
The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adenosine A2A Receptors and Parkinson’s Disease
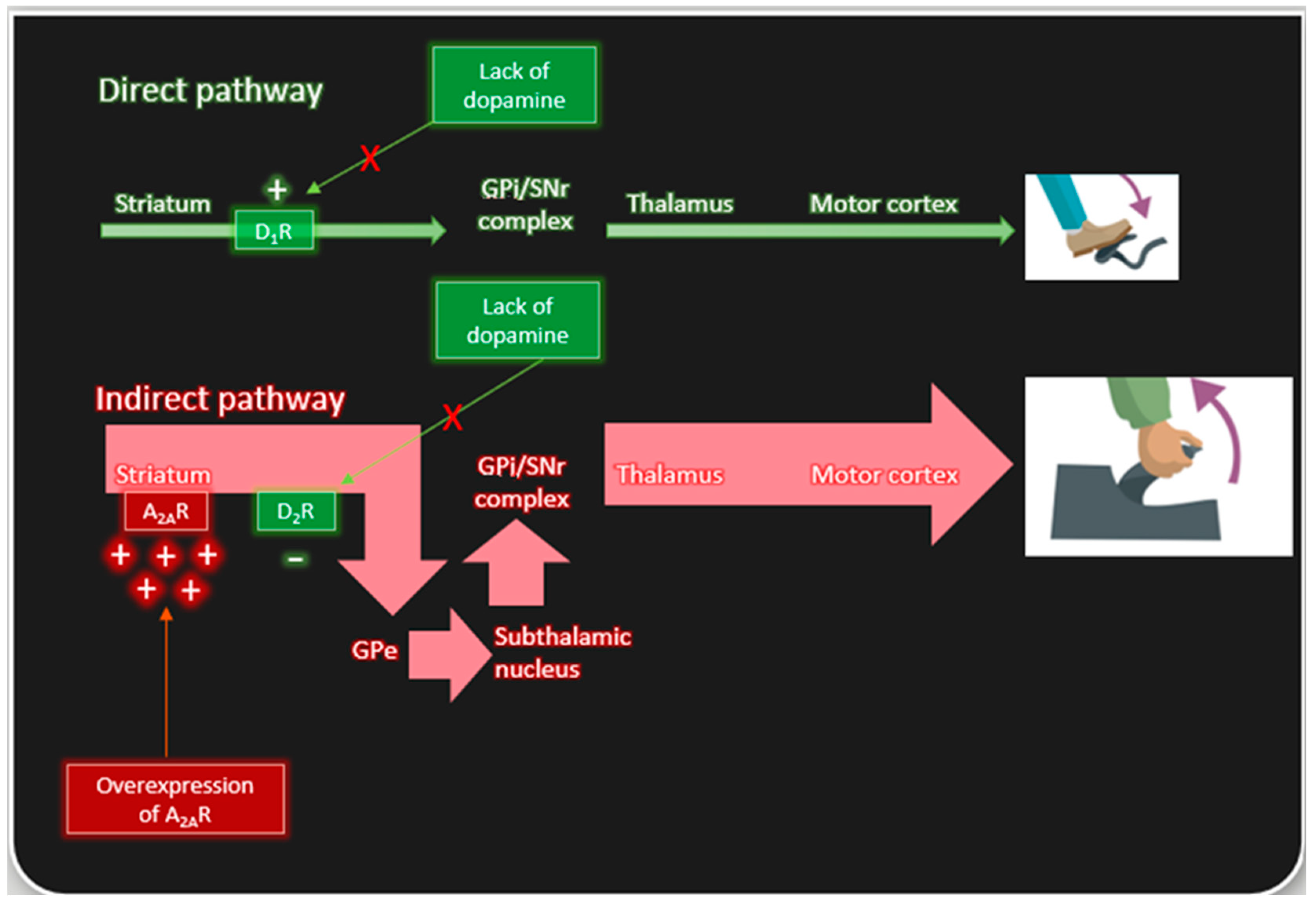
2.1. Basal Ganglia Expression of A2A Receptors
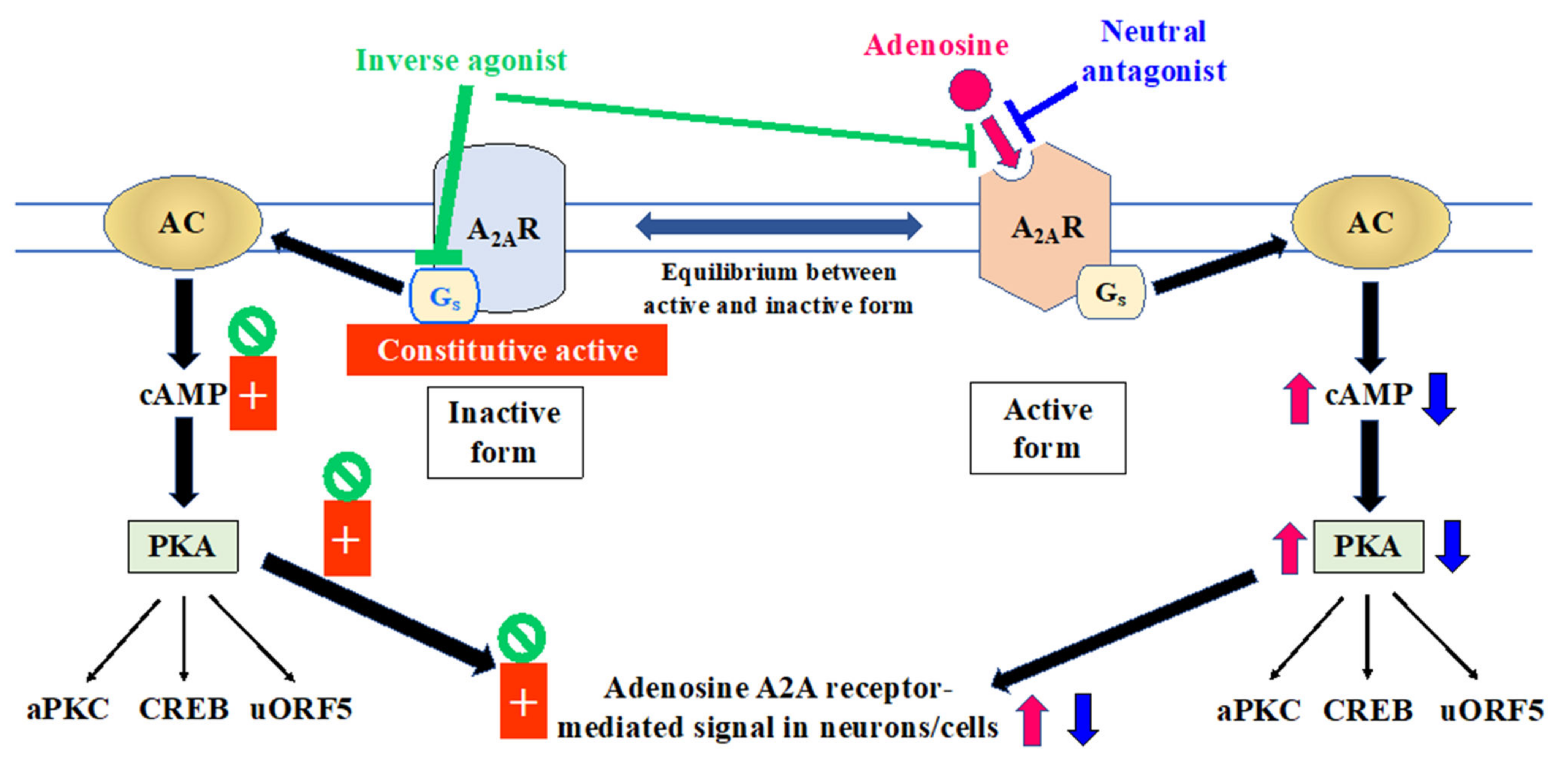
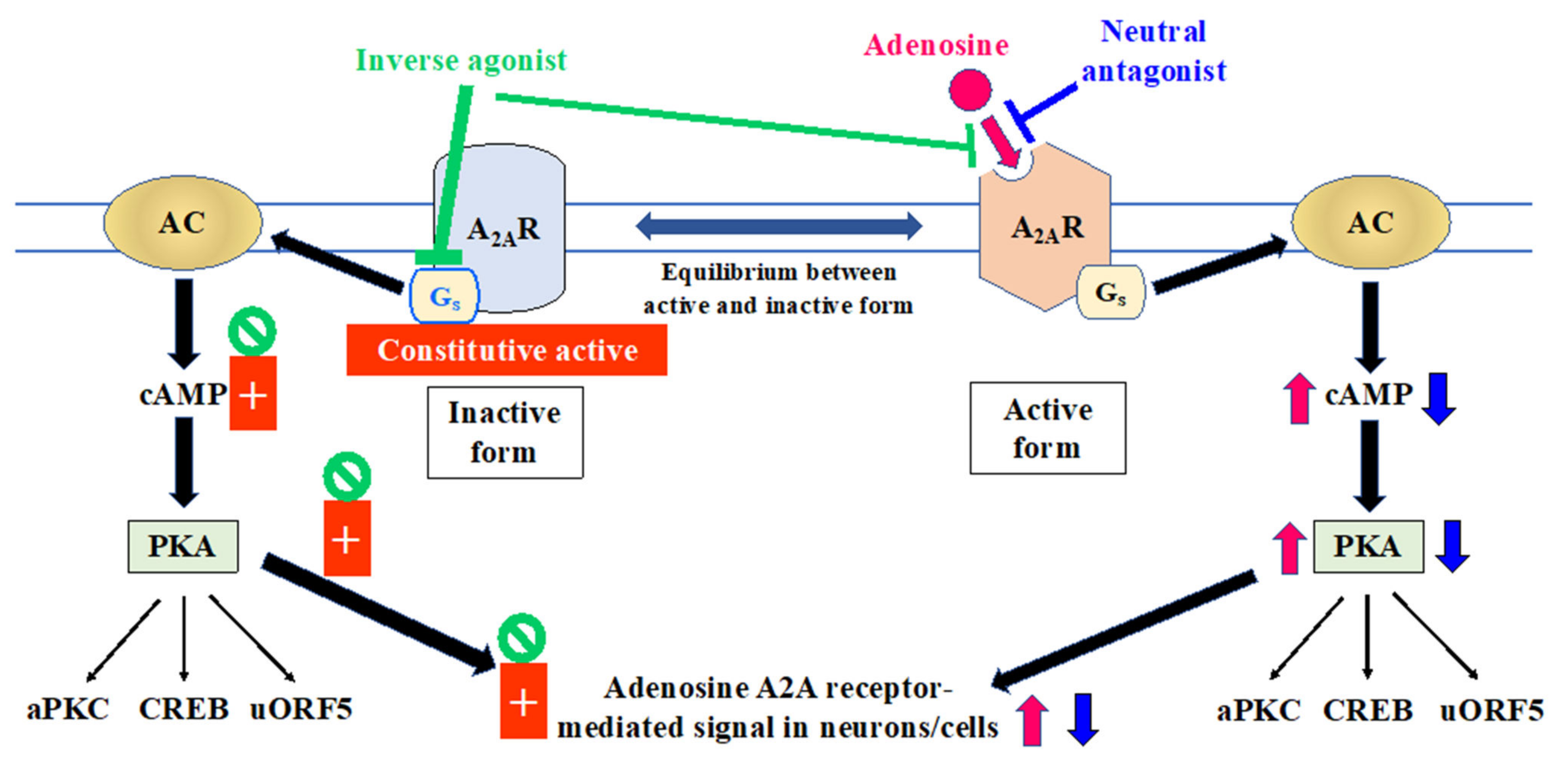
2.2. Relevance of A2A Receptor State to Mechanism of Action of Adenosinergic Agents in Managing the Motor Symptoms of Parkinson’s Disease
3. Clinical Actions of Adenosine A2A Antagonists in Parkinson’s Disease
3.1. Motor Symptoms
3.2. Non-Motor Symptoms
3.2.1. Role of A2A Receptors in Sleep–Wake Cycle Disturbance
3.2.2. Role of A2A Receptors in Mood Disorders
3.3. Role of A2A Receptors in Cognitive Impairment
4. Potential Neuroprotective Effects of A2A Receptor Antagonism in PD
- (1)
- The effect may not be due to caffeine—however, the consumption of decaffeinated coffee was not associated with a reduced risk of developing PD in the Health Professional Follow-up Study [166].
- (2)
- The effect is not universal in the population—it is strong and consistent in men in the Health Professional Follow-up Study [169] and in post-menopausal women who have never used hormone replacement therapy in the Cancer Prevention Study II Nutrition Cohort but uncertain in women and post-menopausal women who have used hormone replacement therapy at some point [172].
- (3)
- The effect could be due to early premotor features of PD causing a reduction in caffeine consumption—however, the persistence of these predictive associations in lag analyses that exclude PD cases that occurred within 2–6 years from dietary survey suggests this is unlikely.
- (4)
- The effect may reflect genetic predisposition to PD—there is emerging evidence that links dietary [173,174,175] and plasma [176]) caffeine exposure as being more negatively associated with the likelihood of developing PD among carriers of a pathogenic LRRK2 mutation than among PD patients who do not carry LRRK2 mutation [173,176].
5. Summary and Conclusions
- A2A receptors are increased in the spinal cord of patients [192].
- Partial genetic ablation of A2A receptors significantly delayed disease progression in SOD1G93A mice [192].
- The timing by which the alterations of the adenosinergic system occur during ALS pathogenesis in patients and animal models is a key factor to completely understand its contribution to disease progression and to identify the proper therapeutic window for putative treatments.
- A2A receptors are decreased in the striatum of HD patients [200].
- There is a 50% reduction in A2A receptor striatal binding in patients with HD [201].
- A reduction in radiotracer binding for A2A receptors is found in the lesioned side compared with the intact side in a rat model of HD [203].
- A2A receptor knockout mice have worse survival and motor behaviors in the Tg mouse model of HD [202].
- However, the effect of A2A receptors blockade in HD mice appears to be complex and could be detrimental because it compromises the function of BDNF [202].
- A2A receptors are increased in the brain of secondary progressive MS [195].
- A2A receptors are highly expressed on infiltrating macrophages [206].
- A2A receptor antagonist (SCH58261) protects against experimental autoimmune encephalomyelitis (EAE) in mice [207].
- A2A receptor agonist (CGS21680) inhibits the EAE progression in mice [208].
- A2A receptor knockout mice show more severe EAE pathology and neurological deficits in EAE model mice [209].

Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | adenylate cyclase |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| BDI | Beck Depression Inventory |
| EAE | experimental autoimmune encephalo-myelitis |
| ESS | Epworth Sleepiness Scale |
| GPe | external global pallidus |
| GP | globus pallidus |
| GPCR | G protein-coupled receptor |
| HD | Huntington’s disease |
| MCI | mild cognitive impairment |
| mIPSCs | miniature inhibitory postsynaptic currents |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | multiple sclerosis |
| NMDA | N-methyl-D-aspartate |
| NMSS | non-motor symptom scale |
| PD | Parkinson’s disease |
| PDQ-8 | PD Questionnaire 8-items |
| PDSS | PD sleep scale |
| PFC | prefrontal cortex |
| PHQ | Patient Health Questionnaire |
| PKA | protein kinase A |
| SHAPS-J | Snaith–Hamilton Pleasure Scale Japanese version |
| SPN | spiny projection neuron |
| 6-OHDA | 6-hydroxydopamine |
References
- IJzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Patel, R.; Gaba, S.; Singh, G.; Gupta, G.D.; Monga, V. Adenosine receptor antagonists: Recent advances and therapeutic perspective. Eur. J. Med. Chem. 2022, 227, 113907. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xie, N.; Illes, P.; Virgilio, F.D.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Atif, M.; Alsrhani, A.; Naz, F.; Imran, M.; Ullah, M.I.; Alameen, A.A.M.; Gondal, T.A.; Raza, Q. Targeting Adenosine Receptors in Neurological Diseases. Cell. Reprogramming 2021, 23, 57–72. [Google Scholar] [CrossRef]
- Salmaso, V.; Jacobson, K.A. Purinergic Signaling: Impact of GPCR Structures on Rational Drug Design. ChemMedChem 2020, 15, 1958–1973. [Google Scholar] [CrossRef]
- Chen, J.F.; Schwarzschild, M.A. Do caffeine and more selective adenosine A2A receptor antagonists protect against dopaminergic neurodegeneration in Parkinson’s disease? Parkinsonism Relat. Disord. 2020, 80, S45–S53. [Google Scholar] [CrossRef]
- Chen, J.F.; Lee, C.F.; Chern, Y. Adenosine receptor neurobiology: Overview. Int. Rev. Neurobiol. 2014, 119, 1–49. [Google Scholar]
- LeWitt, P.A.; Chaudhuri, K.R. Unmet needs in Parkinson disease: Motor and non-motor. Parkinsonism Relat. Disord. 2020, 80, S7–S12. [Google Scholar] [CrossRef]
- Müller, T.; Mueller, B.K.; Riederer, P. Perspective: Treatment for Disease Modification in Chronic Neurodegeneration. Cells 2021, 10, 873. [Google Scholar] [CrossRef]
- Mori, A.; Cross, B.; Uchida, S.; Walker, J.K.; Ristuccia, R. How Are Adenosine and Adenosine A2A Receptors Involved in the Pathophysiology of Amyotrophic Lateral Sclerosis? Biomedicines 2021, 9, 1027. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Mori, A. How do adenosine A2A receptors regulate motor function? Parkinsonism Relat. Disord. 2020, 80, S13–S20. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine A2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Rodriguez, M.; Lanciego, J.L.; Artieda, J.; Gonzalo, N.; Olanow, C.W. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 2000, 23, S8–S19. [Google Scholar] [CrossRef]
- Obeso, J.A.; Marin, C.; Rodriguez-Oroz, C.; Blesa, J.; Benitez-Temino, B.; Mena-Segovia, J.; Rodriguez, M.; Olanow, C.W. The basal ganglia in Parkinson’s disease: Current concepts and unexplained observations. Ann. Neurol. 2008, 64, S30–S46. [Google Scholar] [CrossRef]
- Svenningsson, P.; Moine, C.L.; Fisone, G.; Fredholm, B.B. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog. Neurobiol. 1999, 59, 355–396. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Aradi, S.D.; Hauser, R.A.; Rascol, O. The challenge of developing adenosine A2A antagonists for Parkinson disease: Istradefylline, preladenant, and tozadenant. Parkinsonism Relat. Disord. 2020, 80, S54–S63. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Aradi, S.D.; Hauser, R.A. Istradefylline—A first generation adenosine A2A antagonist for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2021, 21, 317–333. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Svenningsson, P. Why target brain adenosine receptors? A historical perspective. Parkinsonism Relat. Disord. 2020, 80, S3–S6. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, B.; Lebon, G. Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 2017, 8, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.R.; Kachroo, A.; Schintu, N.; Xu, K.; Schwarzschild, M.A.; Wardas, J.; Morelli, M. Inactivation of neuronal forebrain A2A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurochem. 2009, 111, 1478–1489. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A. The role of adenosine in Alzheimer’s disease. Curr. Neuropharmacol. 2009, 7, 207–216. [Google Scholar] [CrossRef]
- Wiprich, M.T.; Bonan, C.D. Purinergic Signaling in the Pathophysiology and Treatment of Huntingtons Disease. Front. Neurosci. 2021, 15, 657338. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Rukavina, K.; Batzu, L.; Leta, V.; Chaudhuri, K.R. New approaches to treatments for sleep, pain and autonomic failure in Parkinson’s disease—Pharmacological therapies. Neuropharmacology 2022, 208, 108959. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Kanda, T. Can adenosine A2A receptor antagonists be used to treat cognitive impairment, depression or excessive sleepiness in Parkinson’s disease? Parkinsonism Relat. Disord. 2020, 80, S28–S36. [Google Scholar] [CrossRef]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef]
- Barrett, M.J.; Sargent, L.; Nawaz, H.; Weintraub, D.; Price, E.T.; Willis, A.W. Antimuscarinic Anticholinergic Medications in Parkinson Disease: To Prescribe or Deprescribe? Mov. Disord. Clin. Pract. 2021, 8, 1181–1188. [Google Scholar] [CrossRef]
- Isaacson, S.H.; Ballard, C.G.; Kreitzman, D.L.; Coate, B.; Norton, J.C.; Fernandez, H.H.; Ilic, T.V.; Azulay, J.P.; Ferreira, J.J.; Abler, V.; et al. Efficacy results of pimavanserin from a multi-center, open-label extension study in Parkinson’s disease psychosis patients. Parkinsonism Relat. Disord. 2021, 87, 25–31. [Google Scholar] [CrossRef]
- Freitas, M.E.; Fox, S.H. Nondopaminergic treatments for Parkinson’s disease: Current and future prospects. Neurodegener. Dis. Manag. 2016, 6, 249–268. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.Z.; Yuan, X.Z.; Luo, X.; Zhang, S.Y.; Wang, X.P. An Update on Nondopaminergic Treatments for Motor and Non-motor symptoms of Parkinson’s disease. Curr. Neuropharmacol. 2022. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Williams, M. Direct autoradiographic localization of adenosine A2 receptors in the rat brain using the A2-selective agonist, [3H]CGS 21680. Eur. J. Pharmacol. 1989, 168, 243–246. [Google Scholar] [CrossRef]
- Rosin, D.L.; Robeva, A.; Woodard, R.L.; Guyenet, P.G.; Linden, J. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J. Comp. Neurol. 1998, 401, 163–186. [Google Scholar] [CrossRef]
- Svenningsson, P.; Hall, H.; Sedvall, G.; Fredholm, B.B. Distribution of adenosine receptors in the postmortem human brain: An extended autoradiographic study. Synapse 1997, 27, 322–335. [Google Scholar] [CrossRef]
- Ishiwata, K.; Mishina, M.; Kimura, Y.; Oda, K.; Sasaki, T.; Ishii, K. First visualization of adenosine A2A receptors in the human brain by positron emission tomography with [11C]TMSX. Synapse 2005, 55, 133–136. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferre, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, G.E.; Crutcher, M.D. Functional architecture of basal ganglia circuits: Neural substrates of parallel processing. Trends Neurosci. 1990, 13, 266–271. [Google Scholar] [CrossRef]
- DeLong, M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Shindou, T. Modulation of GABAergic transmission in the striatopallidal system by adenosine A2A receptors: A potential mechanism for the antiparkinsonian effects of A2A antagonists. Neurology 2003, 61, S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Jun, S.B.; Jin, X.; Pham, M.D.; Vogel, S.S.; Lovinger, D.M.; Costa, R.M. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 2013, 494, 238–242. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Jacobs, O.; Vanderhaeghen, J.J. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: An in situ hybridization histochemistry study. J. Neurochem. 1991, 57, 1062–1067. [Google Scholar] [CrossRef]
- Shindou, T.; Nonaka, H.; Richardson, P.J.; Mori, A.; Kase, H.; Ichimura, M. Presynaptic adenosine A2A receptors enhance GABAergic synaptic transmission via a cyclic AMP dependent mechanism in the rat globus pallidus. Br. J. Pharmacol. 2002, 136, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Shindou, T.; Mori, A.; Kase, H.; Ichimura, M. Adenosine A2A receptor enhances GABAA-mediated IPSCs in the rat globus pallidus. J. Physiol. 2001, 532, 423–434. [Google Scholar] [CrossRef]
- Morales-Figueroa, G.E.; Rivera-Ramirez, N.; Gonzalez-Pantoja, R.; Escamilla-Sanchez, J.; Garcia-Hernandez, U.; Galvan, E.J.; Arias-Montano, J.A. Adenosine A2A and histamine H3 receptors interact at the cAMP/PKA pathway to modulate depolarization-evoked [3H]-GABA release from rat striato-pallidal terminals. Purinergic Signal. 2019, 15, 85–93. [Google Scholar] [CrossRef]
- Mori, A.; Shindou, T.; Ichimura, M.; Nonaka, H.; Kase, H. The role of adenosine A2A receptors in regulating GABAergic synaptic transmission in striatal medium spiny neurons. J. Neurosci. 1996, 16, 605–611. [Google Scholar] [CrossRef]
- Ferré, S.; Bonaventura, J.; Zhu, W.; Hatcher-Solis, C.; Taura, J.; Quiroz, C.; Cai, N.S.; Moreno, E.; Casadó-Anguera, V.; Kravitz, A.V.; et al. Essential Control of the Function of the Striatopallidal Neuron by Pre-coupled Complexes of Adenosine A2A-Dopamine D2 Receptor Heterotetramers and Adenylyl Cyclase. Front. Pharmacol. 2018, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Quiroz, C.; Woods, A.S.; Cunha, R.; Popoli, P.; Ciruela, F.; Lluis, C.; Franco, R.; Azdad, K.; Schiffmann, S.N. An update on adenosine A2A-dopamine D2 receptor interactions: Implications for the function of G protein-coupled receptors. Curr. Pharm. Des. 2008, 14, 1468–1474. [Google Scholar] [CrossRef] [Green Version]
- Chase, T.N.; Bibbiani, F.; Bara-Jimenez, W.; Dimitrova, T.; Oh-Lee, J.D. Translating A2A antagonist KW6002 from animal models to parkinsonian patients. Neurology 2003, 61, S107–S111. [Google Scholar] [CrossRef]
- Uchida, S.; Tashiro, T.; Kawai-Uchida, M.; Mori, A.; Jenner, P.; Kanda, T. Adenosine A2A-receptor antagonist istradefylline enhances the motor response of L-DOPA without worsening dyskinesia in MPTP-treated common marmosets. J. Pharmacol. Sci. 2014, 124, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Soshiroda, K.; Okita, E.; Kawai-Uchida, M.; Mori, A.; Jenner, P.; Kanda, T. The adenosine A2A receptor antagonist, istradefylline enhances the anti-parkinsonian activity of low doses of dopamine agonists in MPTP-treated common marmosets. Eur. J. Pharmacol. 2015, 747, 160–165. [Google Scholar] [CrossRef]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; Benedetti, P.d.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef] [Green Version]
- Hillion, J.; Canals, M.; Torvinen, M.; Casado, V.; Scott, R.; Terasmaa, A.; Hansson, A.; Watson, S.; Olah, M.E.; Mallol, J.; et al. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem. 2002, 277, 18091–18097. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Agnati, L.F.; Jacobsen, K.; Hillion, J.; Canals, M.; Torvinen, M.; Tinner-Staines, B.; Staines, W.; Rosin, D.; Terasmaa, A.; et al. Receptor heteromerization in adenosine A2A receptor signaling: Relevance for striatal function and Parkinson’s disease. Neurology 2003, 61, S19–S23. [Google Scholar] [CrossRef]
- Gerevich, Z.; Wirkner, K.; Illes, P. Adenosine A2A receptors inhibit the N-methyl-D-aspartate component of excitatory synaptic currents in rat striatal neurons. Eur. J. Pharmacol. 2002, 451, 161–164. [Google Scholar] [CrossRef]
- Higley, M.J.; Sabatini, B.L. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat. Neurosci. 2010, 13, 958–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferre, S.; Karcz-Kubicha, M.; Hope, B.T.; Popoli, P.; Burgueno, J.; Gutierrez, M.A.; Casado, V.; Fuxe, K.; Goldberg, S.R.; Lluis, C.; et al. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: Implications for striatal neuronal function. Proc. Natl. Acad. Sci. USA 2002, 99, 11940–11945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coccurello, R.; Breysse, N.; Amalric, M. Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology 2004, 29, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Orlando, L.R.; Grandy, D.K.; Chen, J.F.; Young, A.B.; Schwarzschild, M.A. Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 10414–10419. [Google Scholar] [CrossRef]
- Lerner, T.N.; Horne, E.A.; Stella, N.; Kreitzer, A.C. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J. Neuroscience 2010, 30, 2160–2164. [Google Scholar] [CrossRef]
- Ferre, S.; Lluis, C.; Justinova, Z.; Quiroz, C.; Orru, M.; Navarro, G.; Canela, E.I.; Franco, R.; Goldberg, S.R. Adenosine-cannabinoid receptor interactions. Implications for striatal function. Br. J. Pharmacol. 2010, 160, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Rosin, D.L.; Hettinger, B.D.; Lee, A.; Linden, J. Anatomy of adenosine A2A receptors in brain: Morphological substrates for integration of striatal function. Neurology 2003, 61, S12–S18. [Google Scholar] [CrossRef]
- Rebola, N.; Rodrigues, R.J.; Lopes, L.V.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A. Adenosine A1 and A2A receptors are co-expressed in pyramidal neurons and co-localized in glutamatergic nerve terminals of the rat hippocampus. Neuroscience 2005, 133, 79–83. [Google Scholar] [CrossRef]
- Ciruela, F.; Casado, V.; Rodrigues, R.J.; Lujan, R.; Burgueno, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neuroscience 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Ramlackhansingh, A.F.; Bose, S.K.; Ahmed, I.; Turkheimer, F.E.; Pavese, N.; Brooks, D.J. Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology 2011, 76, 1811–1816. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.; Paolo, T.D.; Wardas, J.; Calon, F.; Xiao, D.; Schwarzschild, M.A. Role of adenosine A2A receptors in parkinsonian motor impairment and l-DOPA-induced motor complications. Prog. Neurobiol. 2007, 83, 293–309. [Google Scholar] [CrossRef]
- Calon, F.; Dridi, M.; Hornykiewicz, O.; Bedard, P.J.; Rajput, A.H.; Paolo, T.D. Increased adenosine A2A receptors in the brain of Parkinson’s disease patients with dyskinesias. Brain 2004, 127, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Mishina, M.; Ishiwata, K.; Naganawa, M.; Kimura, Y.; Kitamura, S.; Suzuki, M.; Hashimoto, M.; Ishibashi, K.; Oda, K.; Sakata, M.; et al. Adenosine A(2A) receptors measured with [C]TMSX PET in the striata of Parkinson’s disease patients. PLoS ONE 2011, 6, e17338. [Google Scholar] [CrossRef]
- Villar-Menendez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Diaz-Sanchez, S.; Albasanz, J.L.; Ferrer, I.; Martin, M.; Barrachina, M. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol. Dis. 2014, 69, 206–214. [Google Scholar] [CrossRef]
- Mori, A. Mode of action of adenosine A2A receptor antagonists as symptomatic treatment for Parkinson’s disease. Int. Rev. Neurobiol. 2014, 119, 87–116. [Google Scholar]
- Xu, K.; Bastia, E.; Schwarzschild, M. Therapeutic potential of adenosine A2A receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 2005, 105, 267–310. [Google Scholar] [CrossRef]
- Carmo, M.; Gonçalves, F.Q.; Canas, P.M.; Oses, J.P.; Fernandes, F.D.; Duarte, F.V. Enhanced ATP release and CD73-mediated adenosine formation sustain adenosine A2A receptor over-activation in a rat model of Parkinson’s disease. Br. J. Pharmacol. 2019, 176, 3666–3680. [Google Scholar] [CrossRef]
- Meng, F.; Guo, Z.; Hu, Y.; Mai, W.; Zhang, Z.; Zhang, B.; Ge, Q.; Lou, H.; Guo, F.; Chen, J.; et al. CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signalling. Brain 2019, 142, 700–718. [Google Scholar] [CrossRef]
- Rodríguez, D.; Gao, Z.G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Modeling 2015, 55, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.A.; Tehan, B.G.; Lebon, G.; Tate, C.G.; Weir, M.; Marshall, F.H.; Langmead, C.J. Pharmacology and Structure of Isolated Conformations of the Adenosine A2A Receptor Define Ligand Efficacy. Mol. Pharmacol. 2013, 83, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Caliman, A.D.; Miao, Y.; McCammon, J.A. Mapping the allosteric sites of the A2A adenosine receptor. Chem. Biol. Drug Des. 2018, 91, 5–16. [Google Scholar] [CrossRef]
- Amelia, T.; Veldhoven, J.P.D.v.; Falsini, M.; Liu, R.; Heitman, L.H.; Westen, G.J.P.v.; Segala, E.; Verdon, G.; Cheng, R.K.Y.; Cooke, R.M.; et al. Crystal Structure and Subsequent Ligand Design of a Nonriboside Partial Agonist Bound to the Adenosine A2A Receptor. J. Med. Chem. 2021, 64, 3827–3842. [Google Scholar] [CrossRef]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12, 1179069518779829. [Google Scholar] [CrossRef] [Green Version]
- Kyowa Kirin North American Pharmaceutical Pipeline. Available online: https://kkna.kyowakirin.com/wp-content/uploads/Kyowa-Kirin-North-American-Pipeline.pdf (accessed on 30 March 2022).
- Kyowa Hakko Kirin Announces Results of Early Phase 2 Trial of KW-6356 for Parkinson’s Disease at IAPRD. Available online: https://www.kyowakirin.com/media_center/news_releases/2018/e20180820_01.html (accessed on 30 March 2022).
- Pinna, A.; Serra, M.; Marongiu, J.; Morelli, M. Pharmacological interactions between adenosine A2A receptor antagonists and different neurotransmitter systems. Parkinsonism Relat. Disord. 2020, 80, S37–S44. [Google Scholar] [CrossRef]
- Shook, B.C.; Jackson, P.F. Adenosine A2A Receptor Antagonists and Parkinson’s Disease. Acs. Chem. Neurosci. 2011, 2, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Hattori, N.; Fernandez, H.; Isaacson, S.H.; Mochizuki, H.; Rascol, O.; Stocchi, F.; Li, J.; Mori, A.; Nakajima, Y.; et al. Efficacy of Istradefylline, an Adenosine A2A Receptor Antagonist, as Adjunctive Therapy to Levodopa in Parkinson’s Disease: A Pooled Analysis of 8 Phase 2b/3 Trials. J. Parkinsons Dis. 2021, 11, 1663–1675. [Google Scholar] [CrossRef]
- Brooks, D.J.; Papapetropoulos, S.; Vandenhende, F.; Tomic, D.; He, P.; Coppell, A.; O’Neill, G. An open-label, positron emission tomography study to assess adenosine A2A brain receptor occupancy of vipadenant (BIIB014) at steady-state levels in healthy male volunteers. Clin. Neuropharmacol. 2010, 33, 55–60. [Google Scholar] [CrossRef]
- Pawsey, S.; Donaldson, K.; Warrington, S.; Tranter, E. A phase I single and repeated dose pharmacokinetic study of oral V81444, a selective non-xanthine adenosine A2A receptor antagonist. J. Neurol. Sci. 2013, 333, e135. [Google Scholar] [CrossRef]
- Pinna, A. Adenosine A2A receptor antagonists in Parkinson’s disease: Progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hasegawa, K.; Kondo, T.; Kuno, S.; Yamamoto, M.; Japanese Istradefylline Study Group. Clinical efficacy of istradefylline (KW-6002) in Parkinson’s disease: A randomized, controlled study. Mov. Disord. 2010, 25, 1437–1443. [Google Scholar] [CrossRef]
- Fernandez, H.H.; Greeley, D.R.; Zweig, R.M.; Wojcieszek, J.; Mori, A.; Sussman, N.M. Istradefylline as monotherapy for Parkinson disease: Results of the 6002-US-051 trial. Parkinsonism Relat. Disord. 2010, 16, 16–20. [Google Scholar] [CrossRef]
- Stacy, M.; Silver, D.; Mendis, T.; Sutton, J.; Mori, A.; Chaikin, P.; Sussman, N.M. A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology 2008, 70, 2233–2240. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef]
- Hauser, R.A.; Shulman, L.M.; Trugman, J.M.; Roberts, J.W.; Mori, A.; Ballerini, R.; Sussman, N.M. Study of istradefylline in patients with Parkinson’s disease on levodopa with motor fluctuations. Mov. Disord. 2008, 23, 2177–2185. [Google Scholar] [CrossRef]
- Pourcher, E.; Fernandez, H.H.; Stacy, M.; Mori, A.; Ballerini, R.; Chaikin, P. Istradefylline for Parkinson’s disease patients experiencing motor fluctuations: Results of the KW-6002-US-018 study. Parkinsonism Relat. Disord. 2012, 18, 178–184. [Google Scholar] [CrossRef]
- Guttman, M. Efficacy of istradefylline in Parkinson’s disease patients treated with levodopa with motor response complications: Results of the KW-6002 US-018 study. Mov. Disord. 2006, 21, S585. [Google Scholar]
- Hauser, R.A.; Hubble, J.P.; Truong, D.D.; Istradefylline US-001 Study Group. Randomized trial of the adenosine A2A receptor antagonist istradefylline in advanced PD. Neurology 2003, 61, 297–303. [Google Scholar] [CrossRef]
- Hauser, R.A.; Cantillon, M.; Pourcher, E.; Micheli, F.; Mok, V.; Onofrj, M.; Huyck, S.; Wolski, K. Preladenant in patients with Parkinson’s disease and motor fluctuations: A phase 2, double-blind, randomised trial. Lancet Neurol. 2011, 10, 221–229. [Google Scholar] [CrossRef]
- Hauser, R.A.; Stocchi, F.; Rascol, O.; Huyck, S.B.; Capece, R.; Ho, T.W.; Sklar, P.; Lines, C.; Michelson, D.; Hewitt, D. Preladenant as an Adjunctive Therapy with Levodopa in Parkinson Disease: Two Randomized Clinical Trials and Lessons Learned. JAMA Neurol. 2015, 72, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- BRIEF-Acorda Discontinues Tozadenant Development Program. Available online: https://www.reuters.com/article/brief-acorda-discontinues-tozadenant-dev/brief-acorda-discontinues-tozadenant-development-program-idUSFWN1NQ0IG (accessed on 30 March 2022).
- Nourianz (Istradefylline) Tablets, for Oral Use (Prescribing Information). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/022075s000lbl.pdf (accessed on 30 March 2022).
- Report on the Deliberation Results. Available online: https://www.pmda.go.jp/files/000153870.pdf (accessed on 30 March 2022).
- Weintraub, D.; Mamikonyan, E. The Neuropsychiatry of Parkinson Disease: A Perfect Storm. Am. J. Geriatr. Psychiatry 2019, 27, 998–1018. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Aarsland, D.; Chaudhuri, K.R.; Dobkin, R.D.; Leentjens, A.F.; Rodriguez-Violante, M.; Schrag, A. The neuropsychiatry of Parkinson’s disease: Advances and challenges. Lancet Neurol. 2022, 21, 89–102. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Chen, J.F. Caffeine and Parkinson’s Disease: Multiple Benefits and Emerging Mechanisms. Front. Neurosci. 2020, 14, 602697. [Google Scholar] [CrossRef]
- Li, P.; Rial, D.; Canas, P.M.; Yoo, J.H.; Li, W.; Zhou, X.; Wang, Y.; Westen, G.J.v.; Payen, M.P.; Augusto, E.; et al. Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 2015, 20, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- Batalha, V.L.; Pego, J.M.; Fontinha, B.M.; Costenla, A.R.; Valadas, J.S.; Baqi, Y.; Radjainia, H.; Müller, C.E.; Sebastião, A.M.; Lopes, L.V. Adenosine A2A receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol. Psychiatry 2013, 18, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Todorova, A.; Jenner, P.; Chaudhuri, K.R. Non-motor Parkinson’s: Integral to motor Parkinson’s, yet often neglected. Pract. Neurol. 2014, 14, 310–322. [Google Scholar] [CrossRef]
- Chahine, L.M.; Amara, A.W.; Videnovic, A. A systematic review of the literature on disorders of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep Med. Rev. 2017, 35, 33–50. [Google Scholar] [CrossRef]
- Oishi, Y.; Xu, Q.; Wang, L.; Zhang, B.J.; Takahashi, K.; Takata, Y.; Luo, Y.J.; Cherasse, Y.; Schiffmann, S.N.; d’Exaerde, A.d.K.; et al. Slow-wave sleep is controlled by a subset of nucleus accumbens core neurons in mice. Nat. Commun. 2017, 8, 734. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Li, R.; Wang, D.R.; Cherasse, Y.; Zhang, Z.; Zhang, M.Q.; Lavielle, O.; McEown, K.; Schiffmann, S.N.; d’Exaerde, A.d.K.; et al. Adenosine A2A receptors in the olfactory bulb suppress rapid eye movement sleep in rodents. Brain Struct. Funct. 2017, 222, 1351–1366. [Google Scholar] [CrossRef]
- Lazarus, M.; Shen, H.Y.; Cherasse, Y.; Qu, W.M.; Huang, Z.L.; Bass, C.E.; Winsky-Sommerer, R.; Semba, K.; Fredholm, B.B.; Boison, D.; et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J. Neurosci. 2011, 31, 10067–10075. [Google Scholar] [CrossRef] [Green Version]
- Porkka-Heiskanen, T.; Strecker, R.E.; Thakkar, M.; Bjorkum, A.A.; Greene, R.W.; McCarley, R.W. Adenosine: A mediator of the sleep-inducing effects of prolonged wakefulness. Science 1997, 276, 1265–1268. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.L.; Qu, W.M.; Eguchi, N.; Chen, J.F.; Schwarzschild, M.A.; Fredholm, B.B.; Urade, Y.; Hayaishi, O. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat. Neurosci. 2005, 8, 858–859. [Google Scholar] [CrossRef]
- Suzuki, K.; Miyamoto, M.; Miyamoto, T.; Uchiyama, T.; Watanabe, Y.; Suzuki, S.; Kadowaki, T.; Fujita, H.; Matsubara, T.; Sakuramoto, H.; et al. Istradefylline improves daytime sleepiness in patients with Parkinson’s disease: An open-label, 3-month study. J. Neurol. Sci. 2017, 380, 230–233. [Google Scholar] [CrossRef]
- Matsuura, K.; Kajikawa, H.; Tabei, K.I.; Satoh, M.; Kida, H.; Nakamura, N.; Tomimoto, H. The effectiveness of istradefylline for the treatment of gait deficits and sleepiness in patients with Parkinson’s disease. Neurosci. Lett. 2018, 662, 158–161. [Google Scholar] [CrossRef]
- Aarsland, D.; Påhlhagen, S.; Ballard, C.G.; Ehrt, U.; Svenningsson, P. Depression in Parkinson disease—Epidemiology, mechanisms and management. Nat. Reviews Neurol. 2011, 8, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Remy, P.; Doder, M.; Lees, A.; Turjanski, N.; Brooks, D. Depression in Parkinson’s disease: Loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005, 128, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, G.; Vitale, C.; Picillo, M.; Cuoco, S.; Moccia, M.; Pezzella, D.; Erro, R.; Longo, K.; Vicidomini, C.; Pellecchia, M.T.; et al. Apathy and striatal dopamine transporter levels in de-novo, untreated Parkinson’s disease patients. Parkinsonism Relat. Disord. 2015, 21, 489–493. [Google Scholar] [CrossRef]
- Heron, C.L.; Plant, O.; Manohar, S.; Ang, Y.S.; Jackson, M.; Lennox, G.; Hu, M.T.; Husain, M. Distinct effects of apathy and dopamine on effort-based decision-making in Parkinson’s disease. Brain 2018, 141, 1455–1469. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant-like activity of the adenosine A(2A) receptor antagonist, istradefylline (KW-6002), in the forced swim test and the tail suspension test in rodents. Pharmacol. Biochem. Behav. 2013, 114, 23–30. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, M.; Shiozaki, S.; Ohta, T.; Mori, A.; Jenner, P.; Kanda, T. Antidepressant activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002) on learned helplessness in rats. Psychopharmacology 2014, 231, 2839–2849. [Google Scholar] [CrossRef]
- Wei, C.J.; Augusto, E.; Gomes, C.A.; Singer, P.; Wang, Y.; Boison, D.; Cunha, R.A.; Yee, B.K.; Chen, J.F. Regulation of fear responses by striatal and extrastriatal adenosine A2A receptors in forebrain. Biol. Psychiatry 2014, 75, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Fukutake, S.; Odake, S.; Kawada, J.; Iwanaga, S.; Kamei, T. Clinical Efficacy of Istradefylline for Depression in Parkinson’s Disease. J. Neurol. Neurosci. 2018, 9, 261–263. [Google Scholar] [CrossRef]
- Nagayama, H.; Kano, O.; Murakami, H.; Ono, K.; Hamada, M.; Toda, T.; Sengoku, R.; Shimo, Y.; Hattori, N. Effect of istradefylline on mood disorders in Parkinson’s disease. J. Neurol. Sci. 2019, 396, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Hameleers, P.A.; Boxtel, M.P.V.; Hogervorst, E.; Riedel, W.J.; Houx, P.J.; Buntinx, F.; Jolles, J. Habitual caffeine consumption and its relation to memory, attention, planning capacity and psychomotor performance across multiple age groups. Hum. Psychopharmacol. 2000, 15, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanboxtel, M.; Schmitt, J.; Bosma, H.; Jolles, J. The effects of habitual caffeine use on cognitive change: A longitudinal perspective. Pharmacol. Biochem. Behav. 2003, 75, 921–927. [Google Scholar] [CrossRef]
- Lindsay, J.; Laurin, D.; Verreault, R.; Hebert, R.; Helliwell, B.; Hill, G.B.; McDowell, I. Risk factors for Alzheimer’s disease: A prospective analysis from the Canadian Study of Health and Aging. Am. J. Epidemiol. 2002, 156, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelder, B.M.v.; Buijsse, B.; Tijhuis, M.; Kalmijn, S.; Giampaoli, S.; Nissinen, A.; Kromhout, D. Coffee consumption is inversely associated with cognitive decline in elderly European men: The FINE Study. Eur. J. Clin. Nutr. 2007, 61, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, K.; Carrière, I.; Portet, F.; Mendonca, A.D.; Dartigues, J.F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.L. The neuro-protective effects of caffeine: A prospective population study (the Three City Study). Neurology 2007, 69, 536–545. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Cirrito, J.R.; Lin, X.; Wang, L.; Verges, D.K.; Dickson, A.; Mamcarz, M.; Zhang, C.; Mori, T.; Arendash, G.W.; et al. Caffeine suppresses amyloid-beta levels in plasma and brain of Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2009, 17, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.H.; Choi, S.M.; Kim, J.T.; Kim, B.C. Association of coffee consumption and non-motor symptoms in drug-naive, early-stage Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 50, 42–47. [Google Scholar] [CrossRef]
- Zhou, S.J.; Zhu, M.E.; Shu, D.; Du, X.P.; Song, X.H.; Wang, X.T.; Zheng, R.Y.; Cai, X.H.; Chen, J.F.; He, J.C. Preferential enhancement of working memory in mice lacking adenosine A(2A) receptors. Brain Res. 2009, 1303, 74–83. [Google Scholar] [CrossRef]
- Wei, C.J.; Singer, P.; Coelho, J.; Boison, D.; Feldon, J.; Yee, B.K.; Chen, J.F. Selective inactivation of adenosine A2A receptors in striatal neurons enhances working memory and reversal learning. Learn. Mem. 2011, 18, 459–474. [Google Scholar] [CrossRef] [Green Version]
- Mingote, S.; Font, L.; Farrar, A.M.; Vontell, R.; Worden, L.T.; Stopper, C.M.; Port, R.G.; Sink, K.S.; Bunce, J.G.; Chrobak, J.J.; et al. Nucleus accumbens adenosine A2A receptors regulate exertion of effort by acting on the ventral striatopallidal pathway. J. Neurosci. 2008, 28, 9037–9046. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, Y.; Chen, M.; Pu, Z.; Chen, L.; Li, P.; Li, B.; Li, H.; Huang, Z.L.; Li, Z.; et al. Optogenetic Activation of Adenosine A2A Receptor Signaling in the Dorsomedial Striatopallidal Neurons Suppresses Goal-Directed Behavior. Neuropsychopharmacology 2016, 41, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Giménez-Llort, L.; Schiffmann, S.N.; Shmidt, T.; Canela, L.; Camón, L.; Wassholm, M.; Canals, M.; Terasmaa, A.; Fernández-Teruel, A.; Tobeña, A.; et al. Working memory deficits in transgenic rats overexpressing human adenosine A2A receptors in the brain. Neurobiol. Learn. Mem. 2007, 87, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Batalha, V.L.; Ferreira, D.G.; Coelho, J.E.; Valadas, J.S.; Gomes, R.; Temido-Ferreira, M.; Shmidt, T.; Baqi, Y.; Buée, L.; Müller, C.E.; et al. The caffeine-binding adenosine A2A receptor induces age-like HPA-axis dysfunction by targeting glucocorticoid receptor function. Sci. Rep. 2016, 6, 31493. [Google Scholar] [CrossRef]
- Knowlton, B.J.; Mangels, J.A.; Squire, L.R. A neostriatal habit learning system in humans. Science 1996, 273, 1399–1402. [Google Scholar] [CrossRef] [Green Version]
- Kehagia, A.A.; Barker, R.A.; Robbins, T.W. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 2010, 9, 1200–1213. [Google Scholar] [CrossRef]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat. Reviews. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Laar, T.v.; Deyn, P.P.D.; Aarsland, D.; Barone, P.; Galvin, J.E. Effects of cholinesterase inhibitors in Parkinson’s disease dementia: A review of clinical data. CNS Neurosci. Ther. 2011, 17, 428–441. [Google Scholar] [CrossRef]
- Taylor, A.E.; Saint-Cyr, J.A.; Lang, A.E. Parkinson’s disease. Cognitive changes in relation to treatment response. Brain 1987, 110, 35–51. [Google Scholar] [CrossRef]
- Gotham, A.M.; Brown, R.G.; Marsden, C.D. ‘Frontal’ cognitive function in patients with Parkinson’s disease ‘on’ and ‘off’ levodopa. Brain 1988, 111, 299–321. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Robbins, T.W.; Marsden, C.D.; James, M.; Owen, A.M.; Paul, G.M. L-dopa withdrawal in Parkinson’s disease selectively impairs cognitive performance in tests sensitive to frontal lobe dysfunction. Psychopharmacology 1992, 107, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, S.M.; Sucharski, I.L.; Finlay, J.M. Desipramine attenuates working memory impairments induced by partial loss of catecholamines in the rat medial prefrontal cortex. Psychopharmacology 2006, 183, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozoski, T.J.; Brown, R.M.; Rosvold, H.E.; Goldman, P.S. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 1979, 205, 929–932. [Google Scholar] [CrossRef]
- Decamp, E.; Schneider, J.S. Attention and executive function deficits in chronic low-dose MPTP-treated non-human primates. Eur. J. Neurosci. 2004, 20, 1371–1378. [Google Scholar] [CrossRef]
- Horita, T.K.; Kobayashi, M.; Mori, A.; Jenner, P.; Kanda, T. Effects of the adenosine A2A antagonist istradefylline on cognitive performance in rats with a 6-OHDA lesion in prefrontal cortex. Psychopharmacology 2013, 230, 345–352. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Wang, T.; Gao, Y.; Li, F.; Chen, L.; Xue, J.; He, Y.; Li, Y.; Guo, W.; et al. The Corticostriatal Adenosine A2A Receptor Controls Maintenance and Retrieval of Spatial Working Memory. Biol. Psychiatry 2018, 83, 530–541. [Google Scholar] [CrossRef]
- Ko, W.K.D.; Camus, S.M.; Li, Q.; Yang, J.; McGuire, S.; Pioli, E.Y.; Bezard, E. An evaluation of istradefylline treatment on Parkinsonian motor and cognitive deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated macaque models. Neuropharmacology 2016, 110, 48–58. [Google Scholar] [CrossRef]
- He, Y.; Huang, L.; Wang, K.; Pan, X.; Cai, Q.; Zhang, F.; Yang, J.; Fang, G.; Zhao, X.; You, F.; et al. α-Synuclein Selectively Impairs Motor Sequence Learning and Value Sensitivity: Reversal by the Adenosine A2A Receptor Antagonists. Cereb. Cortex 2021, 32, 808–823. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Ascherio, A.; Chen, H.; Schwarzschild, M.A.; Zhang, S.M.; Colditz, G.A.; Speizer, F.E. Caffeine, postmenopausal estrogen, and risk of Parkinson’s disease. Neurology 2003, 60, 790–795. [Google Scholar] [CrossRef]
- Saaksjarvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Mannisto, S. Prospective study of coffee consumption and risk of Parkinson’s disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef]
- Grosso, G.; Godos, J.; Galvano, F.; Giovannucci, E.L. Coffee, caffeine, and health outcomes: An umbrella review. Annu. Rev. Nutr. 2017, 37, 131–156. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Li, S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson’s disease. Geriatr. Gerontol. Int. 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Bakshi, R.; Macklin, E.A.; Hung, A.Y.; Hayes, M.T.; Hyman, B.T.; Wills, A.M.; Gomperts, S.N.; Growdon, J.H.; Ascherio, A.; Scherzer, C.R.; et al. Associations of Lower Caffeine Intake and Plasma Urate Levels with Idiopathic Parkinson’s Disease in the Harvard Biomarkers Study. J. Parkinsons Dis. 2020, 10, 505–510. [Google Scholar] [CrossRef]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.M.; Paing, S.S.; Li, H.; Pavanni, R.; Yuen, Y.; Zhao, Y.; Tan, E.K. Differential effect of caffeine intake in subjects with genetic susceptibility to Parkinson’s Disease. Sci. Rep. 2015, 5, 15492. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.M.; Meng, C.; Marder, K.; Bressman, S.; Saunders-Pullman, R.; Alcalay, R.; Tolosa, E.; Brice, A.; Goldman, S.; Schuele, B.; et al. LRRK2 Cohort-Consortium, Caffeinated Drinks, LRRK2 Genotype and PD [abstract]. Mov. Disord. 2017, 32 (Supp. 2), S398. [Google Scholar]
- Yahalom, G.; Rigbi, A.; Israeli-Korn, S.; Krohn, L.; Rudakou, U.; Ruskey, J.A.; Benshimol, L.; Tsafnat, T.; Gan-Or, Z.; Hassin-Baer, S.; et al. Age at Onset of Parkinson’s Disease Among Ashkenazi Jewish Patients: Contribution of Environmental Factors, LRRK2 p.G2019S and GBA p.N370S Mutations. J. Parkinson’s Dis. 2020, 10, 1123–1132. [Google Scholar] [CrossRef]
- Crotty, G.F.; Wang, J.; Montalban, M.; Davis, S.; Alkabsh, J.; Bakshi, R.; Chen, X.; Ascherio, A.; Macklin, E.; Maciuca, R.; et al. Metabolomic Analysis Identifies Caffeine and its Metabolites as Plasma Markers of Resistance to Parkinson’s Disease Among LRRK2 Mutation Carriers in the LRRK2 Cohort Consortium (LCC) [abstract]. Mov. Disord. Clin. Pract. 2020, 7, S183. [Google Scholar]
- Ikeda, K.; Kurokawa, M.; Aoyama, S.; Kuwana, Y. Neuroprotection by adenosine A2A receptor blockade in experimental models of Parkinson’s disease. J. Neurochem. 2002, 80, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Xu, Y.H.; Chen, J.F.; Schwarzschild, M.A. Caffeine’s neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity shows no tolerance to chronic caffeine administration in mice. Neurosci. Lett. 2002, 322, 13–16. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Nobre, H.V., Jr.; Macedo, D.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Cunha, G.M.; Sousa, F.C.; Viana, G.S. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol. Biochem. Behav. 2006, 84, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.F.d.; Bispo, J.M.M.; Leal, P.C.; Gois, A.M.d.; Santos, J.R.D. Commentary: Adenosine A(2A) Receptor Blockade Prevents Rotenone-Induced Motor Impairment in a Rat Model of Parkinsonism. Front. Behav. Neurosci. 2017, 11, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathalla, A.M.; Soliman, A.M.; Moustafa, A.A. Selective A2A receptors blockade reduces degeneration of substantia nigra dopamine neurons in a rotenone-induced rat model of Parkinson’s disease: A histological study. Neurosci. Lett. 2017, 643, 89–96. [Google Scholar] [CrossRef]
- Kalda, A.; Yu, L.; Oztas, E.; Chen, J.F. Novel neuroprotection by caffeine and adenosine A2A receptor antagonists in animal models of Parkinson’s disease. J. Neurol. Sci. 2006, 248, 9–15. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Uthaythas, S.; Govindarajulu, M.; Ramesh, S.; Parameshwaran, K.; Dhanasekaran, M. Caffeine, a natural methylxanthine nutraceutical, exerts dopaminergic neuroprotection. Neurochem. Int. 2021, 148, 105066. [Google Scholar] [CrossRef]
- Bagga, P.; Chugani, A.N.; Patel, A.B. Neuroprotective effects of caffeine in MPTP model of Parkinson’s disease: A (13)C NMR study. Neurochem. Int. 2016, 92, 25–34. [Google Scholar] [CrossRef]
- Sonsalla, P.K.; Wong, L.Y.; Harris, S.L.; Richardson, J.R.; Khobahy, I.; Li, W.; Gadad, B.S.; German, D.C. Delayed caffeine treatment prevents nigral dopamine neuron loss in a progressive rat model of Parkinson’s disease. Exp. Neurol. 2012, 234, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Kachroo, A.; Schwarzschild, M.A. Adenosine A2A receptor gene disruption protects in an alpha-synuclein model of Parkinson’s disease. Ann. Neurol. 2012, 71, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef]
- Silva, S.V.d.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- Ng, S.K.; Higashimori, H.; Tolman, M.; Yang, Y. Suppression of adenosine 2A receptor (A2AR)-mediated adenosine signaling improves disease phenotypes in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2015, 267, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.H.; Kim, D.G.; Krenz, A.; Chen, J.F.; Bynoe, M.S. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 2012, 188, 5713–5722. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Merighi, S.; Gessi, S.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Varani, K. Multiple sclerosis lymphocytes upregulate A2A adenosine receptors that are antiinflammatory when stimulated. Eur. J. Immunol. 2013, 43, 2206–2216. [Google Scholar] [CrossRef]
- Rissanen, E.; Virta, J.R.; Paavilainen, T.; Tuisku, J.; Helin, S.; Luoto, P.; Parkkola, R.; Rinne, J.O.; Airas, L. Adenosine A2A receptors in secondary progressive multiple sclerosis: A [(11)C]TMSX brain PET study. J. Cereb. Blood Flow Metab. 2013, 33, 1394–1401. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.G.; Lo, I.; Schumacher, H.; Ho, K.; Gill, M.; Guo, W.; Kim, D.H.; Knox, A.; Saito, T.; Saido, T.C.; et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 2018, 110, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Jeugd, A.V.d.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenza, R.L.; Armida, M.; Ferrante, A.; Pèzzola, A.; Matteucci, A.; Puopolo, M.; Popoli, P. Effects of chronic caffeine intake in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2013, 91, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Rei, N.; Rombo, D.M.; Ferreira, M.F.; Baqi, Y.; Müller, C.E.; Ribeiro, J.A.; Sebastião, A.M.; Vaz, S.H. Hippocampal synaptic dysfunction in the SOD1(G93A) mouse model of Amyotrophic Lateral Sclerosis: Reversal by adenosine A2AR blockade. Neuropharmacology 2020, 171, 108106. [Google Scholar] [CrossRef] [PubMed]
- Villar-Menéndez, I.; Blanch, M.; Tyebji, S.; Pereira-Veiga, T.; Albasanz, J.L.; Martín, M.; Ferrer, I.; Pérez-Navarro, E.; Barrachina, M. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. Neuromolecular Med. 2013, 15, 295–309. [Google Scholar] [CrossRef]
- Tavares, A.; Barret, O.; Seibyl, J.; Tamagnan, G. Imaging studies with A2A receptor antagonists. In The Adenosinergic System: A Non-Dopaminergic Target in Parkinson’s Disease; Morelli, M., Simola, N., Wardas, J., Eds.; Springer: New York, NY, USA, 2015; Volume 10, pp. 206–232. [Google Scholar]
- Mievis, S.; Blum, D.; Ledent, C. A2A receptor knockout worsens survival and motor behaviour in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 2011, 41, 570–576. [Google Scholar] [CrossRef]
- Ishiwata, K.; Ogi, N.; Hayakawa, N.; Oda, K.; Nagaoka, T.; Toyama, H.; Suzuki, F.; Endo, K.; Tanaka, A.; Senda, M. Adenosine A2A receptor imaging with [11C]KF18446 PET in the rat brain after quinolinic acid lesion: Comparison with the dopamine receptor imaging. Ann. Nucl. Med. 2002, 16, 467–475. [Google Scholar] [CrossRef]
- Scattoni, M.L.; Valanzano, A.; Pezzola, A.; March, Z.D.; Fusco, F.R.; Popoli, P.; Calamandrei, G. Adenosine A2A receptor blockade before striatal excitotoxic lesions prevents long term behavioural disturbances in the quinolinic rat model of Huntington’s disease. Behav. Brain Res. 2007, 176, 216–221. [Google Scholar] [CrossRef]
- Galluzzo, M.; Pintor, A.; Pèzzola, A.; Grieco, R.; Borsini, F.; Popoli, P. Behavioural and neurochemical characterization of the adenosine A2A receptor antagonist ST1535. Eur. J. Pharmacol. 2008, 579, 149–152. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.X.; Zheng, L.P.; Wang, L.; Liu, Y.F.; Yin, W.Y.; Chen, Y.Y.; Wang, X.S.; Hou, S.T.; Chen, J.F.; et al. The adenosine A2A receptor antagonist SCH58261 reduces macrophage/microglia activation and protects against experimental autoimmune encephalomyelitis in mice. Neurochem. Int. 2019, 129, 104490. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, H.; Zhao, P.; Sun, B.; Wang, J.; Kong, Q.; Mu, L.; Zhao, S.; Wang, G.; Wang, D.; et al. Activation of the adenosine A2A receptor attenuates experimental autoimmune encephalomyelitis and is associated with increased intracellular calcium levels. Neuroscience 2016, 330, 150–161. [Google Scholar] [CrossRef]
- Yao, S.Q.; Li, Z.Z.; Huang, Q.Y.; Li, F.; Wang, Z.W.; Augusto, E.; He, J.C.; Wang, X.T.; Chen, J.F.; Zheng, R.Y. Genetic inactivation of the adenosine A2A receptor exacerbates brain damage in mice with experimental autoimmune encephalomyelitis. J. Neurochem. 2012, 123, 100–112. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, A.; Chen, J.-F.; Uchida, S.; Durlach, C.; King, S.M.; Jenner, P. The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease. Molecules 2022, 27, 2366. https://doi.org/10.3390/molecules27072366
Mori A, Chen J-F, Uchida S, Durlach C, King SM, Jenner P. The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease. Molecules. 2022; 27(7):2366. https://doi.org/10.3390/molecules27072366
Chicago/Turabian StyleMori, Akihisa, Jiang-Fan Chen, Shinichi Uchida, Cecile Durlach, Shelby M. King, and Peter Jenner. 2022. "The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease" Molecules 27, no. 7: 2366. https://doi.org/10.3390/molecules27072366
APA StyleMori, A., Chen, J.-F., Uchida, S., Durlach, C., King, S. M., & Jenner, P. (2022). The Pharmacological Potential of Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease. Molecules, 27(7), 2366. https://doi.org/10.3390/molecules27072366