and is rapidly removed by UV irradiation without any unwanted side reactions in peptide synthesis [5]. However, because Npp-OH is an aliphatic primary alcohol, its homologous ester is easily attacked by nucleophiles. Additionally, the diketopiperazine side reaction (DKP) [22] is already present in Npp-OH in some situations, which has limited further applications.
In order to solve the abovementioned problems, we modified the benzyl site of Npp-OH to transform it into a secondary alcohol and obtained a series of derivatives. Furthermore, photosensitivity, acid–base tolerance, and applicability tests of the Npp-OH derivatives (compounds 4 [19], 5, 12 [19], 13, 16, and 17) as carboxyl-protective groups were carried out. Finally, the fast and stable Npb-OH photolabile protecting group was selected for the third-dimensional protective group of the amino acid side chain and successfully applied in cyclic peptide synthesis. It was also verified that, in addition to playing an important role in the structural modification of drugs, the photoprotective group is a convenient tool for the site-specific modification of polypeptides and the preparation of cyclic peptides.
2. Results and Discussion
2.1. Synthesis of the Photolabile Protecting Groups (PPGs)
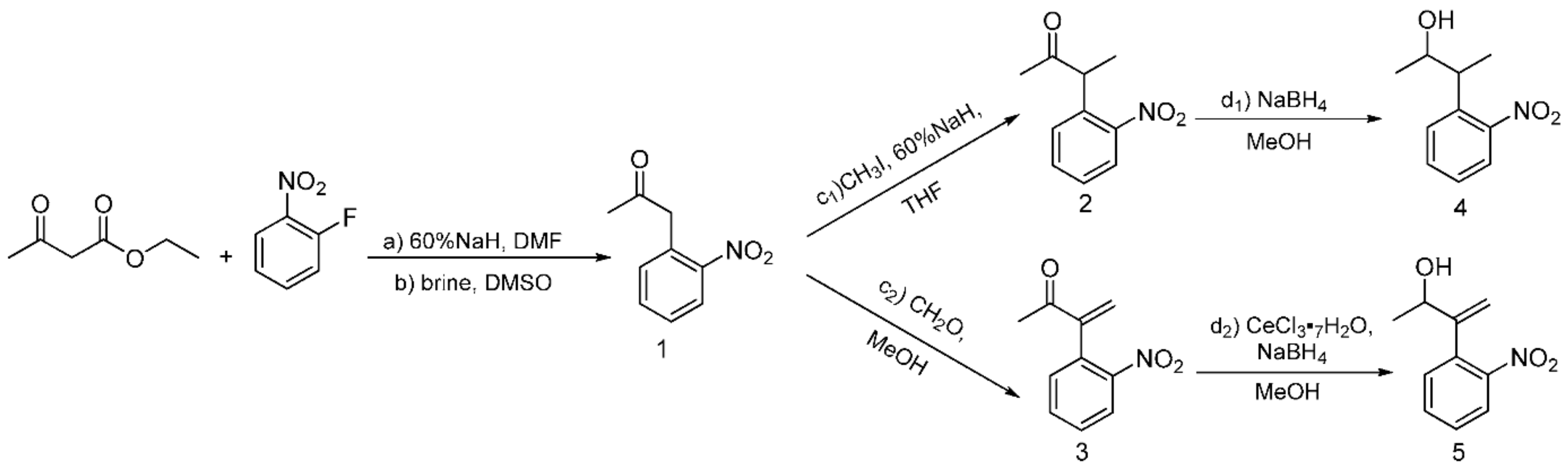
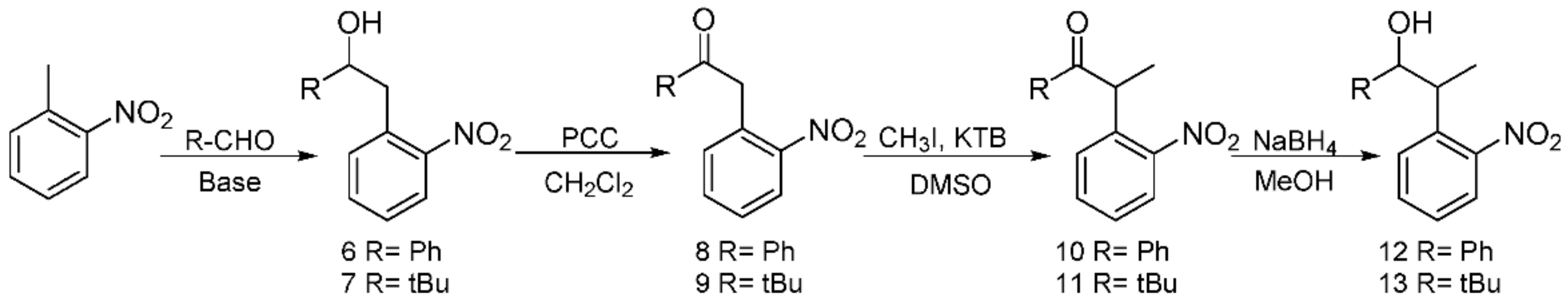
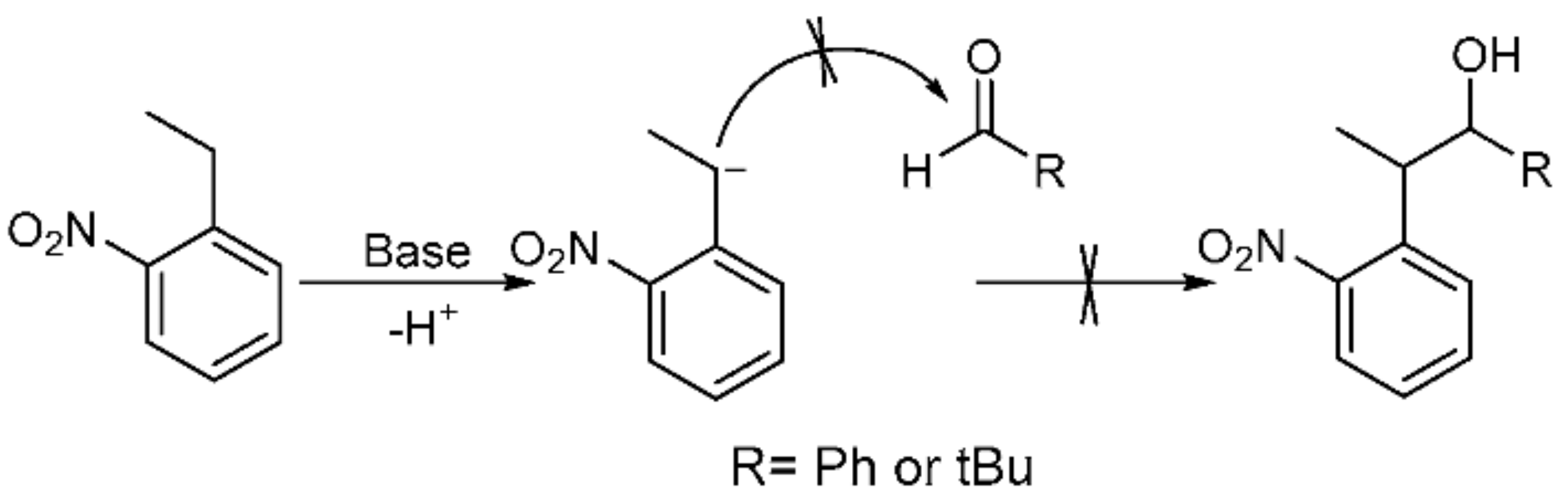
Following previous work based on the Npp-OH photolabile group, six modified derivatives (see 4 [19], 12 [19], 13, 16, 17, and 5) substituted at Npp-OH side chain were synthesized. The derivatives involved the preparation of a series of intermediates and products, ten of them were known compounds including 1 [23], 2 [19], 3 [24], 4, 7 [25], 9 [26], 10 [19], 12 [19], 14 [27] and 15 [27]. The ten compounds were synthesized in different methods from the corresponding literature. For example, the potassium carbonate in the literature was replaced with the sodium hydroxide for the synthesis of 1, and the yield remains at 84.7%. 4 was obtained by reduction of 2 in Scheme 2, and the preparation of 2 used NaH instead of sodium metal, making the reaction conditions milder. Compound 12 was obtained from the reduction of 10 in Scheme 3, and the preparation of 10 used potassium tert-butoxide instead of sodium metal to make the reaction conditions milder. In the literature, 3 was synthesized through two steps with 2-nitrophenyl acetic acid as the material. While in this paper, 3 was synthesized through one step with 1 as the material, and the reaction temperature was reduced from 100 ℃ to 60 ℃. In the synthesis of 7, we tried to replace the potassium hydroxide with potassium tert-butanol, and the yield was slightly reduced. In the synthesis of 9, the starting materials and synthesis route were different from the literature, and the yield was increased from 66% to 79.4%. In the synthesis of 14 and 15, the starting materials and the synthesis route were different from the literature methods with the relatively mild reaction condition of 75 ℃ in tetrahydrofuran replacing the condition of −40 ℃ under nitrogen. The following were the corresponding synthetic routes. The synthesis of PPGs 4 [19] and 5 is illustrated in Scheme 2. Under alkaline conditions, both compounds were subjected to nucleophilic substitution reactions using ethyl acetoacetate and 2-fluoronitrobenzene as starting materials and then hydrolyzed to obtain 1, which was alkylated or alkylidene in the benzylic position with MeI or polyoxymethylene to form the corresponding ketones. The NaBH4 or NaBH4/CeCl3·7H2O reduction of 2 or 3 produced benzylic 1-monosubstituted and 1,2-disubstituted 1-(2-nitrophenyl) propan-2-ol. When CeCl3·7H2O was used as the reducing agent, only the ketone was reduced, which had no effect on the double bond [28]. The structures of the products were assigned with 1H NMR, 13C NMR, and HRMS. Even though compounds 12 and 13 were also modified in the benzylic position, they have different synthetic routes in Scheme 3. We first tried to use 2-nitroethylbenzene as a raw material but found that the addition reaction for the ethyl group in 2-nitroethylbenzene did not proceed normally. Therefore, under base conditions, after inducing a carbon anion addition reaction, we reacted 2-nitrotoluene with benzaldehyde and pivalaldehyde to obtain 6 and 7, which were then converted to the corresponding ketones 8 and 9 in high yields via oxidation with PCC. Subsequently, using CH3I as a raw material, an alkylation reaction was induced at the exocyclic 1-position to obtain 10 and 11. The NaBH4 reduction of 10 and 11 led to high yields of exocyclic 1-monosubstituted 2-(2-nitrophenyl) propanol 12 and 13. Previously, we tried to use the synthesis method using Npp-OH, benzaldehyde, and valeral instead of paraformaldehyde to participate in the reaction and prepare Nppp-OH (12) and Dmnp-OH (13), but we found that the reaction could not be carried out normally. Even when the alkali was replaced with sodium glycolate, potassium tert-butanol, and potassium hydroxide-18-crown-6, the reaction still failed. When sodium glycolate was used as a base, carbanions could not form. Moreover, the use of potassium tert-butanol and potassium hydroxide-18-crown-6 allowed the carbanion to form smoothly, but the subsequent addition reaction could not continue, which was probably caused by the steric hindrance effect of ethyl and aldehyde substituents (Scheme 4). However, we replaced the reaction substrates with 2-nitrotoluene (which presented a deep red color under the influence of alkali) and showed that carbon anions formed; then, benzaldehyde and valeraldehyde were added, and the reaction could occur smoothly. Thus, the failure of the one-step synthesis of 12 and 13 was due to the ethyl in 2-nitroethylbenzene, whose steric-hindrance effect led to the failure of addition.
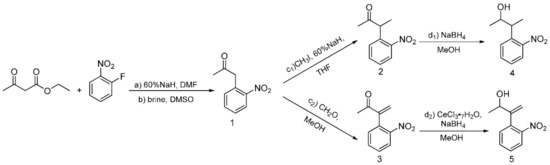
Scheme 2.
Synthesis of the PPGs 3-(o-Nitrophenyl) butan-2-ol 4) and 3-(o-Nitrophenyl)but-3-en-2-ol 5).
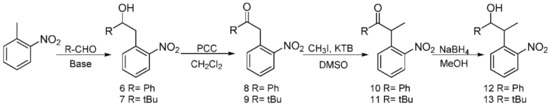
Scheme 3.
Synthesis of the PPGs 2-(2-nitrophenyl)-1-phenylpropan-1-ol 12) and 2,2-dimethyl-4-(2-nitrophenyl)pentan-3-ol 13).
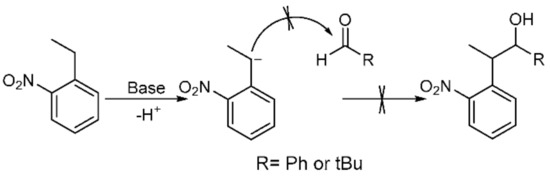
Scheme 4.
Possible mechanism for the failure of one-step synthesis of 12 and 13.
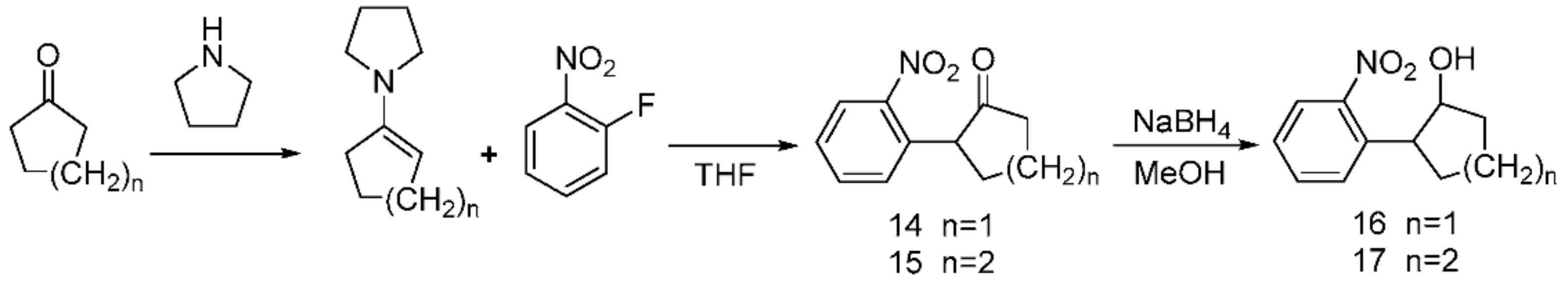
Using cyclones and tetrahydropyrrole as substrates, enamine products were obtained via the alkylation of enamine and purification by vacuum distillation; these products were mixed with 2-fluoronitrobenzene to obtain compounds 14 and 15 via an acylation reaction. A formal exocyclic 1,2-disubstitution was also observed in 4-(2-hydroxypentyl)- and 4-(2-hydroxyhexyl)-substituted 1-dinitrobenzenes 16 and 17, which resulted from 14 and 15 via NaBH4 reduction in the form of the compound shown in Scheme 5.
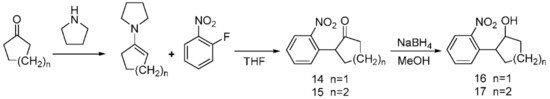
Scheme 5.
Synthesis of the PPGs 2-(2-nitrophenyl)cyclopentan-1-ol 16) and 2-(2-nitrophenyl)cyclohexan-1-ol 17).
2.2. Characterization of PPGs
2.2.1. Acid and Alkali Stability of the PPGs
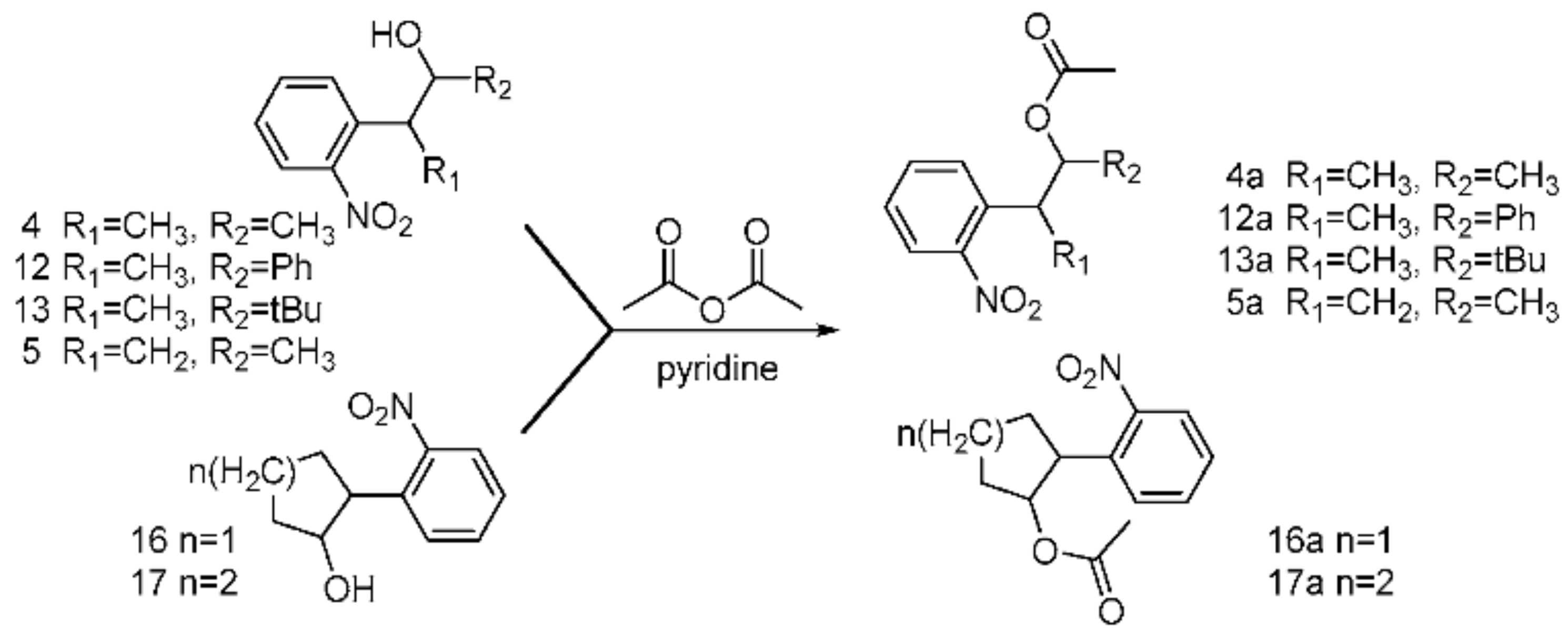
To study the acid–base sensitivity of the newly synthesized o-nitrobenyl-alcohols as multitalented PPGs in peptide chemistry, all the o-nitrobenyl alcohols were acetylated to form the corresponding esters (Scheme 6). Then, all the esters were dissolved in a TFA solution and 20% piperidine–DMF for 24 h under ambient temperature and monitored via TLC. As a result, considering the yield of the esterification and acid–base stability, 4a and 5a exhibited high yields, and 12a, 13a, 16a, and 17a were stable in an alkaline solution and unstable in an acidic solution. These results indicate that the active side chain containing 4a and 5a could be exposed for Fmoc-SPSS under acidolysis-inducing conditions and in piperidine solution (Table 1). Therefore, compounds 4a and 5a are protective groups suitable for peptide synthesis.
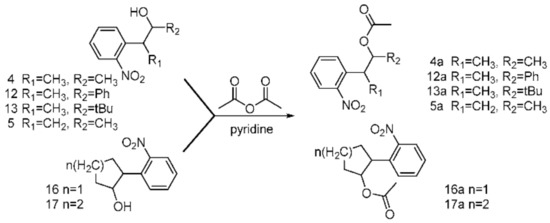
Scheme 6.
The process of the acetylation of o-nitrobenzyl alcohols.

Table 1.
Tolerance test of protecting groups under acidic and alkaline conditions.
2.2.2. Photolysis of Esterified Compounds (4) and (5)
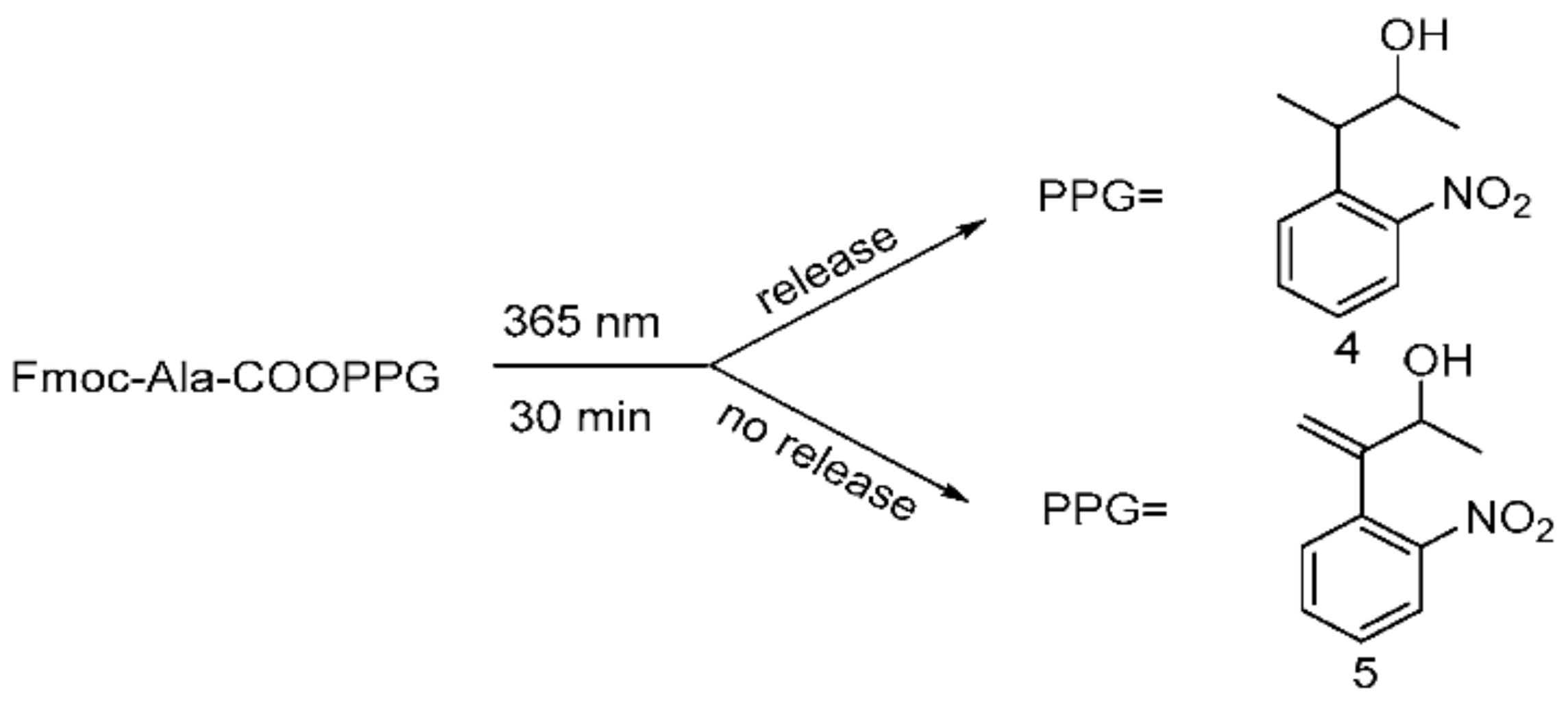
Afterwards, to research the photolability of 4 and 5, we utilized conventional conditions for photolysis experiments. Two o-nitrobenyl-alcohols were separately condensed with Fmoc-Ala-OH to form the corresponding Fmoc-Ala-COPPG esters (Scheme 7), which were dissolved in methanol and irradiated at 365 nm for 30 min using a UV lamp (6 W); the reactions were quantitatively monitored with TLC.
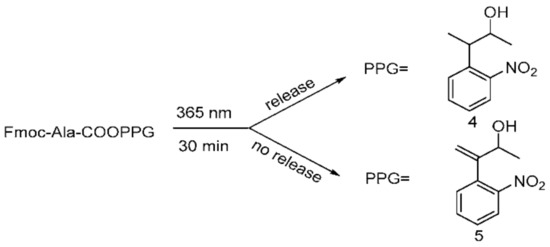
Scheme 7.
Photolysis processes for esterified compounds 4 and 5.
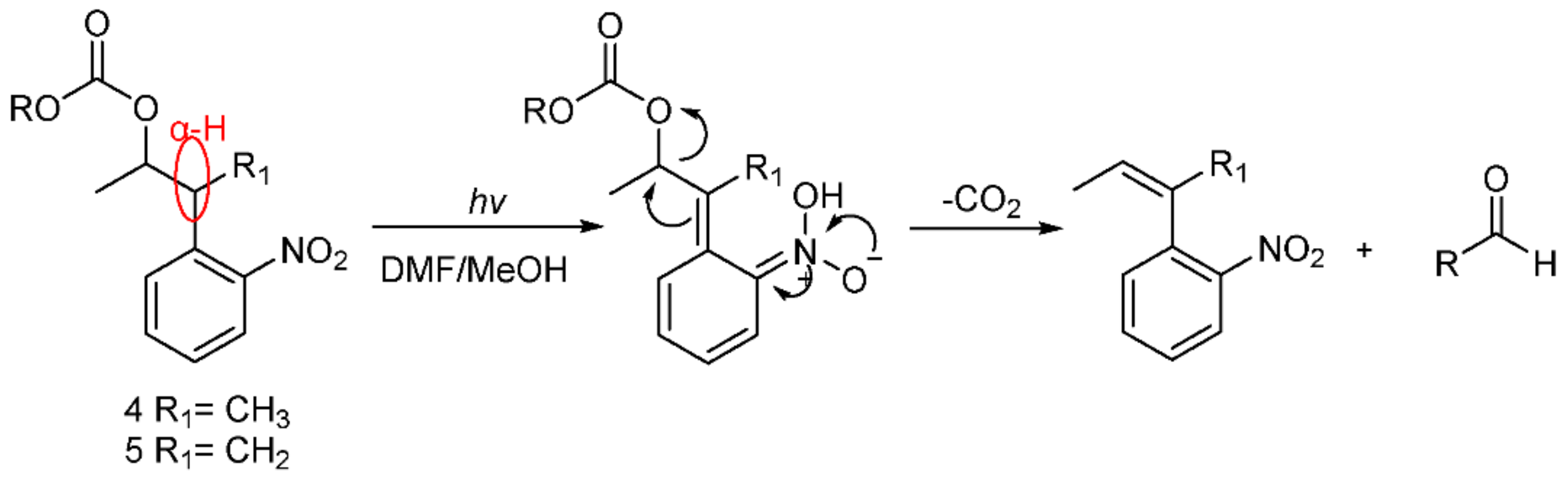
We observed that 5 could not be released, whereas 4 could be completely released, which suggested that 4 has high photolytic efficiency. A possible mechanism of photolysis (Scheme 8), based on that of Npp [16,21], was inferred here. Under the photolysis conditions, compound 4 first underwent hydrogen transfer at the α site, and subsequent β-elimination led to the expected o-nitrostyrene product and the release of the alcohol (ROH) from the carbonate. Of course, compound 5 could not undergo hydrogen transfer due to the absence of hydrogen at the α site. Therefore, compound 4 had a high photolysis rate, and compound 5 had a poor photolysis rate. The results also demonstrate that the presence of α-H at the o-nitrophenyl α site was necessary for the photolability of the o-nitrophenyl propanol compounds.
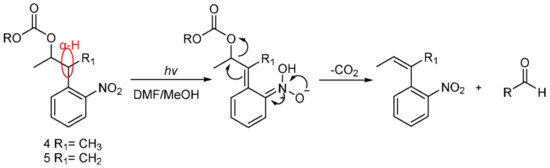
Scheme 8.
Possible mechanism of photolysis.
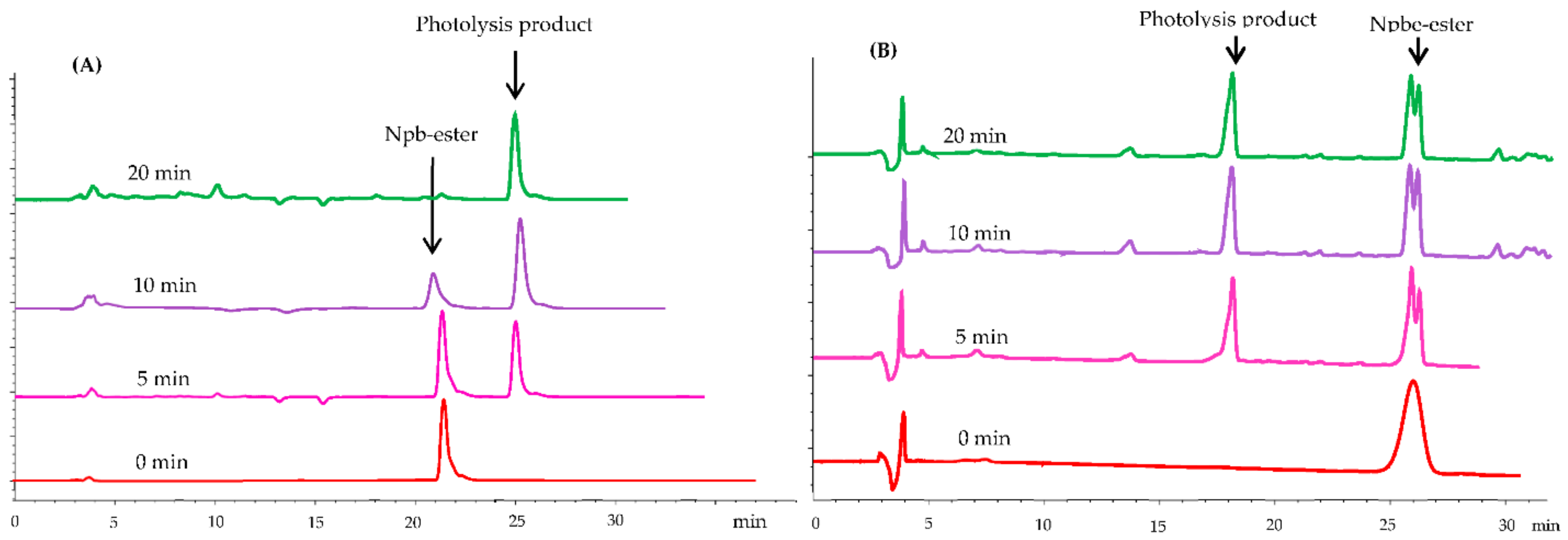
Next, we used RP-HPLC to further investigate the photochemical properties during irradiation at 365 nm. Here, a 0.2 mmol solution of 4a and 5a in DMF–MeOH (3:1, v/v) was prepared to study the progress of photolysis, which was carried out with a LED-UV (365 nm and 350 W) for 0, 5, 10, and 20 min. The analysis was performed on a Venusil ASB C18 column (5 × 150 × 4.6 mm) with CH3CN-H2O (0.1% TFA) as the eluent system (10–60%, 20 min). The photolysis process is shown in Figure 1. During irradiation, the 4a peak gradually disappeared as the reaction proceeded, and it was substituted by a new peak corresponding to the photolysis product (retention time: 25.043 min). After 20 min, 4a was completely photolyzed, but 5a failed to complete photolysis. These results indicate that the photolysis reaction had no side reactions and that 4 had a better photolytic efficiency than 5.
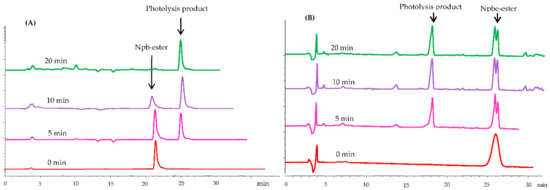
Figure 1.
HPLC traces recorded during irradiation (0, 5, 10, and 20 min, at 365 nm) of 4a in DMF–MeOH (3:1, v/v) with CH3CN–H2O (0.1% TFA) as eluent system (10–60%, 20 min). (A) Npb-ester (4) photolysis analysis spectrum; (B) Npbe-ester (5) photolysis analysis spectrum.
2.2.3. DKP Side Reactions
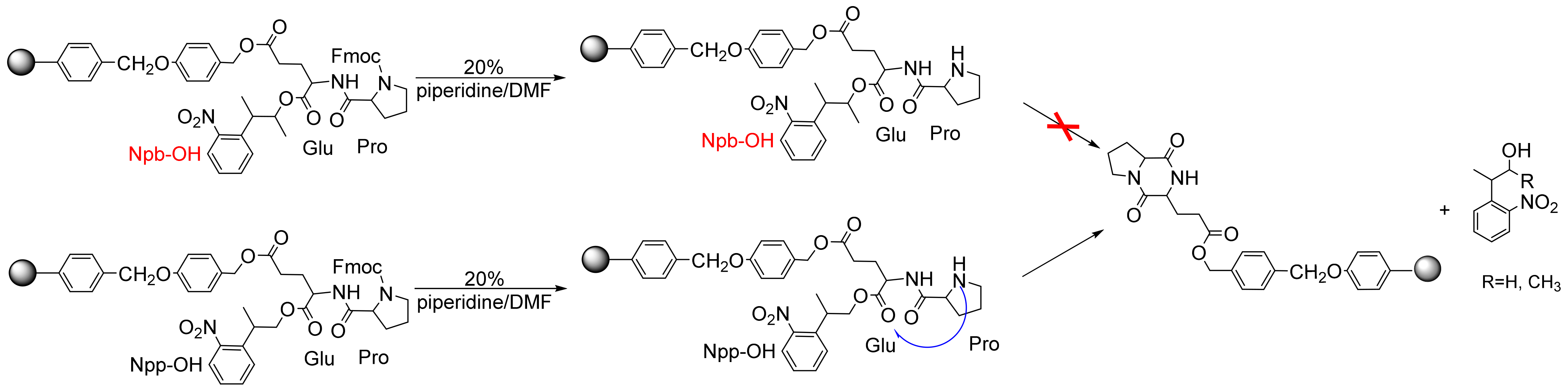
One of the most encountered side reactions in any strategy used for Wang resins is the formation of diketopiperazine (DKP) (2,5-piperazinediones) [29,30,31,32]. The configuration, conformation, and steric hindrance of amino acids all play important roles in SPPS, as DKP formation is especially prone to occur when the solid-phase carrier is an ester junction arm. Especially when the first amino acid is a sterically unhindered amino acid (e.g., glycine), the free NH2 of the second amino acid (e.g., proline) with a cis-amide conformation can easily attack the ester structure, leading to the premature removal of the peptide. Its stable six-membered ring provides the thermodynamic force of the reaction. When we fixed the side chain of amino acids to prepare cyclic peptides, this reaction led to the dissociation of the carboxy-terminal protective group. The side effect resulted in both a lower yield and the crude reaction of several deletion peptides lacking the first amino acids. Thus, we designed the Glu–Pro dipeptide as a template compound and used Npb-ester as a side-chain-protection strategy to avoid the DKP side reaction. We attempted to protect the first Fmoc-glutamic acid side-chain carboxyl group supported on Wang resin with Npb-OH (4) and Npp-OH, then loaded it with Fmoc-Pro-OH, and finally deprotected it with 20% piperidine–DMF. Via TLC monitoring, we found that the Npp-OH PPG was released, but 4 was not liberated. We believe that 4 can effectively counteract the occurrence of DKP side reactions because, in our study, when the C-terminal amino acid was linked via a non-bulky ester group (e.g., Wang resin), the formation of DKP was often accompanied by the deprotection of the second amino acid residue and subsequent acylation, resulting in the release of PPGs (Scheme 9). Therefore, compound 4 is an appropriate PPG for peptide synthesis.
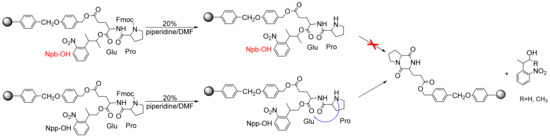
Scheme 9.
Diketopiperazine (DKP) side reaction.
2.2.4. Applicability of PPG 4
We next investigated compound 4′s addition to carboxyl acids with a wide range of esterification protocols. Based on previous studies, the ester was prepared with the DCC–DMAP method (Table 2). The esterification of carboxyl groups by 4 enabled the corresponding esters to be obtained in higher yields and esters to be stable during the acid removal of the side chains, together demonstrating compound 4 to be a carboxyl-protecting group with wide adaptability. Therefore, the Npb-OH photolabile protecting group was considered to be a third-dimensional protective group for Glu residue side-chain protection to be applied in cyclic peptide synthesis.
Table 2.
Carboxyl protection with compound 4.
2.3. Application of PPGs in Cyclic Peptide Synthesis
Anti-inflammatory peptides I [33], II [34], and III [35], all containing Glu residues, were chosen to illustrate the use of the Npb-OH strategy in head-to-tail cyclic peptide I and head-to-side cyclic peptide II and III synthesis (Scheme 10 and Table 3). Firstly, we photo-protected the carboxyl group of the Fmoc-glutamic acid side chain with 4 via an esterification reaction, and then, we removed the t-butyl side chain (which was assembled on the peptide chain via Fmoc SPPS) under standard acidic conditions. The deprotection of the N-terminus of Fmoc was conducted using 20% piperidine–DMF, and the formation of the peptide bond was accomplished with the Fmoc-SPSS strategy. After the completion of peptide assembly, the photo-deprotection reaction was carried out with a LED-UV (365 nm and 350 W) twice for 15 min before the carboxyl group was revealed. The solvent was DMF: MeOH (3:1, v/v), which was degassed under an Ar atmosphere. When we used DMF as a solvent to photolyze the peptide, we needed to add other low-boiling solvents to counter the thermal effects of the UV lamp, so before photolysis, argon gas was pre-bubbled into the solvent to deoxidize. At the same time, 15 min after the first photolysis, the product released via photolysis caused the solution to change from colorless to yellow. The solution was filtered out and replaced with the new solvent, and the second photolysis was carried out for 15 min. The solution remained colorless, indicating that the deprotection reaction had reached the endpoint. Next, the exposed carboxyl group and the N-terminal amino group formed a lactam ring on the resin via a condensation reaction. The cyclic peptide was split from the resin (Scheme 10). The lysate was concentrated, ice diethyl ether was added, and the solid was collected by centrifugation and blow-dried with argon. The purity and structure were confirmed using analytical HPLC (see Supplementary Materials, Figures S1–S3) and Q-FT-ICR–MS (Table 3).
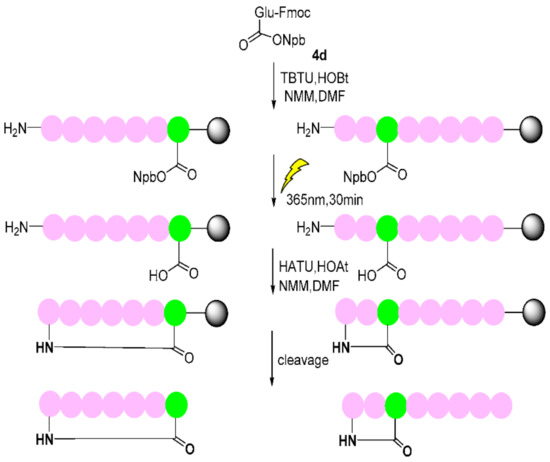
Scheme 10.
The synthesis of head-to-tail cyclic peptide I and head-to-side peptides II and III based on the Npb strategy (Green: glutamic acid; pink: other amino acids in the peptide chain).
Table 3.
Sequences and masses of cyclic peptides used in this study.
3. Materials and Methods
Room temperature (r.t) refers to ambient temperature. Solid-phase synthesis was manually carried out in a polypropylene syringe containing a polyethylene frit. The visualization of the TLC was performed using UV light. The photolysis was performed via a LED-UV (365 nm and 350 W). Flash column chromatography was performed on silica gel (200–300 mesh). The 1H NMR and 13C NMR spectra were recorded with a Bruker AM 400 and AM 500 MHz spectrometer (Palo Alto, CA, USA). The chemical shifts (δ) are reported in ppm, with an internal standard of tetramethylsilane (0 ppm). The coupling constants (J) are provided in hertz. Multiplets were designated as s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). High-resolution mass spectrometry (HRMS) measurements were recorded using an Agilent 1260-G6230A ESI–HQMS spectrometer (Agilent, USA). Analytical high-pressure liquid chromatography (HPLC) was carried out with an Agilent instrument (Agilent1100, Agilent, USA), automatic injector, photodiode array detector, system controller (Empower login, Agilent, USA), and Venusil ASB C18 column (5 mm, 150 × 4.6 mm, Bonna-Agela Technologies, Tianjin, China). UV measurements were recorded at 220 nm.
3.1. Synthesis of the Photolabile Protecting Groups (PPGs)
General Reduction Procedure
NaBH4 (0.40 g, 10.6 mmoL) was slowly added to a suspension of 3-(2-nitrophenyl) butan-2-one 2 (1.02 g, 5.3 mmoL) in MeOH (10 mL) while the temperature was kept below 0 °C for 30 min. After stirring for 3 h at r.t, the mixture was acidified with a HCl solution (2 M, 10 mL), extracted with DCM (3 × 10 mL), and washed with saturated brine. The combined organic layer was dried over MgSO4 and evaporated, and the crude product was purified with flash column chromatography (column chromatography eluent; petroleum ether:EtOAc = 15:1) to yield product 4 [19]. The new compounds 12 [19], 13, 16, 17, and 5 were characterized as follows.
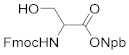
3-(o-Nitrophenyl)butan-2-ol (4): 1.54 g, 80.2% yield. Column chromatography eluent, PE:EA = 10:1. Red-brown oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.75 (d, J = 8.4 Hz, 1H), 7.67–7.60 (m, 2H), 7.36 (t, J = 6.9 Hz, 1H), 4.61 (d, J = 4.9 Hz, 1H), 3.76–3.70 (m, 1H), 2.99 (p, J = 7.1 Hz, 1H), 1.23 (d, J = 7.0 Hz, 3H), 1.04 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 151.0, 138.5, 132.2, 129.0, 126.8, 123.1, 69.7, 40.7, 21.6, 17.7; HRMS (ESI-TOF) calculated for C10H13NO3 [M+Na]+ 218.0793, Found 218.0788. The structure of product 4 was confirmed by the 1H- and 13C-NMR spectra (Figure S4).
3-(o-Nitrophenyl)but-3-en-2-ol (5): 2.34 g, 68.2% yield. Column chromatography eluent, PE:EA = 10:1. Light brown oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.86 (d,1H,Ar-H), δ 7.63 (t, 1H, Ar-H), δ 7.87 (d, J = 8.1 Hz, 1H), 7.64 (t, J = 7.6 Hz, 1H), 7.50 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 5.35 (d, J = 17.3 Hz, 1H), 5.08 (d, J = 5.0 Hz, 1H), 4.92 (d, J = 17.3 Hz, 1H), 4.40–4.34 (m, 1H), 1.07 (d, J = 6.4 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 151.1, 149.3, 135.4, 133.0, 131.7, 129.0, 124.1, 113.6, 68.5, 23.2; HRMS (ESI-TOF) calculated for C10H11NO3 [M+H]+ 194.0817, Found 194.0812. The structure of product 5 was confirmed by the 1H- and 13C-NMR spectra (Figure S5).
2-(2-nitrophenyl)-1-phenylpropan-1-ol (12): 1.59 g, 83.0% yield. Column chromatography eluent, PE:EA = 10:1. Light yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (dd, J = 12.0, 8.1 Hz, 2H), 7.66 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.32–7.22 (m, 5H), 5.32 (s, 1H), 4.59 (d, J = 8.5 Hz, 1H), 3.49–3.42 (m, 1H), 1.05 (d, J = 7.0 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 151.5, 144.8, 138.8, 132.8, 129.3, 128.4, 128.4, 127.7, 127.4, 127.1, 127.1, 123.7, 77.3, 41.2, 18.3; HRMS (ESI-TOF) calculated for C15H15NO3 [M-H]− 256.0974, Found 256.0967. The structure of product 12 was confirmed by the 1H- and 13C-NMR spectra (Figure S9).
2,2-dimethyl-4-(2-nitrophenyl)pentan-3-ol (13): 1.82 g, 85.4% yield. Column chromatography eluent, PE:EA = 6:1. Yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.85 (dd, 1H), δ 7.60 (dd, 1H), δ 7.49 (m, 1H), δ 7.43 (m, 1H), δ 4.56 (d, 1H), δ 3.14 (m, 1H), δ 3.09 (d, 1H), δ 2.72 (dd, 1H), δ 0.88 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 150.9, 138.9, 132.0, 131.8, 127.3, 123.3, 81.8, 36.1, 34.3, 27.2, 27.2 27.2, 23.2; HRMS (ESI-TOF) calculated for C13H19NO3 [M-H]− 236.1285, Found: 236.1289. The structure of product 13 was confirmed by the 1H- and 13C-NMR spectra (Figure S10).
2-(o-Nitrophenyl)cyclopentan-1-ol (16): 0.48 g, 46.2% yield. Column chromatography eluent, PE:EA = 10:1. Yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 8.0 Hz, 1H), 7.67–7.59 (m, 2H), 7.43 (t, J = 7.0 Hz, 1H), 4.82 (s, 1H), 4.11 (q, J = 7.1 Hz, 1H), 3.11 (dt, J = 10.1, 7.9 Hz, 1H), 2.16–2.08 (m, 1H), 2.00–1.92 (m, 1H), 1.79–1.69 (m, 2H), 1.60–1.49 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 151.4, 138.2, 133.17, 128.9, 127.5, 123.8, 78.6, 48.5, 34.7, 32.7, 22.2; HRMS (ESI-TOF) calculated for C11H13NO3 [M+Na]+ 230.0793, Found 230.0785. The structure of product 16 was confirmed by the 1H- and 13C-NMR spectra (Figure S11).
2-(o-Nitrophenyl)cyclohexan-1-ol (17): 1.74 g, 60.8% yield. Column chromatography eluent, PE:EA = 10:1. Yellow oil. 1H-NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.40 (dq, J = 8.4, 4.2 Hz, 1H), 4.52 (d, J = 5.6 Hz, 1H), 3.59–3.53 (m, 1H), 2.78 (td, J = 12.1, 10.0 Hz, 1H), 1.93 (dd, J = 11.8, 4.3 Hz, 1H), 1.84 (dd, J = 12.5, 3.9 Hz, 1H), 1.74–1.67 (m, 2H), 1.54–1.31 (m, 2H), 1.28–1.17 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 151.6, 139.0, 132.9, 128.9, 127.2, 123.8, 72.3, 46.7, 36.6, 33.2, 26.1, 25.0; HRMS (ESI-TOF) calculated for C12H15NO3 [M+Na]+ 244.0950, Found 244.0942. The structure of product 17 was confirmed by the 1H- and 13C-NMR spectra (Figure S12).
3.2. Photolysis of Esterified Compounds (4) and (5)
In a 50 mL round-bottomed quartz bottle, 30 mL of 0.2 mmol 4a and 5a in DMF-MeOH (3:1, v/v) solution were added to study the photolysis process, respectively, which was carried out with a LED-UV (365 nm and 350 W) for 0, 5, 10, and 20 min. The photolysis experiment used LED-UV (365 nm, 700 W), and the actual photolysis power was 50% of the rated power, which was 350 W. The analysis was performed on a Venusil ASB C18 column (5 × 150 × 4.6 mm) with CH3CN-H2O (0.1% TFA) as the eluent system (10–60%, 20 min).
3.3. Applicability of PPG 4
3.3.1. General Esterification Procedure
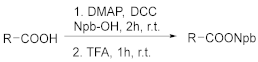
A solution of carboxylic acid (5 mmol), DMAP (0.5 mmol), and Npb-OH (5 mmol) in 50 mL of DCM was cooled while stirring in an ice bath. DCC (6 mmol) was added in small portions, and the reaction mixture was stirred at 0 °C for 30 min and then at r.t for 2 h. The solution was filtered, the filtrate was concentrated to dryness under vacuum, and the residue was purified with flash column chromatography. The new compounds 4b, 4e, 4f, 4g, and 4h were characterized as follows.
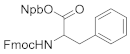
3-(2-nitrophenyl)butan-2-yl(((9H-fluoren-9-yl)methoxy)carbonyl)alaninate (4b). 1.30 g, 83.4% yield. Column chromatography eluent, PE:EA = 6:1. Light yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 7.5 Hz, 2H), 7.79 (d, J = 7.5 Hz, 1H), 7.71–7.67 (m, 3H), 7.65–7.60 (m, 2H), 7.46–7.40 (m, 3H), 7.35–7.28 (m, 2H), 5.06 (m, 1H), 4.29–4.17 (m, 3H), 3.82 (q, J = 7.3 Hz, 1H), 3.24 (q, J = 7.3 Hz, 1H), 1.29 (d, J = 5.6 Hz, 3H), 1.19 (d, J = 5.5 Hz, 3H), 1.17 (dd, J = 8.1, 5.5 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 172.6, 156.2, 151.1, 144.2, 143.0, 141.2, 141.2, 137.1, 133.0, 129.4, 129.0, 129.0, 128.9, 128.9, 128.1, 127.5, 127.5, 125.7, 120.6, 120.6, 74.6, 60.2, 50.1, 47.0, 39.2, 18.7, 18.1, 16.5; HRMS (ESI-TOF) calculated for C28H28N2O6 [M+H]+ 489.2026, Found 489.2019. The structure of 4b was confirmed by the 1H- and 13C-NMR spectra (Figure S13).
3-(2-nitrophenyl)butan-2-yl(((9H-fluoren-9-yl)methoxy)carbonyl)phenylalaninate (4e). 2.28 g, 80.3% yield. Column chromatography eluent, PE: EA = 6:1. Light brown oil. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 7.6 Hz, 2H), 7.82–7.77 (m, 2H), 7.70 (d, J = 8.1 Hz, 1H), 7.66–7.61 (m, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.45–7.38 (m, 3H), 7.31–7.24 (m, 4H), 7.21–7.11 (m, 3H), 5.11 (dq, J = 8.7, 6.0 Hz, 1H), 4.19–4.11 (m, 3H), 3.99 (dt, J = 12.2, 7.9 Hz, 1H), 3.31–3.24 (m, 1H), 2.37 (dd, J = 13.8, 11.0 Hz, 1H), 2.24 (dd, J = 13.7, 3.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H), 1.18 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 171.7, 156.3, 151.2, 144.1, 144.1, 141.1, 141.1, 137.9, 137.0, 133.1, 129.4, 129.4, 129.1, 128.7, 128.7, 128.7, 128.1, 128.1, 127.5, 127.5, 127.0, 125.7, 120.6, 120.6, 75.2, 66.1, 56.1, 47.0, 39.0, 36.0, 18.7, 18.0; HRMS (ESI-TOF) calculated for C34H32N2O6 [M+H]+ 565.2339, Found 565.2333. The structure of 4e was confirmed by the 1H- and 13C-NMR spectra (Figure S16).
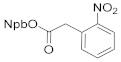
3-(2-nitrophenyl)butan-2-yl 2-(2-nitrophenyl)acetate (4f). 1.68 g, 93.6% yield. Column chromatography eluent, PE:EA = 6:1. Light yellow/brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 7.8Hz, 1H), 7.73 (dd, J = 7.8, 1.4 Hz, 2H), 7.60–7.51 (m, 2H), 7.45 (d, J = 1.4 Hz, 2H), 7.39 (d, J = 7.8 Hz, 1H), 5.03–5.00 (m, 1H), 3.89 (d, J = 7.1 Hz, 2H), 3.26 (t, J = 7.2 Hz, 1H), 1.23 (d, J = 7.0 Hz, 3H), 1.08 (d, J = 6.2 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 169.3, 151.0, 148.5, 136.5, 134.5, 134.1, 132.9, 130.2, 129.3, 129.2, 128.1, 125.3, 123.8, 74.6, 39.4, 38.3, 18.4, 18.0; HRMS (ESI-TOF) calculated for C18H18N2O6 [M+H2O]+ 376.1271, Found: 376.1504. The structure of 4f was confirmed by the 1H- and 13C-NMR spectra (Figure S17).
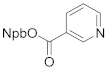
3-(2-nitrophenyl)butan-2-yl nicotinate (4g). 1.35 g, 89.6% yield. Column chromatography eluent, PE:EA = 6:1. Light brown oil. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (d, J = 2.2 Hz, 1H), 8.77 (dd, J = 4.8, 1.7 Hz, 1H), 8.07 (dt, J = 8.0, 2.0 Hz, 1H), 7.78 (dd, J = 8.1, 3.3 Hz, 2H), 7.65 (t, J = 4.8 Hz, 1H), 7.51 (dd, J = 4.8, 1.7 Hz, 1H), 7.41 (t, J = 4.8 Hz, 1H), 5.36 (tq, J = 8.8, 6.2 Hz, 1H), 3.49–3.41 (m, 1H), 1.39 (d, J = 5.4 Hz, 3H), 1.35 (d, J = 5.4 Hz,3H); 13C NMR (150 MHz, DMSO-d6) δ 164.0, 151.0, 150.4, 147.8, 137.8, 136.9, 133.1, 129.5, 128.2, 127.8, 124.9, 123.8, 75.3, 38.9, 18.6, 18.2; HRMS (ESI-TOF) calculated for C16H16N2O4 [M+H]+ 301.1188, Found 301.1185. The structure of 4g was confirmed by the 1H- and 13C-NMR spectra (Figure S18).
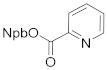
3-(2-nitrophenyl)butan-2-yl picolinate (4h). 1.40 g, 93.3% yield. Column chromatography eluent, PE:EA = 6:1. Light yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (d, J = 4.8 Hz, 1H), 7.94 (t, J = 7.7 Hz, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.77 (t, J = 4.8 Hz, 2H), 7.67–7.58 (m, 2H), 7.42 (t, J = 7.8 Hz, 1H), 5.37 (p, J = 6.3 Hz, 1H), 3.48 (q, J = 7.2 Hz, 1H), 1.37 (d, J = 8.9 Hz, 3H), 1.31 (d, J = 6.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 164.2, 154.2, 151.1, 150.2, 137.0, 136.0, 133.1, 129.0, 128.2, 125.8, 124.3, 123.7, 75.8, 39.1, 18.7, 17.9; HRMS (ESI-TOF) calculated for C16H16N2O4 [M+H]+ 301.1188, Found 301.1183. The structure of 4h was confirmed by the 1H- and 13C-NMR spectra (Figure S19).
3.3.2. General TFA Deprotection Procedure
The tBu-protected Npb-ester (5 mmol) was dissolved in TFA and stirred until the reaction was completed (1 h). The solvent was removed under reduced pressure. The crude residue was purified with flash column chromatography to yield the product as a yellow oil or an amorphous yellow solid. The new compounds 4c and 4d were characterized as follows.
3-(2-nitrophenyl)butan-2-yl(((9H-fluoren-9-yl)methoxy)carbonyl)serinate (4c). 1.82 g, 72.1% yield. Column chromatography eluent, PE:EA = 2:1. Yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 7.5 Hz, 2H), 7.79–7.74 (m, 1H), 7.71 (d, J = 7.5 Hz, 2H), 7.66–7.61 (m, 2H), 7.53–7.50 (m, 1H), 7.46–7.39 (m, 3H), 7.33–7.30 (m, 2H), 5.11–5.02 (m, 1H), 4.28–4.19 (m, 3H), 3.98–3.95 (m, 1H), 3.55 (d, J = 7.5 Hz, 1H), 3.35–3.22 (m, 3H), 1.27 (d, J = 6.2 Hz, 3H), 1.10 (dd, J = 9.3, 5.6 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 170.3, 156.5, 151.0, 144.3, 144.3, 141.2, 141.2, 136.6, 133.0, 128.1, 128.1, 128.1, 127.5, 127.5, 127.5, 127.5, 125.7, 125.7, 120.6, 120.6, 74.4, 66.2, 61.1, 57.4, 47.0, 38.6, 18.4, 18.0; HRMS (ESI-TOF) calculated for C28H28N2O7 [M+Na]+ 527.1794, Found 527.1792. The structure of 4c was confirmed by the 1H- and 13C-NMR spectra (Figure S14).
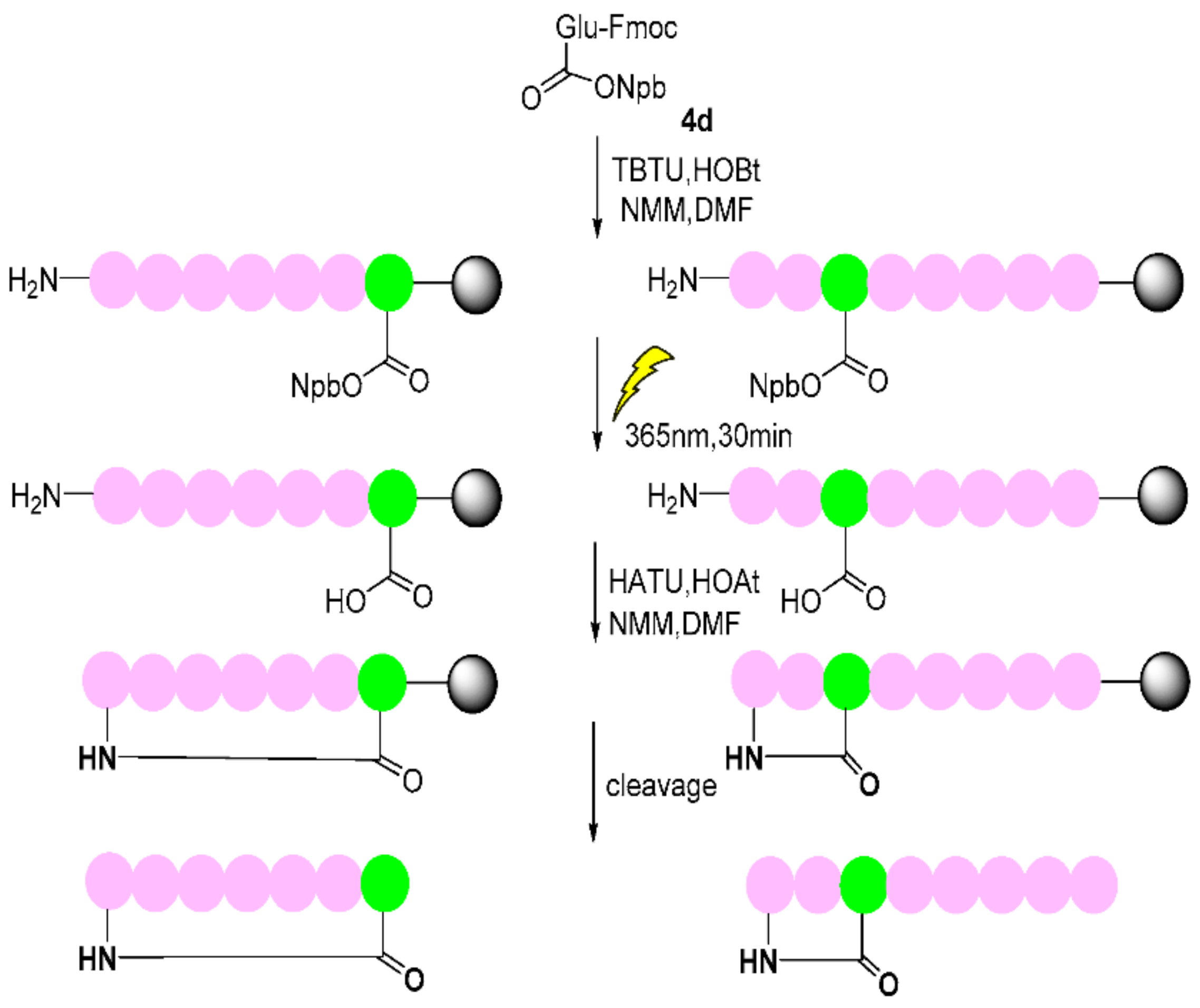
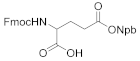
2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-5-((3-(2-nitrophenyl)butan-2-yl)oxy)-5-oxopentanoic acid (4d). 2.48 g, 90.8% yield. Column chromatography eluent, PE:EA = 2:1. Light yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 12.63 (s, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.75–7.68 (m, 3H), 7.66–7.58 (m, 3H), 7.44–7.38 (m, 3H), 7.29 (t, J = 7.5 Hz, 2H), 5.03–4.98 (m, 1H), 4.31–4.20 (m, 3H), 3.90–3.85 (m, 1H), 3.32–3.29 (m, 1H), 2.10–2.21 (m, 2H), 1.82–1.79 (m, 1H), 1.66–1.60 (m, 1H), 1.30 (d, J = 7.0 Hz, 3H), 1.19 (d, J = 6.9 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 173.8, 171.1, 156.4, 151.1, 144.3, 144.2, 141.18, 136.8, 136.3, 132.3, 129.6, 129.2, 128.1, 128.1, 127.5, 127.5, 125.7, 125.7, 123.8, 120.6, 120.6, 74.7, 66.1, 60.2, 47.1, 38.9, 31.2, 21.2, 17.7, 14.6; HRMS (ESI-TOF) calculated for C30H30N2O8 [M-H]− 545.1924, Found 545.1927. The structure of 4d was confirmed by the 1H- and 13C-NMR spectra (Figure S15).
3.4. Application of PPGs in Cyclic Peptide Synthesis
3.4.1. General Procedure for Solid-Phase Reactions
The peptide was synthesized by using 2.005 g of resin (0.11 mmol) as a starting material, followed by (a) Fmoc deprotection with 20% piperidine–DMF (6 mL, 3 equiv.) for 15 min and washing in DMF (6 mL × 3 × 30 s), and (b) establishing peptide-coupling conditions comprising three equivalents of HOBt, three equivalents of TBTU, three equivalents of Fmoc-amino acid, and three equivalents of NMM in DMF (6 mL) for 2 h at r.t, followed by washing in DMF (6 mL × 3 × 30 s). The coupling reaction was monitored with the Kaiser test.
3.4.2. Removal of the Npb Group via Photolysis Experiment
After the last amino acid with the Fmoc group was loaded, and the Fmoc group was removed, the resin-bound linear peptide was transferred to a quartz bottle and suspended in 50 mL of DMF–MeOH (3:1, v/v, 30 mL). The solvent was degassed for 10 min under argon gas to remove the dissolved oxygen. The mixture was then irradiated with a high-pressure Hg lamp (λ max = 365 nm and 250 W) with constant argon bubbling. A reflux condenser was essential because of the heat effect during UV irradiation. After 15 min, the lamp was turned off, and the dark yellow solvent was replaced with fresh solvent mixtures of DMF–MeOH (3:1, v/v, 30 mL), followed by continuous irradiation until the solvent stopped turning yellow. This procedure usually needed 30 min in total. Finally, the photolyzed resin was collected with filtration and then washed with DCM (3 mL × 3 × 30 s) and DMF (3 mL × 3 × 30 s). A synthesis procedure was performed after irradiating for 15 min, as described next.
3.4.3. Cyclization of Peptides I, II, and III
The resin-bound linear peptide was transferred into a reaction vessel and suspended in 6 mL of DMF; then, HATU (3 equiv.), HOAt (3 equiv.), and NMM (3 equiv.) were sequentially added, and the vessel was capped and shaken at r.t until the Kaiser test was negative. Next, the solvent was removed by filtration, and the resin was washed with DMF (6 mL × 3 × 30 s) and MeOH (6 mL × 3 × 30 s). Heterogeneous 5 mL mixtures of TFA/TIS/H2O/ETD/m-Cresol (90:2.5:2.5:2.5:2.5, v/v/v/v/v) solutions were prepared and added to the reaction vessel. The resulting solution was concentrated under vacuum, and a white precipitate appeared soon after the iced ether was added. The cyclic peptides were analyzed using HPLC at 220 nm with CH3CN-H2O (0.1% TFA) as the eluent system (Figures S1–S3 and Table 3) and identified with ESI–TOF–MS.
4. Conclusions
In summary, we synthesized six candidate protective groups with acid–base tolerance, photolability, suitability, and diketopiperazine side-reaction tests. We found that Npb-OH (4) was a photolabile protective group that met our needs in all aspects. In particular, it is more stable than Npp-OH (especially in alkaline conditions) and can avoid the side reaction of diketopiperazine. Compound 4 also has the advantages of a high acid–base tolerance, fast photolysis rate, and wide applicability. The protecting group can be removed without additional reagents, and the photolysis reaction is simple, thorough, rapid, and without side reactions. Moreover, 4 has broad applicability in carboxyl protection and subsequent cyclization with peptide terminal amino groups in situ because it can form a lactam bond after removing protection. Ultimately, compared to Npp-OH, 4 has better chemical tolerance and is more suitable for solid-phase peptide synthesis. Additionally, we can regard the Npb-OH photolabile protective group as a green protective group. Furthermore, we successfully synthesized the anti-inflammatory head-to-tail cyclic peptide I and head-to-side cyclic peptides II and III with the Npb-OH group. The results indicate the feasibility of a cyclic peptide-synthesis strategy based on the Npb-OH PPG, which has the advantages of low cost, simple operation, high yield, and purity. The process was successfully applied to the synthesis of cyclic peptides, facilitating a greener process for SPPS. Further structural modifications based on compound 4 to protect -SH and -NH2 are ongoing.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27072231/s1, including the reaction procedures of the intermediates and table of contents; HPLC chromatograms and identification of cyclic peptide Ⅰ, Ⅱ, Ⅲ (in Figure S1–S3); Figure S4: 1H-NMR and 13C-NMR spectra of 4, Figure S5: 1H-NMR and 13C-NMR spectra of 5, Figure S6: 1H-NMR and 13C-NMR spectra of 7, Figure S7: 1H-NMR and 13C-NMR spectra of 9, Figure S8: 1H-NMR and 13C-NMR spectra of 11, Figure S9: 1H-NMR and 13C-NMR spectra of 12, Figure S10: 1H-NMR and 13C-NMR spectra of 13, Figure S11: 1H-NMR and 13C-NMR spectra of 16, Figure S12: 1H-NMR and 13C-NMR spectra of 17, Figure S13: 1H-NMR and 13C-NMR spectra of 4b, Figure S16: 1H-NMR and 13C-NMR spectra of 4e, Figure S17: 1H-NMR and 13C-NMR spectra of 4f, Figure S18: 1H-NMR and 13C-NMR spectra of 4g, Figure S19: 1H-NMR and 13C-NMR spectra of 4h, Figure S14: 1H-NMR and 13C-NMR spectra of 4c, Figure S15: 1H-NMR and 13C-NMR spectra of 4d [36,37,38,39].
Author Contributions
Conceptualization, L.W., T.C. and G.W.; methodology, L.W., S.Z. and T.P.; performing the chemistry experiments, T.C.; writing—original draft preparation, T.C.; writing—review and editing, L.W., S.Z. and G.W.; validation, H.Y. and J.X.; data collection and analysis, X.W. and L.T.; funding acquisition, Y.S., S.L. and L.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was performed under financial support from National Natural Science Foundation of China (Grant No. 81273431, 21102176).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally Absorbed Cyclic Peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Thapa, P.; Espiritu, M.J.; Menon, V.; Bingham, J.P. From Nature to Creation: Going around in Circles, the Art of Peptide Cyclization. Bioorg. Med. Chem. 2018, 26, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.; Du, J.; Looi, F.Y.; Tang, S.R.; De Veer, S.J.; Bony, A.R.; Rehm, F.B.H.; Xie, J.; Chan, L.Y.; Wang, C.K.; et al. An Environmentally Sustainable Biomimetic Production of Cyclic Disulfide-Rich Peptides. Green Chem. 2020, 22, 5002–5016. [Google Scholar] [CrossRef]
- Al Musaimi, O.; De La Torre, B.G.; Albericio, F. Greening Fmoc/: T Bu Solid-Phase Peptide Synthesis. Green Chem. 2020, 22, 996–1018. [Google Scholar] [CrossRef]
- Wang, G.; Peng, T.; Zhang, S.; Wang, J.; Wen, X.; Yan, H.; Hu, L.; Wang, L. 2-(2-Nitrophenyl) Propyl: A Rapidly Released Photolabile COOH-Protecting Group for Solid-Phase Peptide Synthesis. RSC Adv. 2015, 5, 28344–28348. [Google Scholar] [CrossRef]
- Kaplan, J.H.; Forbush, B.; Hoffman, J.F. Rapid Photolytic Release of Adenosine 5′-Triphosphate from a Protected Analogue: Utilization by the Na:K Pump of Human Red Blood Cell Ghosts. Biochemistry 1978, 17, 1929–1935. [Google Scholar] [CrossRef]
- Engels, J.; Schlaeger, E.J. Synthesis, Structure, and Reactivity of Adenosine Cyclic 3′,5′-Phosphate-Benzyl Triesters. J. Med. Chem. 1977, 20, 907–911. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile Protecting Groups and Linkers. J. Chem. Soc. Perkin Trans. 1 2002, 1, 125–142. [Google Scholar] [CrossRef]
- Ohtsuka, E.; Tanaka, S.; Ikehara, M. Studies on Transfer Ribonucleic Acids and Related Compounds. IX(1) Ribo-Oligonucleotide Synthesis Using a Photosensitive O-Nitrobenzyl Protection at the 2′-Hydroxyl Group. Nucleic Acids Res. 1974, 1, 1351–1358. [Google Scholar] [CrossRef]
- Zehavi, U.; Amit, B.; Patchornik, A. Light-Sensitive Glycosides. I. 6-Nitroveratryl β-D-Glucopyranoside and 2-Nitrobenzyl β-D-Glucopyranoside. J. Org. Chem. 1972, 37, 2281–2285. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Schofield, P. Photosensitive Protecting Groups. Tetrahedron Lett. 1962, 3, 697–699. [Google Scholar] [CrossRef]
- Rich, D.H.; Gurwara, S.K. Preparation of a New O-Nitrobenzyl Resin for Solid-Phase Synthesis of Tert-Butyloxycarbonyl-Protected Peptide Acids. J. Am. Chem. Soc. 1975, 97, 1575–1579. [Google Scholar] [CrossRef]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Convergent Solid-Phase Peptide Synthesis. Tetrahedron 1993, 49, 11065. [Google Scholar] [CrossRef]
- HAMMER, R.P.; ALBERICIO, F.; GERA, L.; BARANY, G. Practical Approach to Solid-phase Synthesis of C-terminal Peptide Amides under Mild Conditions Based on a Photolysable Anchoring Linkage. Int. J. Pept. Protein Res. 1990, 36, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Wöll, D.; Smirnova, J.; Galetskaya, M.; Prykota, T.; Bühler, J.; Stengele, K.P.; Pfleiderer, W.; Steiner, U.E. Intramolecular Sensitization of Photocleavage of the Photolabile 2-(2-Nitrophenyl)Propoxycarbonyl (NPPOC) Protecting Group: Photoproducts and Photokinetics of the Release of Nucleosides. Chem. A Eur. J. 2008, 14, 6490–6497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Lu, W.; Devalankar, D.A.; Ding, Z. Structurally Simple Benzyl-Type Photolabile Protecting Groups for Direct Release of Alcohols and Carboxylic Acids. Org. Lett. 2015, 17, 2114–2117. [Google Scholar] [CrossRef]
- Kretschy, N.; Holik, A.K.; Somoza, V.; Stengele, K.P.; Somoza, M.M. Next-Generation o-Nitrobenzyl Photolabile Groups for Light-Directed Chemistry and Microarray Synthesis. Angew. Chem.-Int. Ed. 2015, 54, 8555–8559. [Google Scholar] [CrossRef] [Green Version]
- Bühler, S.; Lagoja, I.; Giegrich, H.; Stengele, K.P.; Pfleiderer, W. New Types of Very Efficient Photolabile Protecting Groups Based upon the [2-(2-Nitrophenyl)Propoxy]Carbonyl (NPPOC) Moiety. Helv. Chim. Acta 2004, 87, 620–659. [Google Scholar] [CrossRef]
- Hoffmann, J.; Kazmaier, U. Development of a New NPPOC-Derived Photolabile Protecting Group Suitable for Cyclizations via Ring Closing Metathesis. Curr. Org. Synth. 2015, 12, 475–483. [Google Scholar] [CrossRef]
- Hasan, A.; Stengele, K.P.; Giegrich, H.; Cornwell, P.; Isham, K.R.; Sachleben, R.A.; Pfleiderer, W.; Foote, R.S. Photolabile Protecting Groups for Nucleosides: Synthesis and Photodeprotection Rates. Tetrahedron 1997, 53, 4247–4264. [Google Scholar] [CrossRef] [Green Version]
- Goodman, B.M.; Stueben, K.C. Peptide Synthesis via Amino Acid Active Esters. II.1 Some Abnormal Reactions during Peptide Synthesis. J. Am. Chem. Soc. 1962, 84, 1279–1283. [Google Scholar] [CrossRef]
- Yang, H.; Peng, T.; Wen, X.; Chen, T.; Sun, Y.; Liu, S.; Wang, G.; Zhang, S.; Wang, L. A Photolabile Carboxyl Protecting Group for Solid Phase Peptide Synthesis. ChemistryOpen 2021, 10, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Okuro, K.; Gurnham, J.; Alper, H. Ionic Diamine Rhodium Complex Catalyzed Reductive N-Heterocyclization of 2-Nitrovinylarenes. J. Org. Chem. 2011, 76, 4715–4720. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.; Greul, J.N.; Lui, N. Method for Producing Alkenylnitrobenzene Derivatives Unbranched in the 1′-Positon 2010, A1. Available online: https://patentscope2.wipo.int/search/en/detail.jsf?docId=WO2008006576 (accessed on 14 January 2022).
- Gall1, C. Evidence for a Non-Chain SRN1 Reaction Occurring on a Nitroarylhalide. Tetrahedron 1988, 44, 5205–5208. [Google Scholar] [CrossRef]
- Iwama, T.; Birman, V.B.; Kozmin, S.A.; Rawal, V.H. Regiocontrolled Synthesis of Carbocycle-Fused Indoles via Arylation of Siyl Enol Ethers with o-Nitrophenylphenyliodonium Fluoride. Org. Lett. 1999, 1, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.S.; Gu, B.Q.; Zhu, M.Y.; Yu, X.; Deng, W.P. Nonenzymatic Kinetic Resolution of α-Aryl Substituted Allylic Alcohols Catalyzed by Acyl Transfer Catalyst Np-PIQ. Tetrahedron 2015, 71, 1187–1191. [Google Scholar] [CrossRef]
- Gisin, B.F.; Merrifield, R.B. Carboxyl-Catalyzed Intramolecular Aminolysis. A Side Reaction in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1972, 94, 3102–3106. [Google Scholar] [CrossRef]
- Barany, G.; Albericio, F. Three-Dimensional Orthogonal Protection Scheme for Solid-Phase Peptide Synthesis under Mild Conditions. J. Am. Chem. Soc. 1985, 107, 4936–4942. [Google Scholar] [CrossRef]
- Pedroso, E.; Grandas, A.; de las Heras, X.; Eritja, R.; Giralt, E. Diketopiperazine Formation in Solid Phase Peptide Synthesis Using P-Alkoxybenzyl Ester Resins and Fmoc-Amino Acids. Tetrahedron Lett. 1986, 27, 743–746. [Google Scholar] [CrossRef]
- Chiva, C.; Vilaseca, M.; Giralt, E.; Albericio, F. An HPLC-ESMS Study on the Solid-Phase Assembly of C-Terminal Proline Peptides. J. Pept. Sci. 1999, 5, 131–140. [Google Scholar] [CrossRef]
- Moronta, J.; Smaldini, P.L.; Docena, G.H.; Añón, M.C. Peptides of Amaranth Were Targeted as Containing Sequences with Potential Anti-Inflammatory Properties. J. Funct. Foods 2016, 21, 463–473. [Google Scholar] [CrossRef]
- Del Carmen Millán-Linares, M.; Millán, F.; Pedroche, J.; del Mar Yust, M. GPETAFLR: A New Anti-Inflammatory Peptide from Lupinus Angustifolius L. Protein Hydrolysate. J. Funct. Foods 2015, 18, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Nishizawa, M.; Shimizu, Y.; Saeki, H. Anti-Inflammatory Effects of Dulse (Palmaria palmata) Resulting from the Simultaneous Water-Extraction of Phycobiliproteins and Chlorophyll a. Food Res. Int. 2017, 100, 514–521. [Google Scholar] [CrossRef]
- Ruhland, B.; Leclerc, G. Synthesis of 1-Hydroxy-2H,5H-Dihydroisoxazolo[5,4-c]Quinoline. A Novel Heterocyclic Ring System. J. Heterocycl. Chem 1989, 26, 469–471. [Google Scholar] [CrossRef]
- Nykaza, T.v.; Li, G.; Yang, J.; Luzung, M.R.; Radosevich, A.T. P III /P V =O Catalyzed Cascade Synthesis of N-Functionalized Azaheterocycles. Angew. Chem. 2020, 132, 4535–4540. [Google Scholar] [CrossRef]
- Gao, D.M.; Ma, W.L.; Li, T.R.; Huang, L.Z.; Du, Z.T. An Improved Synthesis of 1,2-Diarylethanols under Conventional Heating and Ultrasound Irradiation. Molecules 2012, 17, 10708–10715. [Google Scholar] [CrossRef] [Green Version]
- Coffman, K.C.; Palazzo, T.A.; Hartley, T.P.; Fettinger, J.C.; Tantillo, D.J.; Kurth, M.J. Heterocycle-Heterocycle Strategies: (2-Nitrophenyl)Isoxazole Precursors to 4-Aminoquinolines, 1 H-Indoles, and Quinolin-4(1 H)-Ones. Org. Lett. 2013, 15, 2062–2065. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).