
Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies

 ,
,  , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
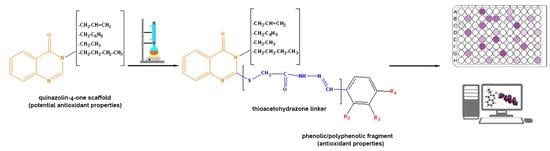
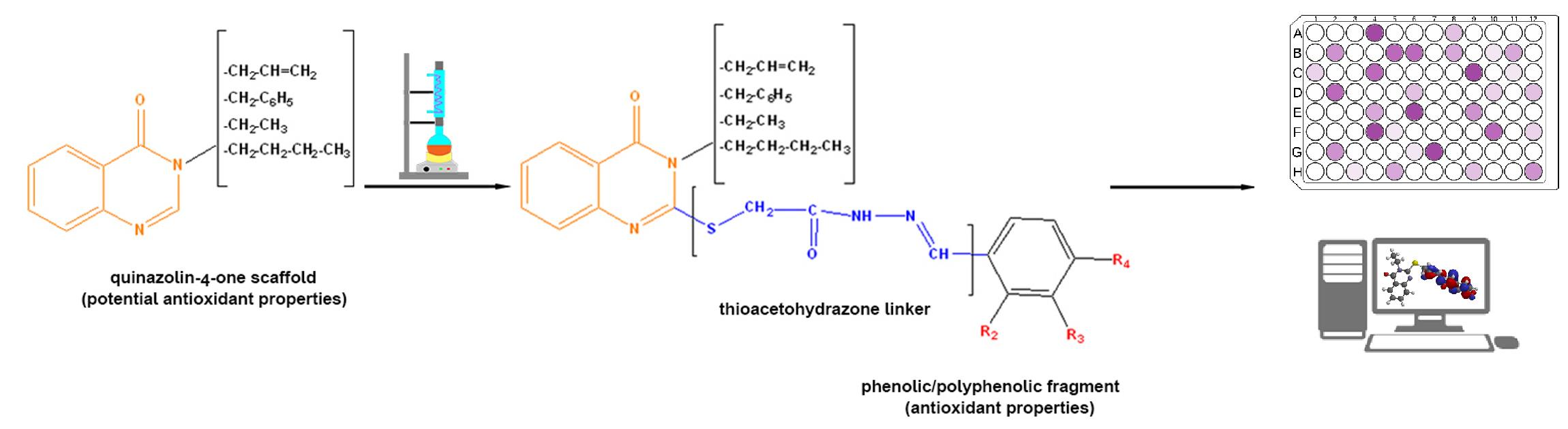
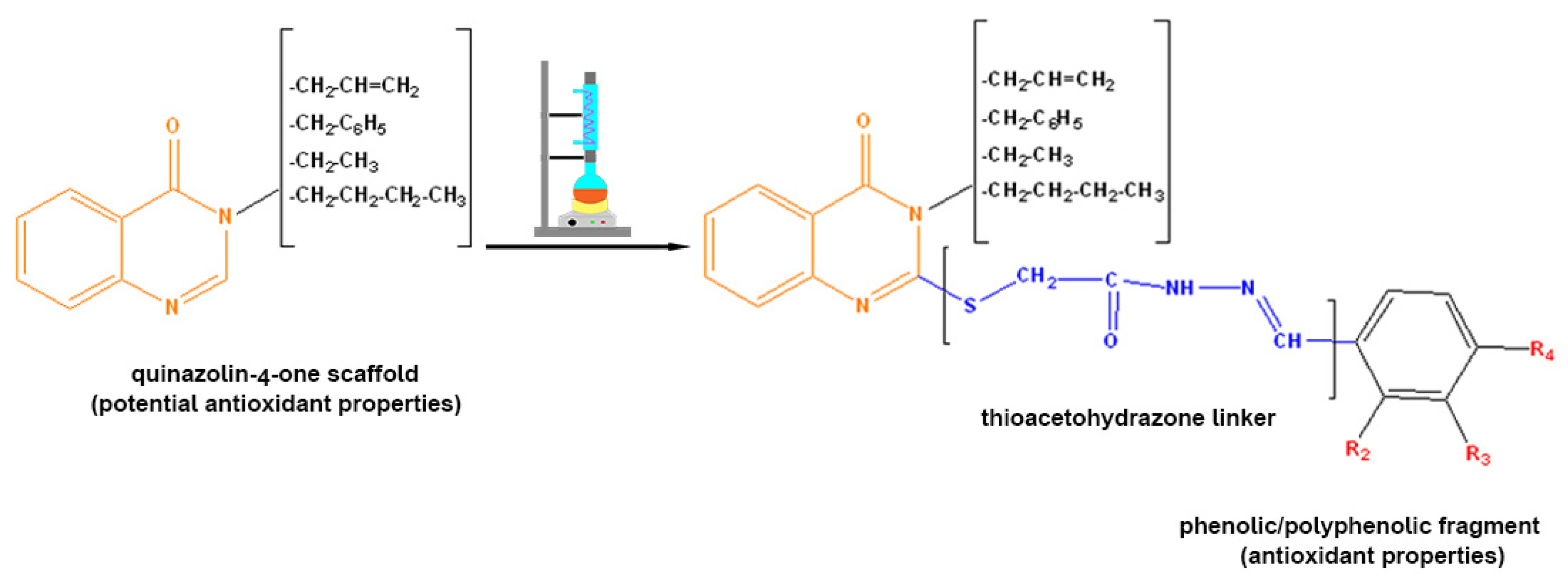
:
1. Introduction
2. Results
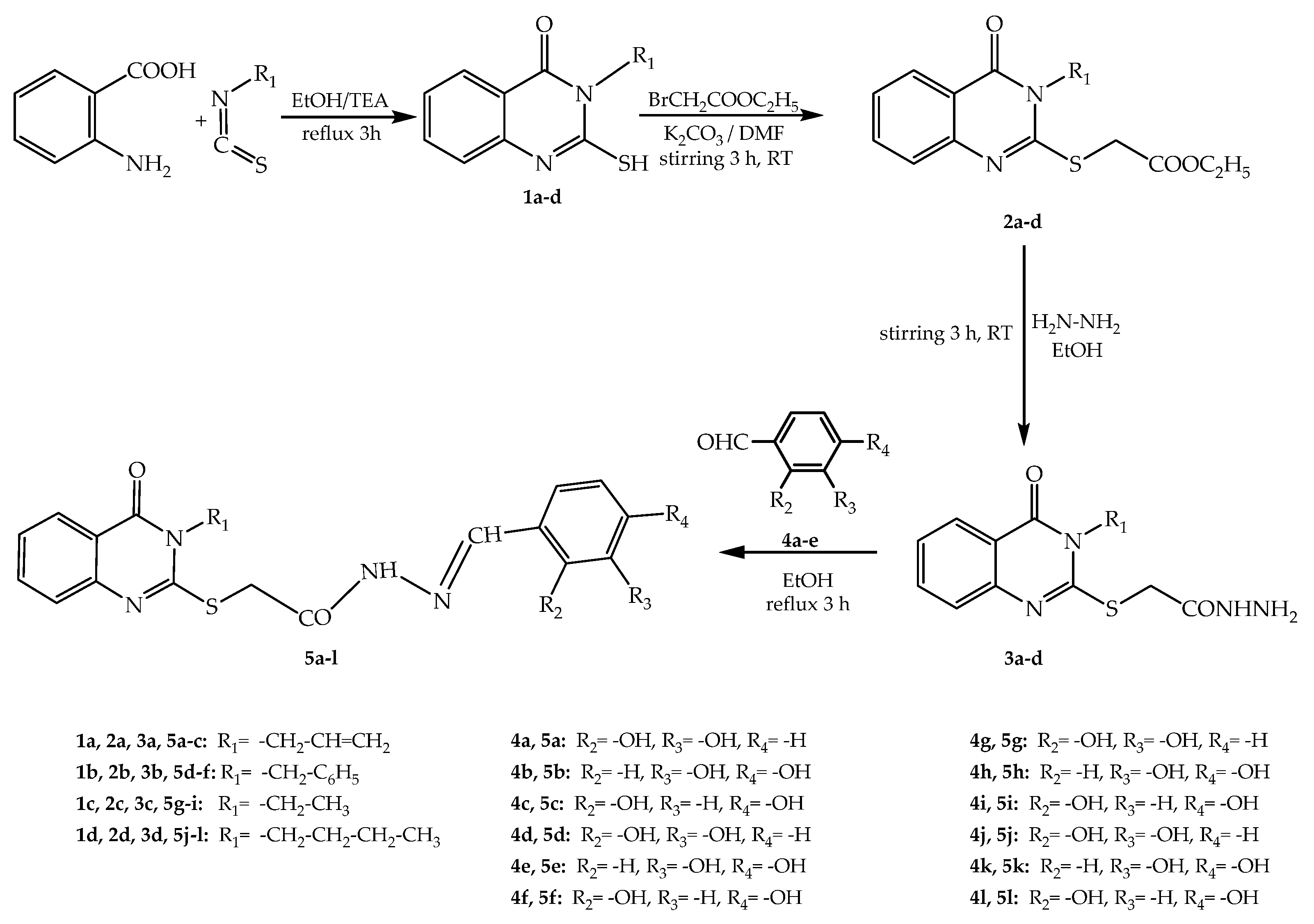
2.1. Chemical Synthesis
2.2. In Vitro Antioxidant, Antiradical and Chelation Assays
2.2.1. Antiradical Assays
ABTS+ Radical Scavenging Assay
DPPH Radical Scavenging Assay
NO Radical Scavenging Assay
2.2.2. Electron Transfer Assays
Ferric Reducing Antioxidant Power (FRAP)
Phosphomolybdate Assay for Total Antioxidant Capacity (TAC)
Reducing Power (RP) Assay
Cupric Reducing Antioxidant Capacity (CUPRAC) Assay
2.2.3. Transition Metal Ions Chelation Assays
Fe2+ Chelation Assay
Cu2+ Chelation Assay
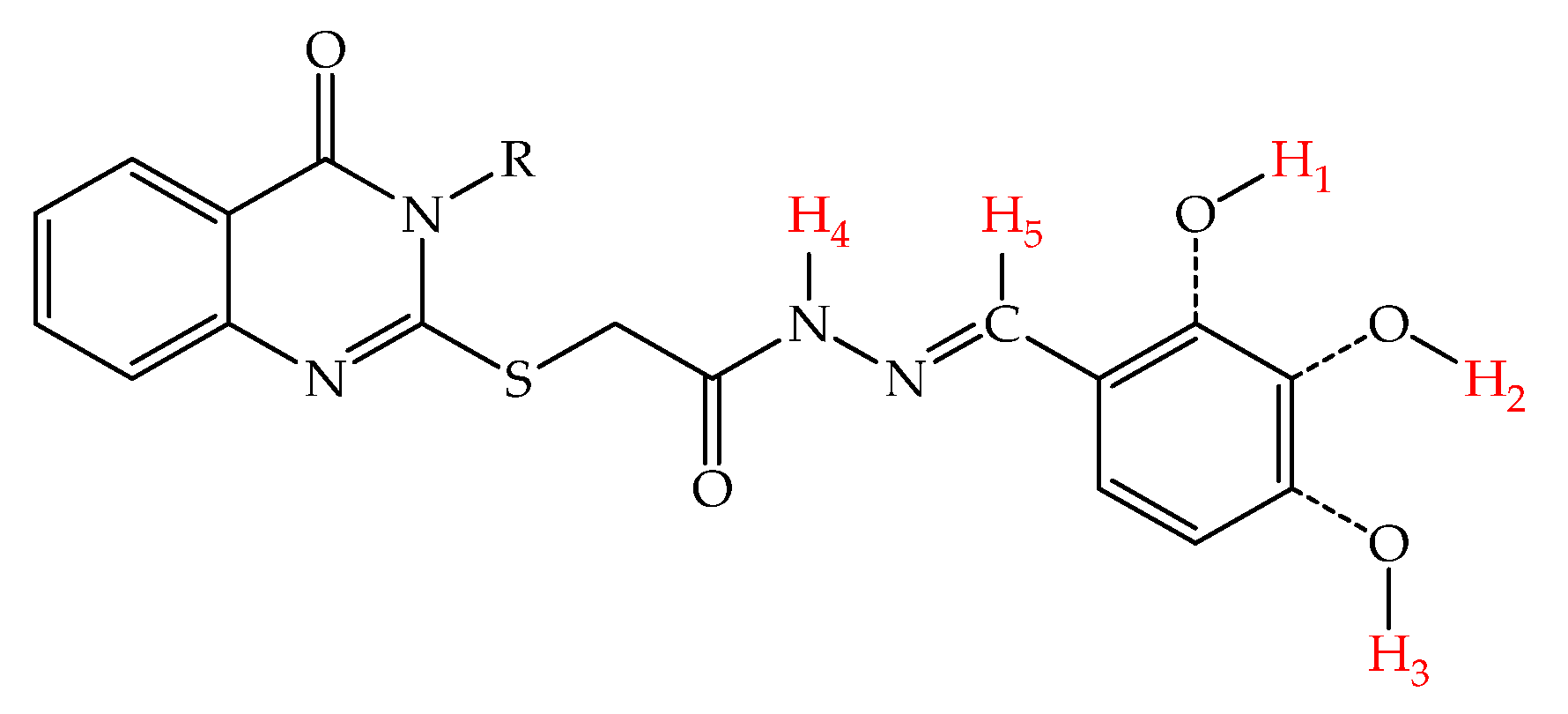
2.3. Theoretical Quantum and Thermodynamical Calculations
3. Discussion
3.1. Chemical Synthesis
3.2. In Vitro Antioxidant, Antiradical and Chelation Assay
3.2.1. Antiradical Assays
3.2.2. Electron Transfer Assays
3.2.3. Transition Metal Ions Chelation Assays
3.3. Theoretical Quantum and Thermodynamical Energy Calculations
4. Materials and Methods
4.1. Chemistry
4.1.1. Synthesis of Compounds 1a–d
4.1.2. Synthesis of Compounds 2a–d
4.1.3. Synthesis of Compounds 3a–d
4.1.4. Synthesis of Compounds 5a–l
4.2. In Vitro Antioxidant, Antiradical and Chelation Assays
4.2.1. Antiradical Assays
ABTS+ Radical Scavenging Assay
DPPH Radical Scavenging Assay
NO Radical Scavenging Assay
4.2.2. Electron Transfer Assays
Ferric Reducing Antioxidant Potential (FRAP)
Phosphomolybdate Assay for Total Antioxidant Capacity (TAC)
Reducing Power (RP) Assay
Cupric Reducing Antioxidant Capacity (CUPRAC) Assay
4.2.3. Transition Metal Ions Chelation Assays
Fe2+ Chelation Assay
Cu2+ Chelation Assay
4.3. Theoretical Quantum and Thermodynamical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Prashanth, M.K.; Revanasiddappa, H.D. Synthesis of some new glutamine linked 2,3-disubstituted quinazolinone derivatives as potent antimicrobial and antioxidant agents. Med. Chem. Res. 2013, 22, 2665–2676. [Google Scholar] [CrossRef]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; El-Gamal, K.M.A.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; Elsohly, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103422. [Google Scholar] [CrossRef] [PubMed]
- Asif, M. Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 2014, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, G.; Alagarsamy, V.; Prakash, C.R. Synthesis, analgesic, anti-inflammatory, and in vitro antimicrobial activities of some novel quinazolin-4(3H)-one derivatives. Med. Chem. Res. 2013, 22, 340–350. [Google Scholar] [CrossRef]
- Chen, J.; Yang, J.; Ma, L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci. Rep. 2020, 10, 2611. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Schluesener, H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res. Rev. 2012, 11, 329–345. [Google Scholar] [CrossRef]
- Tamaian, R.; Moţ, A.; Silaghi-Dumitrescu, R.; Ionuţ, I.; Stana, A.; Oniga, O.; Nastasă, C.; Benedec, D.; Tiperciuc, B. Study of the Relationships between the Structure, Lipophilicity and Biological Activity of Some Thiazolyl-carbonyl-thiosemicarbazides and Thiazolyl-azoles. Molecules 2015, 20, 22188–22201. [Google Scholar] [CrossRef]
- Pandiarajan, D.; Fox, T.; Spingler, B. An N4-Tetradentate Hydrazone Ligand That Binds in a Neutral, Mono- and Bisdeprotonated Form to Iron(II) and Zinc(II) Metal Ions. Crystals 2021, 11, 982. [Google Scholar] [CrossRef]
- Ahmad, F.; Alkahtani, M.D.F.; Taj, M.B.; Alnajeebi, A.M.; Alzahrani, S.O.; Babteen, N.A.; Alelwani, W.; Bannunah, A.M.; Noor, S.; Ayub, R.; et al. Synthesis of New Naphthyl Aceto Hydrazone-Based Metal Complexes: Micellar Interactions, DNA Binding, Antimicrobial, and Cancer Inhibition Studies. Molecules 2021, 26, 1044. [Google Scholar] [CrossRef]
- Wang, C.; Xing, N.; Feng, W.; Guo, S.; Liu, M.; Xu, Y.; You, Z. New mononuclear dioxidomolybdenum(VI) complexes with hydrazone ligands: Synthesis, crystal structures and catalytic performance. Inorg. Chim. Acta 2019, 486, 625–635. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Franchini, A.H.; Vodnar, D.C.; Barta, G.; Tertiş, M.; Şanta, I.; Cristea, C.; Pîrnău, A.; Ciorîţă, A.; et al. Phenolic Thiazoles with Antioxidant and Antiradical Activity. Synthesis, In Vitro Evaluation, Toxicity, Electrochemical Behavior, Quantum Studies and Antimicrobial Screening. Antioxidants 2021, 10, 1707. [Google Scholar] [CrossRef] [PubMed]
- Warad, I.; Ali, O.; Ai Ali, A.; Jaradat, N.A.; Hussein, F.; Abdallah, L.; Al-Zaqri, N.; Alsalme, A.; Alharthi, F.A. Synthesis and Spectral Identification of Three Schiff Bases with a 2-(Piperazin-1-yl)-N-(thiophen-2-yl methylene)ethanamine Moiety Acting as Novel Pancreatic Lipase Inhibitors: Thermal, DFT, Antioxidant, Antibacterial, and Molecular Docking Investigations. Molecules 2020, 25, 2253. [Google Scholar] [CrossRef] [PubMed]
- Antonijević, M.R.; Simijonović, D.M.; Avdović, E.H.; Ćirić, A.; Petrović, Z.D.; Marković, J.D.; Stepanić, V.; Marković, Z.S. Green One-Pot Synthesis of Coumarin-Hydroxybenzohydrazide Hybrids and Their Antioxidant Potency. Antioxidants 2021, 10, 1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, X.; Ouyang, X.; Liu, J.; Liu, Y.; Chen, D. Comparison of Ferroptosis-Inhibitory Mechanisms between Ferrostatin-1 and Dietary Stilbenes (Piceatannol and Astringin). Molecules 2021, 26, 1092. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, S.K.; Elrazaz, E.Z.; Abouzid, K.A.M.; El-Naggar, A.M. Design, synthesis and in silico studies of new quinazolinone derivatives as antitumor PARP-1 inhibitors. RSC Adv. 2020, 10, 29475–29492. [Google Scholar] [CrossRef]
- Hagar, M.; Soliman, S.M.; Ibid, F.; El Ashry, E.S.H. Synthesis, molecular structure and spectroscopic studies of some new quinazolin-4(3H)-one derivatives; an account on the N-versus S-Alkylation. J. Mol. Struct. 2016, 1108, 667–679. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Abdel-Hamide, S.G.; Sayed-Ahmed, M.M.; Hassan, G.S.; El-Hadiyah, T.M.; Al-Shabanah, O.A.; Al-Deeb, O.A.; El-Subbagh, H.I. Novel 4(3H)-quinazolinone analogs: Synthesis and anticonvulsant activity. Med. Chem. Res. 2013, 22, 2815–2827. [Google Scholar] [CrossRef]
- Haghighijoo, Z.; Firuzi, O.; Hemmateenejad, B.; Emami, S.; Edraki, N.; Miri, R. Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer’s disease. Bioorg. Chem. 2017, 74, 126–133. [Google Scholar] [CrossRef]
- Santos, J.S.; Brizola, V.R.A.; Granato, D. High-throughput assay comparison and standardization for metal chelating capacity screening: A proposal and application. Food Chem. 2017, 214, 515–522. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Oniga, S.D.; Pîrnău, A.; Vlase, L.; Oniga, O. New Phenolic Derivatives of Thiazolidine-2,4-dione with Antioxidant and Antiradical Properties: Synthesis, Characterization, In Vitro Evaluation, and Quantum Studies. Molecules 2019, 24, 2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stana, A.; Vodnar, D.C.; Marc, G.; Benedec, D.; Tiperciuc, B.; Tamaian, R.; Oniga, O. Antioxidant activity and antibacterial evaluation of new thiazolin-4-one derivatives as potential tryptophanyl-tRNA synthetase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionuţ, I.; Olteanu, D.; Marc, G.; Clichici, S.; Benedec, D.; Tiperciuc, B.; Oniga, O. Antioxidant Activity Evaluation for Some Novel Chromenyl-Derivatives. Farmacia 2018, 66, 416–420. [Google Scholar] [CrossRef]
- Khodair, A.I.; Alsafi, M.A.; Nafie, M.S. Synthesis, molecular modeling and anti-cancer evaluation of a series of quinazoline derivatives. Carbohydr. Res. 2019, 486, 107832. [Google Scholar] [CrossRef]
- El-Azab, A.S.; Al-Dhfyan, A.; Abdel-Aziz, A.A.M.; Abou-Zeid, L.A.; Alkahtani, H.M.; Al-Obaid, A.M.; Al-Gendy, M.A. Synthesis, anticancer and apoptosis-inducing activities of quinazoline–isatin conjugates: Epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J. Enzym. Inhib. Med. Chem. 2017, 32, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT-Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Bedlovičová, Z.; Strapáč, I.; Baláž, M.; Salayová, A. A Brief Overview on Antioxidant Activity Determination of Silver Nanoparticles. Molecules 2020, 25, 3191. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Method Enzymol. 1999, 299, 15–27. [Google Scholar] [CrossRef]
- Prieto, P.; Pineda, M.; Aguilar, M. Spectrophotometric Quantitation of Antioxidant Capacity through the Formation of a Phosphomolybdenum Complex: Specific Application to the Determination of Vitamin, E. Anal. Biochem. 1999, 269, 337–341. [Google Scholar] [CrossRef]
- Baig, H.; Ahmed, D.; Zara, S.; Aujla, M.I.; Asghar, M.N. In Vitro Evaluation of Antioxidant Properties of Different Solvent Extracts of Rumex acetosella Leaves. Orient. J. Chem. 2011, 27, 1509–1516. [Google Scholar]
- Alam, M.N.; Bristi, N.J.; Rafiquzzaman, M. Review on in vivo and in vitro methods evaluation of antioxidant activity. Saudi Pharm. J. 2013, 21, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shopska, V.; Denkova-Kostova, R.; Dzhivoderova-Zarcheva, M.; Teneva, D.; Denev, P.; Kostov, G. Comparative Study on Phenolic Content and Antioxidant Activity of Different Malt Types. Antioxidants 2021, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Syrpas, M.; Valanciene, E.; Augustiniene, E.; Malys, N. Valorization of Bilberry (Vaccinium myrtillus L.) Pomace by Enzyme-Assisted Extraction: Process Optimization and Comparison with Conventional Solid-Liquid Extraction. Antioxidants 2021, 10, 773. [Google Scholar] [CrossRef] [PubMed]
- Manojlovic, N.T.; Vasiljevic, P.J.; Maskovic, P.Z.; Juskovic, M.; Bogdanovic-Dusanovic, G. Chemical Composition, Antioxidant, and Antimicrobial Activities of Lichen Umbilicaria cylindrica (L.) Delise (Umbilicariaceae). Evid. -Based Complement. Altern. Med. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mic, M.; Pîrnău, A.; Floare, C.G.; Marc, G.; Franchini, A.H.; Oniga, O.; Vlase, L.; Bogdan, M. Synthesis and molecular interaction study of a diphenolic hidrazinyl-thiazole compound with strong antioxidant and antiradical activity with HSA. J. Mol. Struct. 2021, 1244, 131278. [Google Scholar] [CrossRef]
- Cesari, L.; Mutelet, F.; Canabady-Rochelle, L. Antioxidant properties of phenolic surrogates of lignin depolymerisation. Ind. Crops Prod. 2019, 129, 480–487. [Google Scholar] [CrossRef]
- Wu, H.-C.; Shiau, C.-Y.; Chen, H.-M.; Chiou, T.-K. Antioxidant activities of carnosine, anserine, some free amino acids and their combination. J. Food Drug Anal. 2003, 11, 148–153. [Google Scholar] [CrossRef]
- Świsłocka, R.; Regulska, E.; Karpińska, J.; Świderski, G.; Lewandowski, W. Molecular Structure and Antioxidant Properties of Alkali Metal Salts of Rosmarinic Acid. Experimental and DFT Studies. Molecules 2019, 24, 2645. [Google Scholar] [CrossRef] [Green Version]
- Grozav, A.; Porumb, I.-D.; Găină, L.; Filip, L.; Hanganu, D. Cytotoxicity and Antioxidant Potential of Novel 2-(2-((1H-indol-5yl)methylene)-hydrazinyl)-thiazole Derivatives. Molecules 2017, 22, 260. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % of ABTS+ Scavenging | IC50 (µg/mL) | |||||
|---|---|---|---|---|---|---|---|
| 3.33 µg/mL | 4.99 µg/mL | 6.66 µg/mL | 9.99 µg/mL | 13.32 µg/mL | 16.65 µg/mL | ||
| 5a | 14.13 | 21.94 | 30.87 | 44.61 | 60.39 | 75.09 | 11.09 |
| 5b | 52.73 | 60.97 | 71.00 | 88.62 | + | + | 2.87 |
| 5c | 32.20 | 59.31 | 88.59 | + | + | + | 4.40 |
| 5d | 49.82 | 56.51 | 63.76 | 76.99 | 86.60 | + | 3.17 |
| 5e | 28.63 | 38.66 | 49.82 | 65.81 | 82.66 | + | 7.07 |
| 5f | 44.01 | 55.27 | 66.73 | 83.76 | + | + | 4.15 |
| 5g | 42.49 | 66.39 | 92.31 | + | + | + | 3.85 |
| 5h | 60.48 | 72.18 | 80.92 | + | + | + | 1.54 |
| 5i | 33.09 | 45.73 | 59.07 | 84.39 | + | + | 5.52 |
| 5j | 27.51 | 42.01 | 53.16 | 70.44 | 90.53 | + | 6.55 |
| 5k | 60.34 | 72.03 | 83.81 | + | + | + | 1.86 |
| 5l | 57.62 | 66.81 | 74.19 | 91.08 | + | + | 1.73 |
| Ascorbic acid | 60.97 | 73.24 | 87.73 | + | + | + | 2.01 |
| Trolox | 38.66 | 53.16 | 66.54 | 94.57 | + | + | 4.66 |
| Compound | % of DPPH Scavenging | IC50 (µg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| 2.5 µg/mL | 3.75 µg/mL | 5 µg/mL | 7.5 µg/mL | 10 µg/mL | 12.5 µg/mL | 15 µg/mL | ||
| 5a | − | − | − | − | 6.16 | 19.89 | 28.55 | >15 |
| 5b | 49.14 | 58.34 | 71.82 | 90.78 | + | + | + | 2.30 |
| 5c | 10.73 | 19.89 | 28.14 | 41.61 | 56.11 | 69.67 | 83.89 | 9.02 |
| 5d | − | − | − | − | 8.45 | 23.32 | 33.12 | >15 |
| 5e | 5.58 | 20.03 | 30.70 | 50.23 | 75.09 | 94.62 | + | 7.32 |
| 5f | 27.37 | 38.48 | 46.70 | 61.71 | 80.84 | + | + | 5.60 |
| 5g | − | − | − | − | − | 6.16 | 17.60 | >15 |
| 5h | 49.65 | 58.40 | 64.90 | 75.62 | + | + | + | 2.30 |
| 5i | 47.36 | 55.37 | 58.80 | 69.95 | 81.03 | + | + | 2.88 |
| 5j | − | − | − | − | − | 6.04 | 18.75 | >15 |
| 5k | 49.65 | 60.36 | 71.55 | 90.16 | + | + | + | 2.47 |
| 5l | 22.18 | 32.54 | 46.42 | 64.28 | 81.69 | + | + | 5.82 |
| Ascorbic acid | 47.45 | 55.71 | 64.21 | 79.16 | 94.39 | + | + | 2.83 |
| Trolox | 28.53 | 40.42 | 53.87 | 75.85 | 94.85 | + | + | 4.68 |
| Compound | % of NO Scavenged |
|---|---|
| 5a | 37.84 |
| 5b | 42.60 |
| 5c | 18.72 |
| 5d | 46.79 |
| 5e | 44.12 |
| 5f | 30.72 |
| 5g | 43.47 |
| 5h | 50.53 |
| 5i | 32.14 |
| 5j | 50.28 |
| 5k | 42.70 |
| 5l | 23.09 |
| Gentisic acid | 48.14 |
| Compound | % of Activity of Ascorbic Acid | % of Activity of Trolox | ||||||
|---|---|---|---|---|---|---|---|---|
| FRAP | TAC | RP | CUPRAC | FRAP | TAC | RP | CUPRAC | |
| 5a | 25.59 | 58.40 | 38.94 | 46.72 | 29.43 | 112.81 | 56.74 | 44.53 |
| 5b | 71.09 | 37.66 | 60.11 | 155.11 | 81.75 | 72.75 | 87.58 | 147.84 |
| 5c | 16.18 | 46.38 | 32.82 | 6.96 | 18.60 | 89.59 | 47.82 | 6.63 |
| 5d | 19.76 | 65.42 | 36.71 | 91.61 | 22.73 | 126.38 | 53.49 | 87.32 |
| 5e | 24.83 | 20.91 | 51.08 | 182.84 | 28.56 | 40.39 | 74.43 | 174.28 |
| 5f | 8.38 | 51.91 | 15.07 | 9.74 | 9.63 | 100.28 | 21.97 | 9.28 |
| 5g | 51.17 | 89.39 | 67.74 | 56.00 | 58.85 | 172.68 | 98.70 | 53.37 |
| 5h | 71.99 | 33.71 | 71.76 | 99.24 | 82.78 | 65.12 | 104.56 | 94.59 |
| 5i | 14.08 | 39.31 | 39.23 | 17.94 | 16.19 | 75.94 | 57.17 | 17.10 |
| 5j | 26.64 | 72.86 | 30.16 | 45.95 | 30.64 | 140.75 | 43.94 | 43.80 |
| 5k | 63.38 | 48.33 | 67.42 | 165.05 | 72.89 | 93.36 | 98.24 | 157.31 |
| 5l | 10.80 | 55.33 | 18.66 | 32.59 | 12.42 | 106.88 | 27.19 | 31.07 |
| Compound | Chelation Capacity (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 17.70 µg/mL | 20.59 µg/mL | 29.41 µg/mL | 44.11 µg/mL | 58.82 µg/mL | 88.23 µg/mL | 117.64 µg/mL | 257.46 µg/mL | 343.28 µg/mL | |
| 5a | − | − | − | − | − | − | − | − | 15.85 |
| 5b | − | − | − | − | − | − | − | − | − |
| 5c | − | − | − | − | − | − | − | − | − |
| 5d | − | − | − | − | 9.01 | 15.69 | 24.50 | 36.86 | 55.57 |
| 5e | − | − | − | − | − | − | − | − | − |
| 5f | − | − | − | − | − | − | − | − | − |
| 5g | − | − | − | − | − | − | − | − | 6.45 |
| 5h | − | − | − | − | − | − | − | − | − |
| 5i | − | − | − | − | − | − | − | − | − |
| 5j | − | − | 10.98 | 15.85 | 19.47 | 27.02 | 34.29 | 47.28 | 57.41 |
| 5k | − | − | − | − | − | − | − | − | − |
| 5l | − | − | − | − | − | − | − | − | − |
| EDTA-Na2 | 1.32 | 20.59 | 42.89 | 95.10 | + | + | + | + | + |
| Compound | Chelation Capacity (%) | ||
|---|---|---|---|
| 3.36 µg/mL | 8.40 µg/mL | 16.80 µg/mL | |
| 5a | 5.90 | 12.00 | 21.80 |
| 5b | 5.87 | 11.11 | 18.01 |
| 5c | 7.60 | 15.24 | 33.04 |
| 5d | 5.39 | 14.99 | 31.86 |
| 5e | 4.21 | 9.12 | 16.01 |
| 5f | 5.68 | 13.68 | 23.71 |
| 5g | 2.26 | 14.63 | 33.32 |
| 5h | 0.49 | 5.90 | 13.56 |
| 5i | 5.20 | 13.99 | 23.89 |
| 5j | 2.02 | 8.60 | 18.86 |
| 5k | 4.11 | 8.74 | 14.86 |
| 5l | 4.50 | 8.73 | 15.72 |
| EDTA-Na2 | 10.39 | 22.68 | 44.51 |
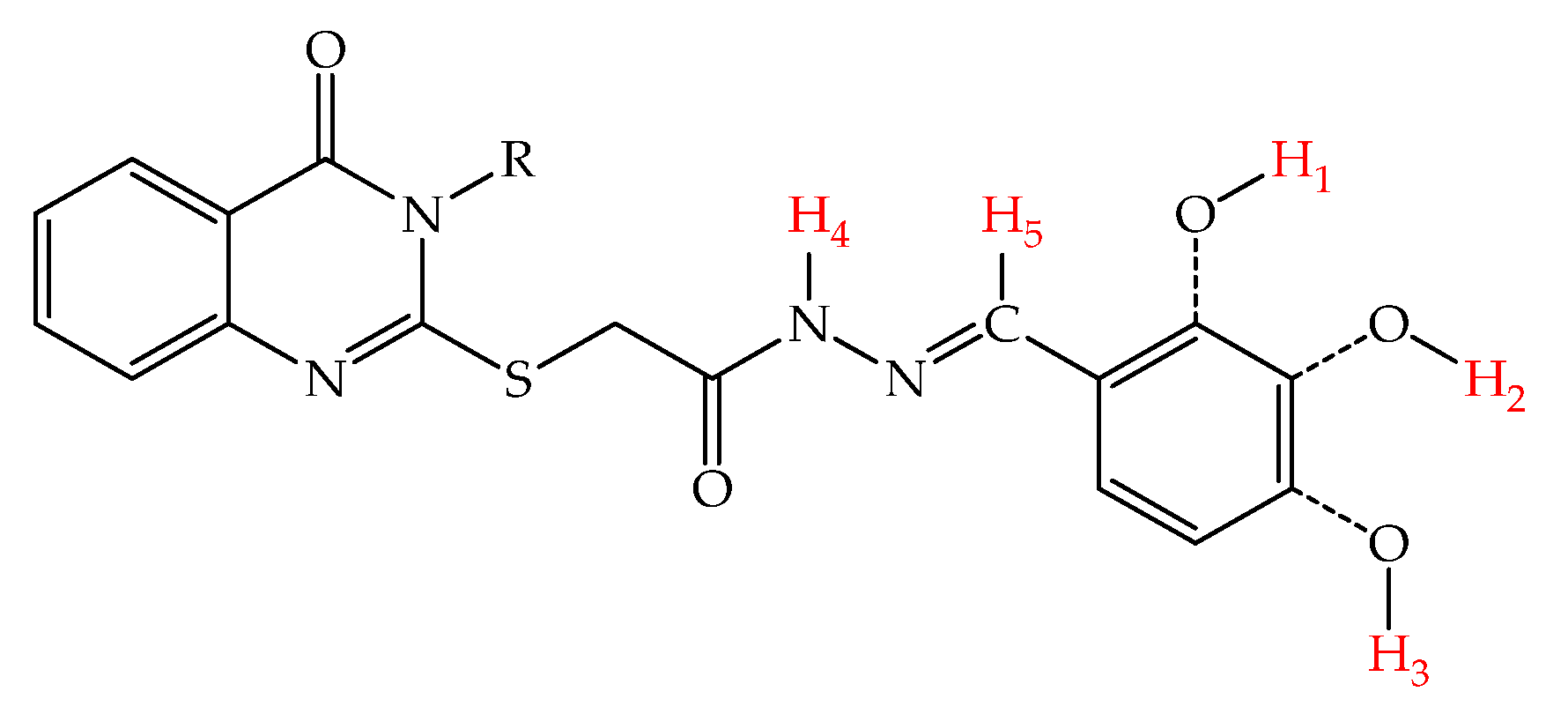
| Compound | Frontier Orbitals (eV) | X-H BDE (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | Gap | H1 | H2 | H3 | H4 | H5 | |
| 5a | −5.54 | −1.66 | 3.88 | 81.54 | 80.20 | N/A | 89.57 | 101.16 |
| 5b | −5.45 | −1.53 | 3.92 | N/A | 77.50 | 72.19 | 97.22 | 99.87 |
| 5c | −5.69 | −1.42 | 4.27 | 88.04 | N/A | 77.00 | 81.23 | 100.36 |
| 5d | −5.45 | −1.67 | 3.78 | 81.60 | 80.23 | N/A | 88.08 | 101.25 |
| 5e | −5.45 | −1.55 | 3.90 | N/A | 77.44 | 72.00 | 88.82 | 99.78 |
| 5f | −5.50 | −1.62 | 3.88 | 85.56 | N/A | 85.25 | 86.38 | 104.97 |
| 5g | −5.54 | −1.67 | 3.87 | 81.49 | 80.17 | N/A | 88.15 | 101.18 |
| 5h | −5.45 | −1.50 | 3.95 | N/A | 77.45 | 72.17 | 90.60 | 99.80 |
| 5i | −5.68 | −1.36 | 4.32 | 80.25 | N/A | 76.86 | 81.08 | 102.56 |
| 5j | −5.53 | −1.61 | 3.92 | 81.56 | 80.14 | N/A | 87.98 | 101.20 |
| 5k | −5.43 | −1.49 | 3.94 | N/A | 77.40 | 72.07 | 88.96 | 99.84 |
| 5l | −5.58 | −1.28 | 4.30 | 82.70 | N/A | 79.22 | 82.64 | 105.04 |
| Compound | Conformation | Compound | Conformation |
|---|---|---|---|
| 5a |  | 5g |  |
| 5b |  | 5h |  |
| 5c |  | 5i |  |
| 5d |  | 5j |  |
| 5e |  | 5k |  |
| 5f |  | 5l |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pele, R.; Marc, G.; Stana, A.; Ionuț, I.; Nastasă, C.; Tiperciuc, B.; Oniga, I.; Pîrnău, A.; Vlase, L.; Oniga, O. Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies. Molecules 2022, 27, 2599. https://doi.org/10.3390/molecules27082599
Pele R, Marc G, Stana A, Ionuț I, Nastasă C, Tiperciuc B, Oniga I, Pîrnău A, Vlase L, Oniga O. Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies. Molecules. 2022; 27(8):2599. https://doi.org/10.3390/molecules27082599
Chicago/Turabian StylePele, Raluca, Gabriel Marc, Anca Stana, Ioana Ionuț, Cristina Nastasă, Brîndușa Tiperciuc, Ilioara Oniga, Adrian Pîrnău, Laurian Vlase, and Ovidiu Oniga. 2022. "Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies" Molecules 27, no. 8: 2599. https://doi.org/10.3390/molecules27082599
APA StylePele, R., Marc, G., Stana, A., Ionuț, I., Nastasă, C., Tiperciuc, B., Oniga, I., Pîrnău, A., Vlase, L., & Oniga, O. (2022). Synthesis of New Phenolic Derivatives of Quinazolin-4(3H)-One as Potential Antioxidant Agents—In Vitro Evaluation and Quantum Studies. Molecules, 27(8), 2599. https://doi.org/10.3390/molecules27082599