
Ergothioneine, Ovothiol A, and Selenoneine—Histidine-Derived, Biologically Significant, Trace Global Alkaloids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ergothioneine (10)
2.1. Occurrence
2.2. Biology
2.3. Biosynthesis
2.4. Genetic Studies
2.5. Ergothioneine Production
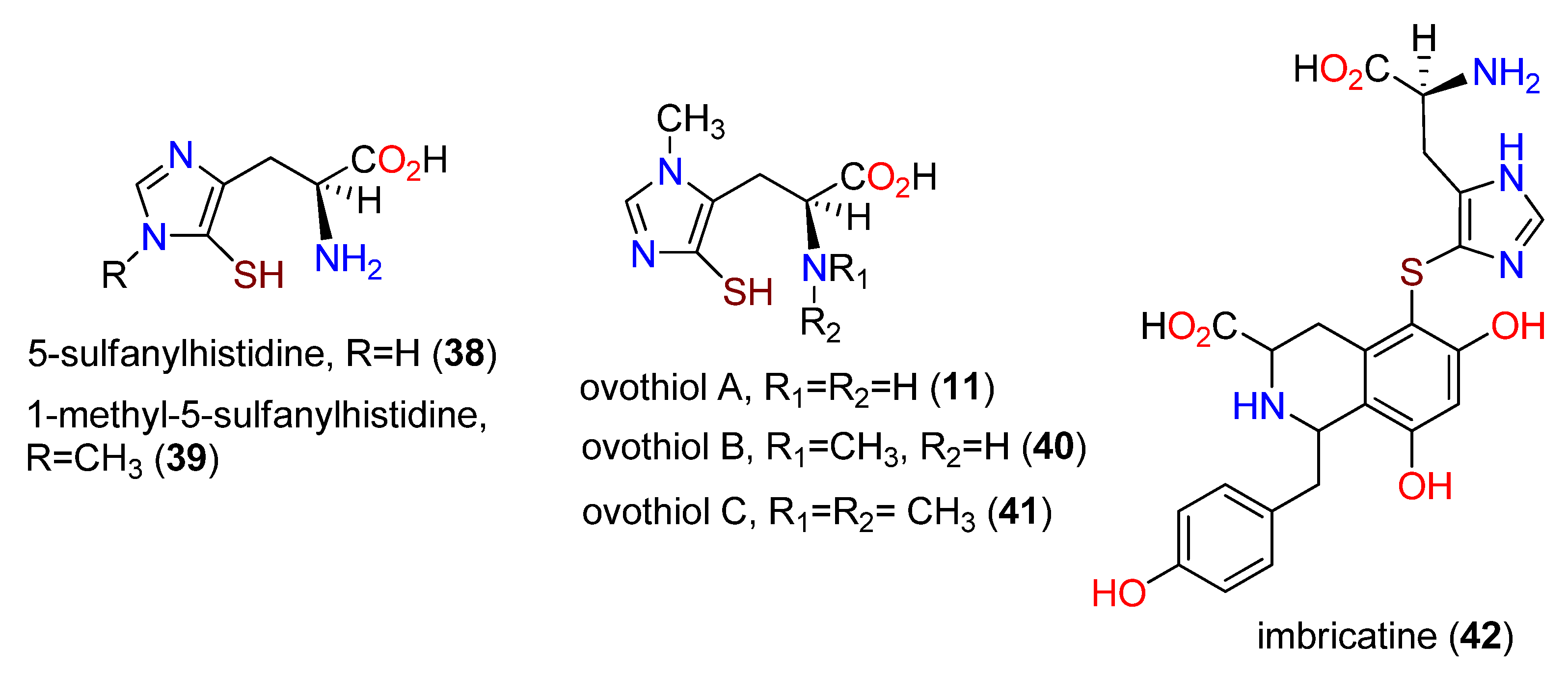
3. Ovothiols
3.1. Occurrence
3.2. Biology
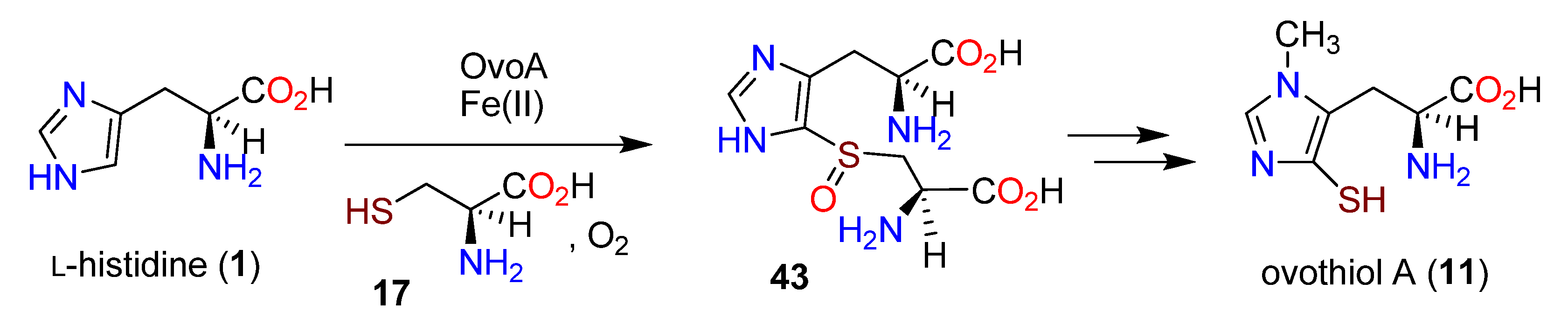
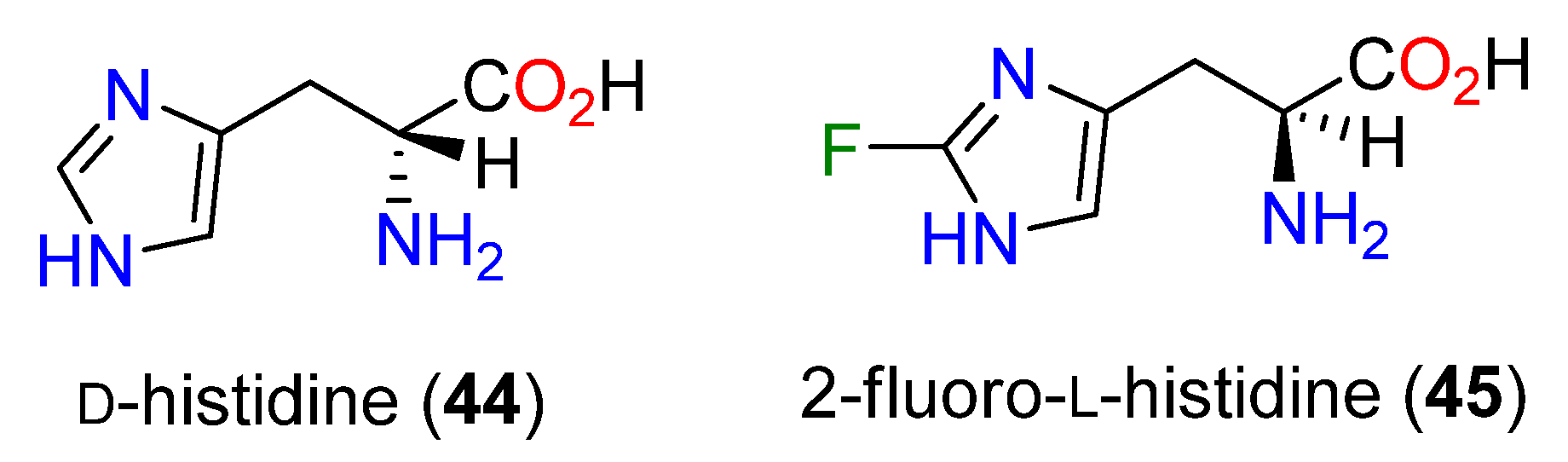
3.3. Biosynthesis
3.4. Anaerobic Synthesis of Ergothioneine
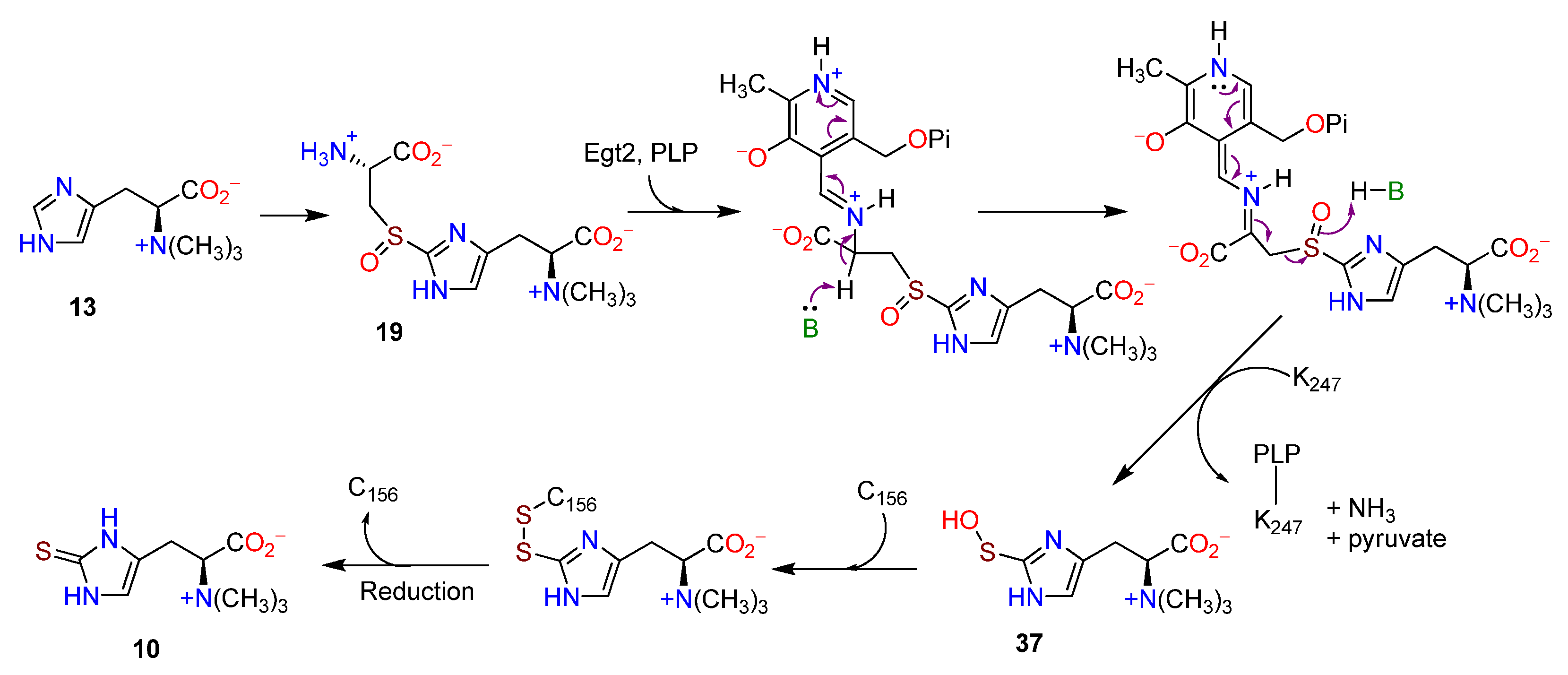
3.5. Final Pathway Steps
3.6. Additional Sources
4. Selenoneine
4.1. Introduction
4.2. Biosynthesis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Funayama, S.; Cordell, G.A. Alkaloids—A Treasury of Poisons and Medicines; Academic Press: New York, NY, USA, 2015; p. 157. [Google Scholar]
- Santos, A.P.; Moreno, P.R.H. Alkaloids derived from histidine: Imidazole (pilocarpine, pilosine). In Handbook of Natural Products; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 861–882. [Google Scholar]
- Rosenberg, H.; Paul, A.G. Dolichotheline, an imidazole alkaloid from Dolichothele sphaerica. Tetrahedron Lett. 1969, 10, 1039–1042. [Google Scholar] [CrossRef] [Green Version]
- Ferrigni, N.R.; Meyer, B.N.; McLaughlin, J.L. Cactus alkaloids. LVI. Carbon-13 and proton NMR of the imidazoles, Nα,Nα-dimethylhistamine and dolichotheline. Rev. Latinoamer. Quim. 1984, 14, 131–133. [Google Scholar]
- Rosenberg, H.; Paul, A.G. Biosynthesis of dolichotheline in Dolichothele sphaerica. Lloydia 1971, 34, 372–376. [Google Scholar]
- Andrade, P.; Willoughby, R.; Pomponi, S.A.; Kerr, R.G. Biosynthetic studies of the alkaloid, stevensine, in a cell culture of the marine sponge Teichaxinella morchella. Tetrahedron Lett. 1999, 40, 4775–4778. [Google Scholar] [CrossRef]
- Albizati, K.F.; Faulkner, D.J. Stevensine, a novel alkaloid of an unidentified marine sponge. J. Org. Chem. 1985, 50, 4163–4164. [Google Scholar] [CrossRef]
- Chilton, M.-D.; Drummond, M.H.; Merlo, D.J.; Sciaky, D.; Montoya, A.L.; Gordon, M.P.; Nester, E.W. Stable incorporation of plasmid DNA into higher plant cells: The molecular basis of crown gall tumorigenesis. Cell 1977, 11, 263–271. [Google Scholar] [CrossRef]
- Isogai, A.; Fukuchi, N.; Hayashi, M.; Kamada, H.; Harada, H.; Suzuki, A. Structure of a new opine, mikimopine, in hairy root induced by Agrobacterium rhizogenes. Agric. Biol. Chem. 1988, 52, 3235–3237. [Google Scholar] [CrossRef]
- Isogai, A.; Fukuchi, N.; Hayashi, M.; Kamada, H.; Harada, H.; Suzuki, A. Mikimopine, an opine in hairy roots of tobacco induced by Agrobacterium rhizogenes. Phytochemistry 1990, 29, 3131–3134. [Google Scholar] [CrossRef]
- Tanret, C. Sur une base nouvelle retirée du seigle ergote, l’ergothioneine. Compt. Rend. Acad. Sci. 1909, 149, 222–224. [Google Scholar]
- Barger, G.; Ewins, A.J. The constitution of ergothioneine: A betaine related to histidine. J. Chem. Soc. Trans. 1911, 99, 2336–2341. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.; Eagles, B.A. The isolation from blood of a hitherto unknown substance, and its bearing on present methods for the estimation of uric acid. J. Biol. Chem. 1925, 65, 623–641. [Google Scholar] [CrossRef]
- Hunter, G.; Eagles, B.A. Non-protein sulphur compounds of blood. I. Sympectothion. J. Biol. Chem. 1927, 72, 123–132. [Google Scholar] [CrossRef]
- Benedict, S.A.; Newton, E.B.; Behre, J.A. A new sulfur-containing compound (thiasine) in the blood. J. Biol. Chem. 1926, 67, 267–277. [Google Scholar] [CrossRef]
- Newton, E.B.; Benedict, S.R.; Dakin, H.D. The chemical constitution of thiasine. Science 1926, 64, 602. [Google Scholar] [CrossRef] [PubMed]
- Newton, E.B.; Benedict, S.R.; Dakin, H.D. On thiasine, its structure and identification with ergothioneine. J. Biol. Chem. 1927, 62, 367–373. [Google Scholar] [CrossRef]
- Hunter, G. A new test for ergothioneine upon which is based a method for its estimation in simple solution and blood-filtrates. Biochem. J. 1928, 22, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Sotgia, S.; Mangoni, A.A.; Forteschi, M.; Murphy, R.B.; Elliot, D.; Sotgiu, E.; Pintus, G.; Carru, C.; Zinellu, A. Identification of the main intermediate precursor of L-ergothioneine biosynthesis in human biological specimens. Molecules 2016, 21, 1298. [Google Scholar] [CrossRef] [Green Version]
- Tokuhiro, S.; Yamada, R.; Chang, C.; Suzuki, A.; Kochi, Y.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Ohtsuki, M.; Ono, M.; et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet. 2003, 35, 341–348. [Google Scholar] [CrossRef]
- Peltekova, V.D.; Wintle, R.F.; Rubin, L.A.; Amos, C.I.; Huang, Q.; Gu, X.; Newman, B.; Oene, M.V.; Cescon, D.; Greenberg, G.; et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004, 36, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Eagles, B.A.; Vars, H.M. The physiology of ergothioneine. J. Biol. Chem. 1928, 80, 615–622. [Google Scholar] [CrossRef]
- Ramirez-Martinez, A.; Wesolek, N.; Yadan, J.-C.; Moutet, M.; Roudot, A.-C. Intake assessment of L-ergothioneine in some European countries and in the United States. Hum. Ecol. Risk Assess. 2016, 22, 667–677. [Google Scholar] [CrossRef]
- Melville, D.B.; Eich, S. The occurrence of ergothioneine in plant material. J. Biol. Chem. 1956, 218, 647–651. [Google Scholar] [CrossRef]
- Ey, J.; Schömig, E.; Taubert, D. Dietary sources and antioxidant effects of ergothioneine. J. Agric. Food Chem. 2007, 55, 6466–6474. [Google Scholar] [CrossRef] [PubMed]
- Dubost, N.; Ou, B.; Beelman, R. Quantification of polyphenols and ergothioneine in cultivated mushrooms and correlation to total antioxidant capacity. Food Chem. 2007, 105, 727–735. [Google Scholar] [CrossRef]
- Kalaras, M.D.; Richie, J.P.; Calcagnotto, A.; Beelman, R.B. Mushrooms: A rich source of the antioxidants ergothioneine and glutathione. Food Chem. 2017, 233, 429–433. [Google Scholar] [CrossRef]
- Beelman, R.B.; Kalaras, M.D.; Phillips, A.T.; Richie, J.P. Is ergothioneine a ‘longevity vitamin’ limited in the American diet? J. Nutr. Sci. 2020, 9, e52. [Google Scholar] [CrossRef]
- Servillo, L.; D’Onofrio, N.; Balestrieri, M.L. Ergothioneine antioxidant function: From chemistry to cardiovascular therapeutic potential. J. Cardiovasc. Pharmacol. 2017, 69, 183–191. [Google Scholar] [CrossRef]
- Cumming, B.M.; Chinta, K.C.; Reddy, V.P.; Steyn, A.J.C. Role of ergothioneine in microbial physiology and pathogenesis. Antioxid. Redox Signal. 2018, 28, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Cheah, I.K.; Tang, R.M.Y. Ergothioneine—A diet-derived antioxidant with therapeutic potential. FEBS Letts. 2018, 592, 3357–3366. [Google Scholar] [CrossRef] [Green Version]
- Cheah, I.K.; Halliwell, B. Ergothioneine, recent developments. Redox Biol. 2021, 42, 101868. [Google Scholar] [CrossRef]
- Heath, H.; Lawson, A.; Rimington, C. Synthesis of ergothioneine. Nature 1950, 166, 106. [Google Scholar] [CrossRef] [PubMed]
- Heath, H.; Lawson, A.; Rimington, C. 2-Mercaptoglyoxalines. Part I. The synthesis of ergothioneine. J. Chem. Soc. 1951, 2215–2217. [Google Scholar] [CrossRef]
- Sunko, D.E.; Wolf, G. Synthesis of DL-2-mercaptohistidine-α-C14 and DL-ergothioneine-α-C14. J. Am. Chem. Soc. 1958, 80, 4405–4406. [Google Scholar] [CrossRef]
- Xu, J.; Yadan, J.C. Synthesis of L-(+)-ergothioneine. J. Org. Chem. 1995, 60, 6296–6301. [Google Scholar] [CrossRef]
- Khonde, P.L.; Jardine, A. Improved synthesis of the super antioxidant, ergothioneine, and its biosynthetic pathway intermediates. Org. Biomol. Chem. 2015, 13, 1415–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naowarojna, N.; Cheng, R.; Chen, L.; Quill, M.; Xu, M.; Zhao, C.; Liu, P. Mini-review: Ergothioneine and ovothiol biosyntheses, an unprecedented trans-sulfur strategy in natural product biosynthesis. Biochemistry 2018, 57, 3309–3325. [Google Scholar] [CrossRef] [Green Version]
- Borodina, I.; Kenny, L.C.; McCarthy, C.M.; Paramasivan, K.; Pretorius, E.; Roberts, T.J.; van der Hoek, S.A.; Kell, D.B. The biology of ergothioneine, an antioxidant nutraceutical. Nutr. Res. Rev. 2020, 33, 190–217. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Cheah, I.K.; Drum, C.L. Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem. Biophys. Res. Commun. 2016, 470, 245–250. [Google Scholar] [CrossRef]
- Lam-Sidun, D.; Peters, K.M.; Borradaile, N.M. Mushroom-derived medicine? Preclinical studies suggest potential benefits of ergothioneine for cardiometabolic health. J. Mol. Sci. 2021, 2, 32446. [Google Scholar] [CrossRef]
- Melville, D.B.; Horner, W.H.; Lubschez, R. Tissue ergothioneine. J. Biol. Chem. 1954, 206, 221–228. [Google Scholar] [CrossRef]
- Shires, T.K.; Brummel, M.C.; Pulido, J.S.; Stegink, L.D. Ergothioneine distribution in bovine and porcine ocular tissues. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1997, 117, 117–120. [Google Scholar] [CrossRef]
- Leone, E.; Mann, T. Ergothioneine in the seminal vesicle secretion. Nature 1951, 168, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Heath, H.; Rimington, C.; Mann, T. Further studies on seminal ergothioneine of the pig. Biochem. J. 1957, 65, 369–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundemann, D.; Harlfinger, S.; Golz, S.; Geerts, A.; Lazar, A.; Berkels, R.; Jung, N.; Rubbert, A.; Schomig, E. Discovery of the ergothioneine transporter. Proc. Natl. Acad. Sci. USA 2005, 102, 5256–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschirka, J.; Kreisor, M.; Betz, J.; Gründemann, D. Substrate selectivity check of the ergothioneine transporter. Drug Metab. Disp. 2018, 46, 779–785. [Google Scholar] [CrossRef]
- Kato, Y.; Kubo, Y.; Iwata, D.; Kato, S.; Sudo, T.; Sugiura, T.; Kagaya, T.; Wakayama, T.; Hirayama, A.; Sugimoto, M.; et al. Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm. Res. 2010, 27, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.; Snyder, S. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 2010, 17, 1134. [Google Scholar] [CrossRef] [Green Version]
- Cheah, I.K.; Halliwell, B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I.; Whiteman, M.; England, T.G.; Halliwell, B. Antioxidant action of ergothioneine: Assessment of its ability to scavenge peroxynitrite. Biochem. Biophys. Res. Commun. 1997, 231, 389–391. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Spencer, J.P.E.; Mahmood, N. Protection against oxidative damage and cell death by the natural antioxidant ergothioneine. Food Chem. Toxicol. 1999, 37, 1043–1053. [Google Scholar] [CrossRef]
- Kaneko, I.; Takeuchi, Y.; Yamaoka, Y.; Tanaka, Y.; Fukuda, T.; Fukumori, Y.; Mayumi, T.; Hama, T. Quantitative determination of ergothioneine in plasma and tissues by TLC densitometry. Chem. Pharm. Bull. 1980, 28, 3093–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, I. Ergothioneine in the central nervous system. J. Neurochem. 1972, 19, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Crossland, J.; Mitchell, J.; Woodruff, G.N. The presence of ergothioneine in the central nervous system and its probable identity with the cerebellar factor. J. Physiol. 1966, 182, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncaster, J.A.; Walsh, D.T.; Gentleman, S.M.; Jen, L.-S.; Aruoma, O.I. Ergothioneine treatment protects neurons against N-methyl-D-aspartate excitotoxicity in an in vivo rat retinal model. Neurosci. Lett. 2002, 328, 55–59. [Google Scholar] [CrossRef]
- Cheah, I.K.; Feng, L.; Tang, R.M.Y.; Lim, K.H.C.; Halliwell, B. Ergothioneine levels in an elderly population decrease with age and incidence of cognitive decline; a risk factor for neurodegeneration? Biochem. Biophys. Res. Commun. 2016, 478, 162–167. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Smith, E.; Ottosson, F.; Hellstrand, S.; Ericson, U.; Orho-Melander, M.; Fernandez, C.; Melander, O. Ergothioneine is associated with reduced mortality and decreased risk of cardiovascular disease. Heart 2020, 106, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G. On ergothioneine in blood and diazo-reacting substances in maize. Biochem. J. 1951, 48, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Melville, D.B.; Homer, W.H. Blood ergothioneine in the germ-free chicken. J. Biol. Chem. 1953, 202, 187–191. [Google Scholar] [CrossRef]
- Heath, H. The metabolism of 35S-labelled 2-thiolhistidine and ergothioneine in the rat. Biochem. J. 1953, 54, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Melville, D.B.; Horner, W.H.; Otken, C.C.; Ludwig, M.L. Studies on the origin of ergothioneine in animals. J. Biol. Chem. 1955, 213, 61–68. [Google Scholar] [CrossRef]
- Baldridge, R.C.; Lewis, H.B. Diet and the ergothioneine content of blood. J. Biol. Chem. 1953, 202, 169–176. [Google Scholar] [CrossRef]
- Baldridge, R.C. Blood ergothioneine and dietary oats. J. Nutr. 1955, 56, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Melville, D.B.; Genghof, D.S.; Inamine, E.; Kovalenko, V. Ergothioneine in microorganisms. J. Biol. Chem. 1956, 223, 9–17. [Google Scholar] [CrossRef]
- Heath, H.; Wildy, J. The biosynthesis of ergothioneine and histidine by Claviceps purpurea. I. The incorporation of [2-14C]-acetate. Biochem. J. 1956, 64, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Wildy, J.; Heath, H. Biosynthesis of ergothioneine by Claviceps purpurea. II. Incorporation of [35S]methionine and the non-utilization of [2(ring)-14C]histamine. Biochem. J. 1957, 65, 220–222. [Google Scholar] [CrossRef]
- Heath, H.; Wildy, J. Biosynthesis of ergothioneine. Nature 1957, 179, 196–197. [Google Scholar] [CrossRef]
- Heath, H.; Wildy, J. Biosynthesis of ergothioneine by Claviceps purpurea. III. The incorporation of labelled histidine. Biochem. J. 1958, 68, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Melville, D.B.; Eich, S.; Ludwig, M.L. The biosynthesis of ergothioneine. J. Biol. Chem. 1957, 224, 871–877. [Google Scholar] [CrossRef]
- Melville, D.B.; Ludwig, M.L.; Inamine, E.; Rachele, J.R. Transmethylation in the biosynthesis of ergothioneine. J. Biol. Chem. 1959, 234, 1195–1198. [Google Scholar] [CrossRef]
- Askari, A.; Melville, D.B. The reaction sequence in ergothioneine biosynthesis: Hercynine as an intermediate. J. Biol. Chem. 1962, 237, 1615–1618. [Google Scholar] [CrossRef]
- Kutscher, F. Die basischen Extraktstoffe des Champignons (Agaricus campestris). Z. Für Unters. Der Nahr.-Und Genußmittel Sowie Der Gebrauchsgegenstände 1911, 21, 535–540. [Google Scholar]
- Genghof, D.S. The production of ergothioneine by Mycobacterium tuberculosis. Bacteriol. Proc. 1960, 190. [Google Scholar]
- Genghof, D.S.; Van Damme, O. Biosynthesis of ergothioneine and hercynine by mycobacteria. J. Bacteriol. 1964, 87, 852–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genghof, D.S. Biosynthesis of ergothioneine and hercynine by fungi and Actinomycetales. J. Bacteriol. 1970, 103, 475–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.H.; Audley, B.G. Ergothioneine and hercynine in Hevea brasiliensis latex. Phytochemistry 1968, 7, 109–118. [Google Scholar] [CrossRef]
- Park, E.-J.; Lee, W.Y.; Kim, S.T.; Ahn, J.K.; Bae, E.K. Ergothioneine accumulation in a medicinal plant Gastrodia elata. J. Med. Plants Res. 2010, 4, 1141–1147. [Google Scholar]
- Pfeiffer, C.; Bauer, T.; Surek, B.; Schömig, E.; Gründemann, D. Cyanobacteria produce high levels of ergothioneine. Food Chem. 2011, 129, 1766–1769. [Google Scholar] [CrossRef]
- Ferrazzano, G.F.; Papa, C.; Pollio, A.; Ingenito, A.; Sangianantoni, G.; Cantile, T. Cyanobacteria and microalgae as sources of functional foods to improve human general and oral health. Molecules 2020, 25, 5164. [Google Scholar] [CrossRef]
- Genghof, D.S.; Van Damme, O. Biosynthesis of ergothioneine from endogenous hercynine in Mycobacterium smegmatis. J. Bacteriol. 1968, 95, 340–344. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, V.N.; Ishikawa, Y.; Melville, D.B. Conversion of histidine to hercynine by Neurospora crassa. J. Bacteriol. 1970, 101, 881–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Melville, D.B. The enzymatic α-N-methylation of histidine. J. Biol. Chem. 1970, 245, 5967–5973. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Israel, S.E.; Melville, D.B. Participation of an intermediate sulfoxide in the enzymatic thiolation of the imidazole ring of hercynine to form ergothioneine. J. Biol. Chem. 1974, 249, 4420–4427. [Google Scholar] [CrossRef]
- Seebeck, F.P. In Vitro reconstitution of mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc. 2010, 132, 6632–6633. [Google Scholar] [CrossRef]
- Harth, G.; Masleša-Galić, S.; Tullius, M.V.; Horwitz, M.A. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Mol. Microbiol. 2005, 58, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, J.A.; Dodson, G.; Duggleby, H.J.; Moody, P.C.; Smith, J.L.; Tomchick, D.R.; Murzin, A.G. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 1995, 378, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Vit, A.; Mashabela, G.; Blankenfeldt, W.; Seebeck, F.P. Structure of the ergothioneine-biosynthesis amidohydrolase EgtC. ChemBioChem 2015, 16, 1490–1496. [Google Scholar] [CrossRef] [Green Version]
- Goncharenko, K.V.; Vit, A.; Blankenfeldt, W.; Seebeck, F.P. Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway. Angew. Chem. Int Ed. Engl. 2015, 54, 2821–2824. [Google Scholar] [CrossRef] [Green Version]
- Vit, A.; Misson, L.; Blankenfeldt, W.; Seebeck, F.P. Ergothioneine biosynthetic methyltransferase EgtD reveals the structural basis of aromatic amino acid betaine biosynthesis. ChemBioChem 2015, 16, 119–125. [Google Scholar] [CrossRef]
- Liao, C.; Seebeck, F.P. Convergent evolution of ergothioneine biosynthesis in cyanobacteria. ChemBioChem 2017, 18, 2115–2118. [Google Scholar] [CrossRef]
- Liscombe, D.K.; Louie, G.V.; Noel, J.P. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 2012, 29, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Misson, L.; Burn, R.; Vit, A.; Hildesheim, J.; Beliaeva, M.A.; Blankenfeldt, W.; Seebeck, F.P. Inhibition and regulation of the ergothioneine biosynthetic methyltransferase EgtD. ACS Chem. Biol. 2018, 13, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Richard-Greenblatt, M.; Bach, H.; Adamson, J.; Peña-Diaz, S.; Li, W.; Steyn, A.J.C.; Av-Gay, Y. Regulation of ergothioneine biosynthesis and its effect on Mycobacterium tuberculosis growth and infectivity. J. Biol. Chem. 2015, 290, 23064–23076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, L.; Owens, R.A.; Dolan, S.K.; O’Keeffe, G.; Schrettl, M.; Kavanagh, K.; Jones, G.W.; Doyle, S. The Aspergillus fumigatus protein GliK protects against oxidative stress and is essential for gliotoxin biosynthesis. Eukaryot. Cell 2012, 11, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, P.; Buchmeier, N.; Newton, G.L.; Rawat, M.; Fahey, R.C. Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J. Bacteriol. 2011, 193, 1981–1990. [Google Scholar] [CrossRef] [Green Version]
- Bello, M.H.; Barrera-Perez, V.; Morin, D.; Epstein, L. The Neurospora crassa mutant NcΔEgt-1 identifies an ergothioneine biosynthetic gene and demonstrates that ergothioneine enhances conidial survival and protects against peroxide toxicity during conidial germination. Fungal Genet. Biol. 2012, 49, 160–172. [Google Scholar] [CrossRef]
- Sheridan, K.J.; Lechner, B.E.; O’Keeffe, G.; Keller, M.A.; Werner, E.R.; Lindner, H.; Jones, G.W.; Haas, H.; Doyle, S. Ergothioneine biosynthesis and functionality in the opportunistic fungal pathogen, Aspergillus fumigatus. Sci. Rep. 2016, 6, 35306. [Google Scholar] [CrossRef] [Green Version]
- Pluskal, T.; Ueno, M.; Yanagida, M. Genetic and metabolomic dissection of the ergothioneine and selenoneine biosynthetic pathway in the fission yeast, S. pombe, and construction of an overproduction system. PLoS ONE 2014, 9, e97774. [Google Scholar] [CrossRef] [Green Version]
- Braunshausen, A.; Seebeck, F.P. Identification and characterization of the first ovothiol biosynthetic enzyme. J. Am. Chem. Soc. 2011, 133, 1757–1759. [Google Scholar] [CrossRef]
- Mashabela, G.T.M.; Seebeck, F.P. Substrate specificity of an oxygen dependent sulfoxide synthase in ovothiol biosynthesis. Chem. Commun. 2013, 49, 7714–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.J.; Siegbahn, P.E.; Liao, R.Z. Theoretical study of the mechanism of the non-heme iron enzyme EgtB. Inorg. Chem. 2017, 56, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Su, H.; Liu, Y. Mechanism of sulfoxidation and C–S bond formation involved in the biosynthesis of ergothioneine catalyzed by ergothioneine synthase (EgtB). ACS Catal. 2018, 8, 5875–5889. [Google Scholar] [CrossRef]
- Naowarojna, N.; Irani, S.; Hu, W.; Cheng, R.; Zhang, L.; Li, X.; Chen, J.; Zhang, Y.J.; Liu, P. Crystal structure of the ergothioneine sulfoxide synthase from Candidatus Chloracidobacterium thermophilum and structure-guided engineering to modulate its substrate selectivity. ACS Catal. 2019, 9, 6955–6961. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Leninger, M.; Lee, N.; Liu, P. Regioselectivity of the oxidative C–S bond formation in ergothioneine and ovothiol biosyntheses. Org. Lett. 2013, 15, 4854–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Hu, W.; Naowarojna, N.; Her, A.S.; Wang, S.; Desai, R.; Qin, L.; Chen, X.; Liu, P. Mechanistic studies of a novel C-S lyase in ergothioneine biosynthesis: The involvement of a sulfenic acid intermediate. Sci. Rep. 2015, 5, 11870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irani, S.; Naowarojna, N.; Tang, Y.; Kathuria, K.R.; Wang, S.; Dhembi, A.; Lee, N.; Yan, W.; Lyu, H.; Costello, C.E.; et al. Snapshots of CS cleavage in Egt2 reveals substrate specificity and reaction mechanism. Cell Chem. Biol. 2018, 25, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.W.; Doyle, S.; Fitzpatrick, D.A. The evolutionary history of the genes involved in the biosynthesis of the antioxidant ergothioneine. Gene 2014, 549, 161–170. [Google Scholar] [CrossRef]
- Kuettner, E.B.; Hilgenfeld, R.; Weiss, M.S. The active principle of garlic at atomic resolution. J. Biol. Chem. 2002, 277, 46402–46407. [Google Scholar] [CrossRef] [Green Version]
- Stampfli, A.R.; Blankenfeldt, W.; Seebeck, F.P. Structural basis of ergothioneine biosynthesis. Curr. Opin. Struct. Biol. 2020, 65, 1–8. [Google Scholar] [CrossRef]
- Osawa, T.; Kamide, T.; Satoh, Y.; Kawano, Y.; Ohtsu, I.; Dairi, T. Heterologous and high production of ergothioneine in Escherichia coli. J. Agric. Food Chem. 2018, 66, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Kawano, Y.; Satoh, Y.; Dairi, T.; Ohtsu, I. Gramscale fermentative production of ergothioneine driven by overproduction of cysteine in Escherichia coli. Sci. Rep. 2019, 9, 1895. [Google Scholar] [CrossRef] [PubMed]
- Takusagawa, S.; Satoh, Y.; Ohtsu, I.; Dairi, T. Ergothioneine production with Aspergillus oryzae. Biosci. Biotechnol. Biochem. 2019, 83, 181–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Hoek, S.A.; Darbani, B.; Zugaj, K.E.; Prabhala, B.K.; Biron, M.B.; Randelovic, M.; Medina, J.B.; Kell, D.B.; Borodina, I. Engineering the yeast Saccharomyces cerevisiae for the production of L-(+)-ergothioneine. Front. Bioeng. Biotechnol. 2019, 7, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-Y.; Pan, H.-Y.; Guo, L.-Q.; Lin, J.-F.; Liao, H.-L.; Li, H.-Y. Successful biosynthesis of natural antioxidant ergothioneine in Saccharomyces cerevisiae required only two genes from Grifola frondosa. Microb. Cell Fact. 2020, 19, 164. [Google Scholar] [CrossRef]
- Nardi, G.; Palumbo, A.; Prota, G. The role of the white bodies in the biosynthesis of adenochrome. Comp. Biochem. Phvsiol. 1982, 71B, 297–300. [Google Scholar] [CrossRef]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Isolation and structure of a new sulphur-containing aminoacid from sea urchin eggs. Tetrahedron Lett. 1982, 23, 3207–3208. [Google Scholar] [CrossRef]
- Turner, E.; Klevit, R.; Hopkins, P.B.; Shapiro, B.M. Ovothiol: A novel thiohistidine compound from sea urchin eggs that confers NAD(P)H-O2 oxidoreductase activity on ovoperoxidase. J. Biol. Chem. 1986, 261, 13056–13063. [Google Scholar] [CrossRef]
- Turner, E.; Klevit, R.; Hager, L.J.; Shapiro, B.M. Ovothiols, a family of redox-active mercaptohistidine compounds from marine invertebrate eggs. Biochemistry 1987, 26, 4028–4036. [Google Scholar] [CrossRef]
- Steenkamp, D.J.; Spies, H.S. Identification of a major low-molecular-mass thiol of the trypanosomatid Crithidia fasciculata as ovothiol A. Facile isolation and structural analysis of the bimane derivative. Eur. J. Biochem. 1994, 223, 43–50. [Google Scholar] [CrossRef]
- Spies, H.S.; Steenkamp, D.J. Thiols of intracellular pathogens. Identification of ovothiol A in Leishmania donovani and structural analysis of a novel thiol from Mycobacterium bovis. Eur. J. Biochem. 1994, 224, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Holler, T.P.; Salpenstein, A.; Turner, E.; Klevit, R.E.; Shapiro, B.M.; Hopkins, P.B. Synthesis and structure reassignment of mercaptohistidines of marine origin. Syntheses of L-ovothiols A and C. J. Org. Chem. 1987, 52, 4420–4421. [Google Scholar] [CrossRef]
- Holler, T.P.; Ruan, F.; Salpenstein, A.; Hopkins, P.B. Total synthesis of marine mercaptohistidines: Ovothiols A, B, and C. J. Org. Chem. 1989, 54, 4570–4575. [Google Scholar] [CrossRef]
- Mirzahosseini, A.; Hosztafi, S.; Tóth, G.; Noszál, B. A cost-effective synthesis of enantiopure ovothiol A from L-histidine, its natural precursor. Arkivoc 2014, vi, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pathirana, C.; Andersen, R.J. Imbricatine, an unusual benzyltetrahydro-isoquinoline alkaloid isolated from the starfish Dermasterias imbricata. J. Am. Chem. Soc. 1986, 108, 8288–8289. [Google Scholar] [CrossRef]
- Burgoyne, D.L.; Miao, S.; Pathirana, C.; Andersen, R.J.; Ayer, W.A.; Singer, P.P.; Kokke, W.C.M.C.; Ross, D.M. The structure and partial synthesis of imbricatine, a benzyltetrahydroisoquinoline alkaloid from the starfish Dermasterias imbricata. Can. J. Chem. 1991, 69, 20–27. [Google Scholar] [CrossRef]
- Shapiro, B.M.; Hopkins, P.B. Ovothiols: Biological and chemical perspectives. Adv. Enzymol. Relat. Areas Mol. Biol. 1991, 64, 291–316. [Google Scholar]
- Castellano, I.; Seebeck, F.P. On ovothiol biosynthesis and biological roles: From life in the ocean to therapeutic potential. Nat. Prod. Rep. 2018, 35, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Turner, E.; Hager, L.J.; Shapiro, B.M. Ovothiol replaces glutathione peroxidase as a hydrogen peroxide scavenger in sea urchin eggs. Science 1988, 242, 939–941. [Google Scholar] [CrossRef]
- Holler, T.P.; Hopkins, P.B. Ovothiols as free-radical scavengers and the mechanism of ovothiol-promoted NAD(P)H-O2 oxidoreductase activity. Biochemistry 1990, 29, 1953–1961. [Google Scholar] [CrossRef]
- Marjanovic, B.; Simic, M.G.; Jovanovic, S.V. Heterocyclic thiols as antioxidants: Why ovothiol C is a better antioxidant than ergothioneine. Free Radic. Biol. Med. 1995, 18, 679–685. [Google Scholar] [CrossRef]
- Castellano, I.; Migliaccio, O.; D’Aniello, S.; Merlino, A.; Napolitano, A.; Palumbo, A. Shedding light on ovothiol biosynthesis in marine metazoans. Sci. Rep. 2016, 6, 21506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osik, N.A.; Zelentsova, E.A.; Tsentalovich, Y.P. Kinetic studies of antioxidant properties of ovothiol A. Antioxidants 2021, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Cerio, O.; Reina, L.; Squatrito, V.; Etxebarria, N.; Gonzalez-Gaya, B.; Cancio, I. Gametogenesis-related fluctuations in ovothiol levels in the mantle of mussels from different estuaries: Fighting oxidative stress for spawning in polluted waters. Biomolecules 2020, 10, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauth-Siegel, R.L.; Leroux, A.E. Low-molecular-mass antioxidants in parasites. Antioxid. Redox Signal. 2012, 17, 583–607. [Google Scholar] [CrossRef] [PubMed]
- Ariyanayagam, M.R.; Fairlamb, A.H. Ovothiol and trypanothione as antioxidants in trypanosomatids. Mol. Biochem. Parasitol. 2001, 115, 189–198. [Google Scholar] [CrossRef]
- Gonçalves, C.; Costa, P.M. Histochemical detection of free thiols in glandular cells and tissues of different marine Polychaeta. Histochem. Cell Biol. 2020, 154, 315–325. [Google Scholar] [CrossRef]
- Yanshole, V.V.; Yanshole, L.V.; Zelentsova, E.A.; Tsentalovich, Y.P. Ovothiol A is the main antioxidant in fish lens. Metabolites 2019, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Milito, A.; Orefice, I.; Smerilli, A.; Castellano, I.; Napolitano, A.; Brunet, C.; Palumbo, A. Insights into the light response of Skeletonema marinoi: Involvement of ovothiol. Mar. Drugs 2020, 18, 477. [Google Scholar] [CrossRef]
- O’Neill, E.C.; Trick, M.; Hill, L.; Rejzek, M.; Dusi, R.G.; Hamilton, C.J.; Zimba, P.V.; Henrissat, B.; Field, R.A. The transcriptome of Euglena gracilis reveals unexpected metabolic capabilities for carbohydrate and natural product biochemistry. Mol. BioSyst. 2015, 11, 2808–2820. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.P.; Lin, Z.; Watkins, M.; Salcedo, P.F.; Baskin, R.P.; Elhabian, S.; Safavi-Hemami, H.; Taylor, D.; Tun, J.; Concepcion, G.P. Small-molecule mimicry hunting strategy in the imperial cone snail, Conus imperialis. Sci. Adv. 2021, 7, eabf2704. [Google Scholar] [CrossRef] [PubMed]
- Röhl, I.; Schneider, B.; Schmidt, B.; Zeeck, E. L-Ovothiol A: The egg release pheromone of the marine polychaete Platynereis dumerilii: Annelida: Polychaeta. Z. Nat. C 1999, 54, 1145–1174. [Google Scholar] [CrossRef]
- Castellano, I.; Di Tomo, P.; Di Pietro, N.; Mandatori, D.; Pipino, C.; Formoso, G.; Napolitano, A.; Palumbo, A. Anti-inflammatory activity of marine ovothiol A in an in vitro model of endothelial dysfunction induced by hyperglycemia. Oxid. Med. Cell. Longev. 2018, 2018, 2087373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancaccio, M.; D’Argenio, G.; Lembo, V.; Palumbo, A.; Castellano, I. Antifibrotic effect of marine ovothiol in an in vivo model of liver fibrosis. Oxid. Med. Cell. Longev. 2018, 2018, 5045734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milito, A.; Brancaccio, M.; D’Argenio, G.; Castellano, I. Natural sulfur-containing compounds: An alternative therapeutic strategy against liver fibrosis. Cells 2019, 8, 1356. [Google Scholar] [CrossRef] [Green Version]
- Russo, G.L.; Russo, M.; Castellano, I.; Napolitano, A.; Palumbo, A. Ovothiol isolated from sea urchin oocytes induces autophagy in the Hep-G2 cell line. Mar. Drugs 2014, 12, 4069–4085. [Google Scholar] [CrossRef] [Green Version]
- Brancaccio, M.; Russo, M.; Masullo, M.; Palumbo, A.; Russo, G.L.; Castellano, I. Sulfur-containing histidine compounds inhibit γ-glutamyl transpeptidase activity in human cancer cells. J. Biol. Chem. 2019, 294, 14603–14614. [Google Scholar] [CrossRef]
- Milito, A.; Brancaccio, M.; Lisurek, M.; Masullo, M.; Palumbo, A.; Castellano, I. Probing the interactions of sulfur-containing histidine compounds with human gamma-glutamyl transpeptidase. Mar. Drugs 2019, 17, 650. [Google Scholar] [CrossRef] [Green Version]
- Steenkamp, D.J.; Weldrick, D.; Spies, H.S. Studies on the biosynthesis of ovothiol A. Identification of 4-mercaptohistidine as an intermediate. Eur. J. Biochem. 1996, 242, 557–566. [Google Scholar] [CrossRef]
- Vogt, R.N.; Spies, H.S.; Steenkamp, D.S. The biosynthesis of ovothiol A (N-methyl-4-mercaptohistidine). Identification of S-(4′-L-histidyl)-L-cysteine sulfoxide as an intermediate and the products of the sulfoxide lyase reaction. Eur. J. Biochem. 2001, 268, 5229–5241. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Her, A.S.; Raso, F.; Zhen, Z.; Huo, Y.; Liu, P. Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Org. Lett. 2014, 16, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Naowarojna, N.; Chen, B.; Xu, M.; Quill, M.; Wang, J.; Deng, Z.; Zhao, C.; Liu, P. Mechanistic studies of a nonheme iron enzyme OvoA in ovothiol biosynthesis using a tyrosine analogue, 2-amino-3-(4-hydroxy-3-(methoxyl)phenyl) propanoic acid (MeOTyr). ACS Catal. 2018, 9, 253–258. [Google Scholar] [CrossRef]
- Chen, L.; Naowarojna, N.; Song, H.; Wang, S.; Wang, J.; Deng, Z.; Zhao, C.; Liu, P. Use of a tyrosine analogue to modulate the two activities of a nonheme iron enzyme OvoA in ovothiol biosynthesis, cysteine oxidation versus oxidative C–S bond formation. J. Am. Chem. Soc. 2018, 140, 4604–4612. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Song, H.; Her, A.S.; Bak, D.W.; Naowarojna, N.; Elliott, S.J.; Qin, L.; Chen, X.; Liu, P. Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus Neurospora crassa ergothioneine biosynthetic pathway. Org. Lett. 2014, 16, 5382–5385. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, H.J.; Morris, J.H.; Ferrin, T.E.; Babbitt, P.C. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE 2009, 4, e4345. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Burn, R.; Misson, L.; Meury, M.; Seebeck, F.P. Anaerobic origin of ergothioneine. Angew. Chem. Int. Ed. 2017, 56, 12508–12511. [Google Scholar] [CrossRef]
- Ruszczycky, M.W.; Liu, H.W. The surprising history of an antioxidant. Nature 2017, 551, 37–38. [Google Scholar] [CrossRef]
- Cheng, R.; Wu, L.; Lai, R.; Peng, C.; Naowarojna, N.; Hu, W.; Li, X.; Whelan, S.A.; Lee, N.; Lopez, J.; et al. Single-step replacement of an unreactive C–H bond by a C–S bond using polysulfide as the direct sulfur source in the anaerobic ergothioneine biosynthesis. ACS Catal. 2020, 10, 8981–8994. [Google Scholar] [CrossRef]
- Muramatsu, H.; Matsuo, H.; Okada, N.; Ueda, M.; Yamamoto, H.; Kato, S.-I.; Nagata, S. Characterization of ergothionase from Burkholderia sp. HME13 and its application to enzymatic quantification of ergothioneine. Appl. Microbiol. Biotechnol. 2013, 97, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Black, K.A.; Dos Santos, P.C. Shared-intermediates in the biosynthesis of thio-cofactors: Mechanism and functions of cysteine desulfurases and sulfur acceptors. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.M.; White, R.H.; Cash, V.L.; Jack, R.F.; Dean, D.R. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc. Natl. Acad. Sci. USA 1993, 90, 2754–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.; Neumann, P.; Ficner, R.; Tittmann, K. Observation of a stable carbene at the active site of a thiamin enzyme. Nat. Chem. Biol. 2013, 9, 488–490. [Google Scholar] [CrossRef]
- Miller, B.G.; Wolfenden, R. Catalytic proficiency: The unusual case of OMP decarboxylase. Annu. Rev. Biochem. 2002, 71, 847–885. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.; Lai, R.; Peng, C.; Lopez, J.; Li, Z.; Naowarojna, N.; Li, K.; Wong, C.; Lee, N.; Whelan, S.A.; et al. Implications for an imidazole-2-yl carbene intermediate in the rhodanase-catalyzed C-S bond formation reaction of anaerobic ergothioneine biosynthesis. ACS Catal. 2021, 11, 3319–3334. [Google Scholar] [CrossRef] [PubMed]
- Turrini, N.G.; Kroepfl, N.; Jensen, K.B.; Reiter, T.C.; Francesconi, K.A.; Schwerdtle, T.; Kroutil, W.; Kuehnelt, D. Biosynthesis and isolation of selenoneine from genetically modified fission yeast. Metallomics 2018, 10, 1532–1538. [Google Scholar] [CrossRef]
- Goncharenko, K.V.; Flückiger, S.; Liao, C.; Lim, D.; Stampfli, A.R.; Seebeck, F.P. Selenocysteine as a substrate, an inhibitor and a mechanistic probe for bacterial and fungal iron-dependent sulfoxide synthases. Chem.-Eur. J. 2020, 26, 1328–1334. [Google Scholar] [CrossRef]
- Naowarojna, N.; Huang, P.; Cai, Y.; Song, H.; Wu, L.; Cheng, R.; Li, Y.; Wang, S.; Lyu, H.; Zhang, L.; et al. In Vitro reconstitution of the remaining steps in ovothiol A biosynthesis: C–S lyase and methyltransferase reactions. Org. Lett. 2018, 20, 5427–5430. [Google Scholar] [CrossRef]
- Kathuria, K.R.; Irani, S.; Liu, P.; Zhang, Y. Examining the mechanism of Egt2 in ergothioneine biosynthesis. FASEB J. 2017, 31, 606–608. [Google Scholar]
- Gerdol, M.; Sollitto, M.; Pallavicini, A.; Castellano, I. The complex evolutionary history of sulfoxide synthase in ovothiol biosynthesis. Proc. R. Soc. B 2019, 286, 1812. [Google Scholar] [CrossRef] [PubMed]
- Milito, A.; Castellano, I.; Burn, R.; Seebeck, F.P.; Brunet, C.; Palumbo, A. First evidence of ovothiol biosynthesis in marine diatoms. Free Rad. Biol. Med. 2020, 152, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Roncalli, V.; Lauritano, C.; Carotenuto, Y. First report of OvoA gene in marine arthropods: A new candidate stress biomarker in copepods. Marine Drugs 2021, 19, 647. [Google Scholar] [CrossRef]
- Brancaccio, M.; Tangherlini, M.; Danovaro, R.; Castellano, I. Metabolic adaptations to marine environments: Molecular diversity and evolution of ovothiol biosynthesis in bacteria. Genome Biol. Evol. 2021, 13, evab169. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Dong, X. Proteiniphilum acetatigenes gen. nov., sp. nov., from a UASB reactor treating brewery wastewater. Int. J. Syst. Evol. Microbiol. 2005, 55, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Shao, M.F.; Zhang, T. Non-contiguous finished genome sequence and description of Sulfurimonas hongkongensis sp. nov., a strictly anaerobic denitrifying, hydrogen- and sulfur-oxidizing chemolithoautotrophy isolated from marine sediment. Stand Genom. Sci. 2014, 9, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Leisinger, F.; Burn, R.; Meury, M.; Lukat, P.; Seebeck, F.P. Structural and mechanistic basis for anaerobic ergothioneine biosynthesis. J. Am. Chem. Soc. 2019, 141, 6906–6914. [Google Scholar] [CrossRef]
- Yamashita, M.; Imamura, S.; Yabu, T.; Ishihara, K.; Yamashita, Y. Selenium in seafood: Potential nutritional and physiological functions of selenoneine. Biomed. Res. Trace Elem. 2013, 24, 176–184. [Google Scholar]
- Alhasan, R.; Nasim, M.J.; Jacob, C.; Gaucher, C. Selenoneine: A unique reactive selenium species from the blood of tuna with implications for human diseases. Curr. Pharmacol. Rep. 2019, 5, 163–173. [Google Scholar] [CrossRef]
- Kurokawa, S.; Berry, M.J. Selenium. Role of the essential metalloid in health. Metal Ions Life Sci. 2013, 13, 499–534. [Google Scholar]
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef] [PubMed]
- Schrauzer, G.N. Nutritional selenium supplements: Product types, quality, and safety. J. Am. Coll. Nutr. 2001, 20, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.R.; Salt, D.E. Plants, selenium and human health. Curr. Opin. Plant Biol. 2003, 6, 273–279. [Google Scholar] [CrossRef]
- Tinggi, U. Selenium: Its role as antioxidant in human health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Wise, L.A. Environmental selenium and human health: An update. Curr. Environ. Health Rep. 2018, 5, 464–485. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef]
- Zou, K.; Liu, G.; Wu, T.; Du, L. Selenium for preventing Kashin-Beck osteoarthropathy in children: A meta-analysis. Osteoarthr. Cartilage 2009, 17, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Papp, L.V.; Holmgren, A.; Khanna, K.K. Selenium and selenoproteins in health and disease. Antioxid. Redox Signal. 2010, 12, 793–795. [Google Scholar] [CrossRef]
- Hira, C.K.; Partal, K.; Dhillon, K. Dietary selenium intake by men and women in high and low selenium areas of Punjab. Public Health Nutr. 2004, 7, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Rayman, M.P. Selenium intake, status, and health: A complex relationship. Hormones 2020, 19, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plateau, P.; Saveanu, C.; Lestini, R.; Dauplais, M.; Decourty, L.; Jacquier, A.; Blanquet, S.; Lazard, M. Exposure to selenomethionine causes selenocysteine misincorporation and protein aggregation in Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 44761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazard, M.; Dauplais, M.; Blanquet, S.; Plateau, P. Recent advances in the mechanism of selenoamino acids toxicity in eukaryotic cells. Biomol. Concepts 2017, 8, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.C.; Hoffmann, P.R. Selenium, selenoproteins, and immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, Y.; Yamashita, M. Identification of a novel selenium-containing compound, selenoneine, as the predominant chemical form of organic selenium in the blood of bluefin tuna. J. Biol. Chem. 2010, 285, 18134–18138. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.; Yabu, T.; Yamashita, M. Discovery of the strong antioxidant selenoneine in tuna and selenium redox metabolism. World J. Biol. Chem. 2010, 1, 144–150. [Google Scholar] [CrossRef]
- Yamashita, M.; Yamashita, Y.; Ando, T.; Wakamiya, J.; Akiba, S. Identification and determination of selenoneine, 2-selenyl-Nα,Nα,Nα-trimethyl-L-histidine, as the major organic selenium in blood cells in a fish-eating population on remote Japanese islands. Biol. Trace Elem. Res. 2013, 156, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Little, M.; Achouba, A.; Dumas, P.; Ouellet, N.; Ayotte, P.; Lemire, M. Determinants of selenoneine concentration in red blood cells of Inuit from Nunavik (Northern Québec, Canada). Environ. Int. 2019, 127, 243–252. [Google Scholar] [CrossRef]
- Achouba, A.; Dumas, P.; Ouellet, N.; Little, M.; Lemire, M.; Ayotte, P. Selenoneine is a major selenium species in beluga skin and red blood cells of Inuit from Nunavik. Chemosphere 2019, 229, 549–558. [Google Scholar] [CrossRef]
- Yamashita, Y.; Amlund, H.; Suzuki, T.; Hara, T.; Hossain, M.A.; Yabu, T.; Touhata, K.; Yamashita, M. Selenoneine, total selenium, and total mercury content in the muscle of fishes. Fish. Sci. 2011, 77, 679–686. [Google Scholar] [CrossRef]
- Kroepfl, N.; Francesconi, K.A.; Schwerdtle, T.; Kuehnelt, D. Selenoneine and ergothioneine in human blood cells determined simultaneously by HPLC/ICP-QQQ-MS. J. Anal. Atom. Spectrom. 2019, 34, 127–134. [Google Scholar] [CrossRef]
- Seko, T.; Uchida, H.; Yamashita, Y.; Yamashita, M. Novel method for separating selenoneine reduced monomer and ergothioneine from fission yeast extracts. Sep. Purif. Technol. 2021, 254, 117607. [Google Scholar] [CrossRef]
- Lim, D.; Gründemann, D.; Seebeck, F.P. Total synthesis and functional characterization of selenoneine. Angew. Chem. Int. Ed. 2019, 58, 15026–15030. [Google Scholar] [CrossRef] [PubMed]
- Rohn, I.; Kroepfl, N.; Aschner, M.; Bornhorst, J.; Kuehnelt, D.; Schwerdtle, T. Selenoneine ameliorates peroxide-induced oxidative stress in C. elegans. J. Trace Elem. Med. Biol. 2019, 55, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tohfuku, T.; Ando, H.; Morishita, N.; Yamashita, M.; Kondo, M. Dietary intake of selenoneine enhances antioxidant activity in the muscles of the amberjack Seriola dumerili grown in aquaculture. Mar. Biotechnol. 2021, 23, 847–853. [Google Scholar] [CrossRef]
- Yamashita, M.; Yamashita, Y.; Suzuki, T.; Kani, Y.; Mizusawa, N.; Imamura, S.; Takemoto, K.; Hara, T.; Hossain, M.A.; Yaki, T.; et al. Selenoneine, a novel selenium-containing compound, mediates detoxification mechanisms against methylmercury accumulation and toxicity in zebrafish embryo. Mar. Biotechnol. 2013, 15, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Masuda, J.; Umemura, C.; Yokozawa, M.; Yamauchi, K.; Seko, T.; Yamashita, M.; Yamashita, Y. Dietary supplementation of selenoneine-containing tuna dark muscle extract effectively reduces pathology of experimental colorectal cancers in mice. Nutrients 2018, 10, 1380. [Google Scholar] [CrossRef] [Green Version]
- Seko, T.; Imamura, S.; Ishihara, K.; Yamashita, Y.; Yamashita, M. Selenoneine suppresses melanin synthesis by inhibiting tyrosinase in murine B16 melanoma cells and 3D-cultured human melanocytes. Fish. Sci. 2020, 86, 171–179. [Google Scholar] [CrossRef]
- Seko, T.; Imamura, S.; Ishihara, K.; Yamashita, Y.; Yamashita, M. Inhibition of angiotensin-converting enzyme by selenoneine. Fish. Sci. 2019, 85, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Drobyshev, E.; Raschke, S.; Glabonjat, R.A.; Bornhorst, J.; Ebert, F.; Kuehnelt, D.; Schwerdtle, T. Capabilities of selenoneine to cross the in vitro blood-brain barrier model. Metallomics 2021, 13, mfaa007. [Google Scholar] [CrossRef]
- Seko, T.; Imamura, S.; Ishihara, K.; Yamashita, Y.; Yamashita, M. Antioxidative effects of selenium containing imidazole compound, selenoneine, on human leukemic K562 cells. FEBS Open Bio 2018, 8, 358. [Google Scholar]




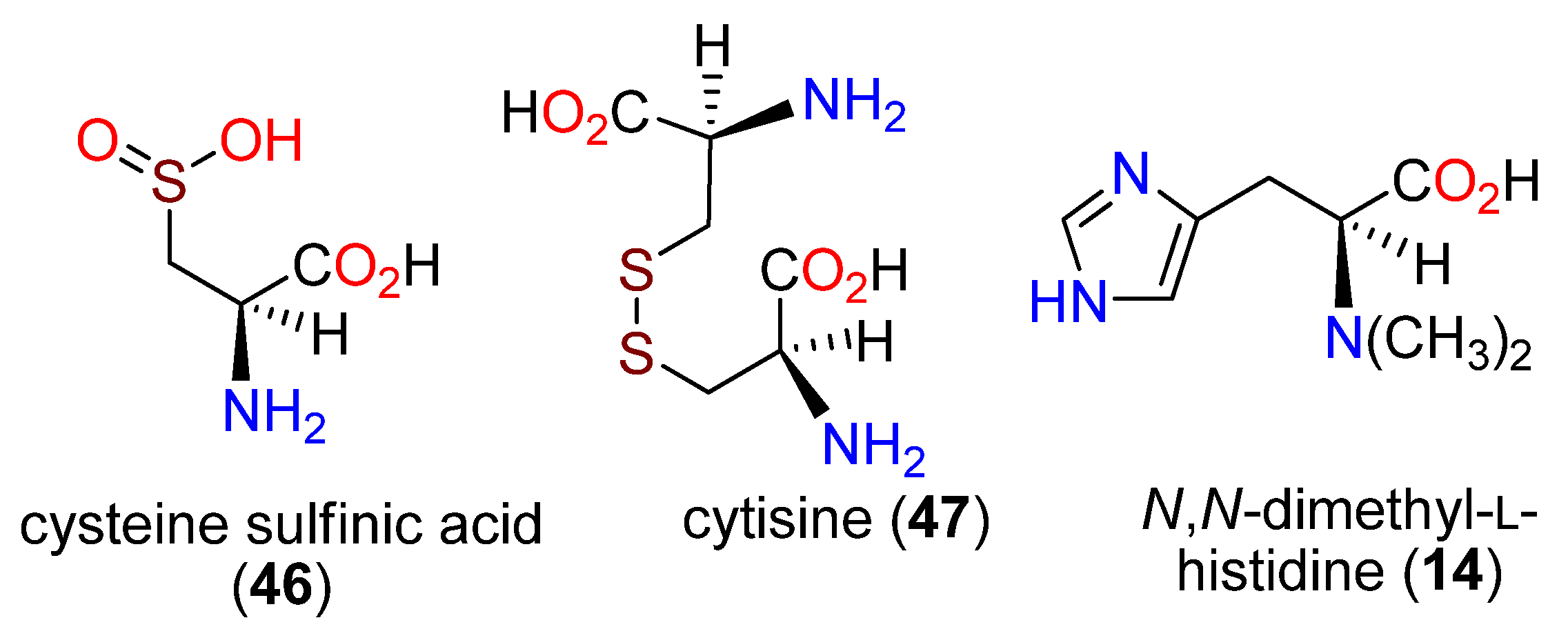
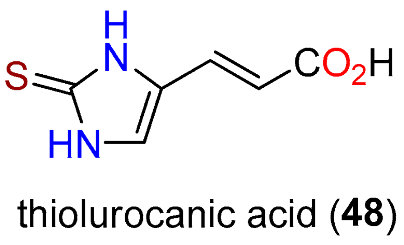

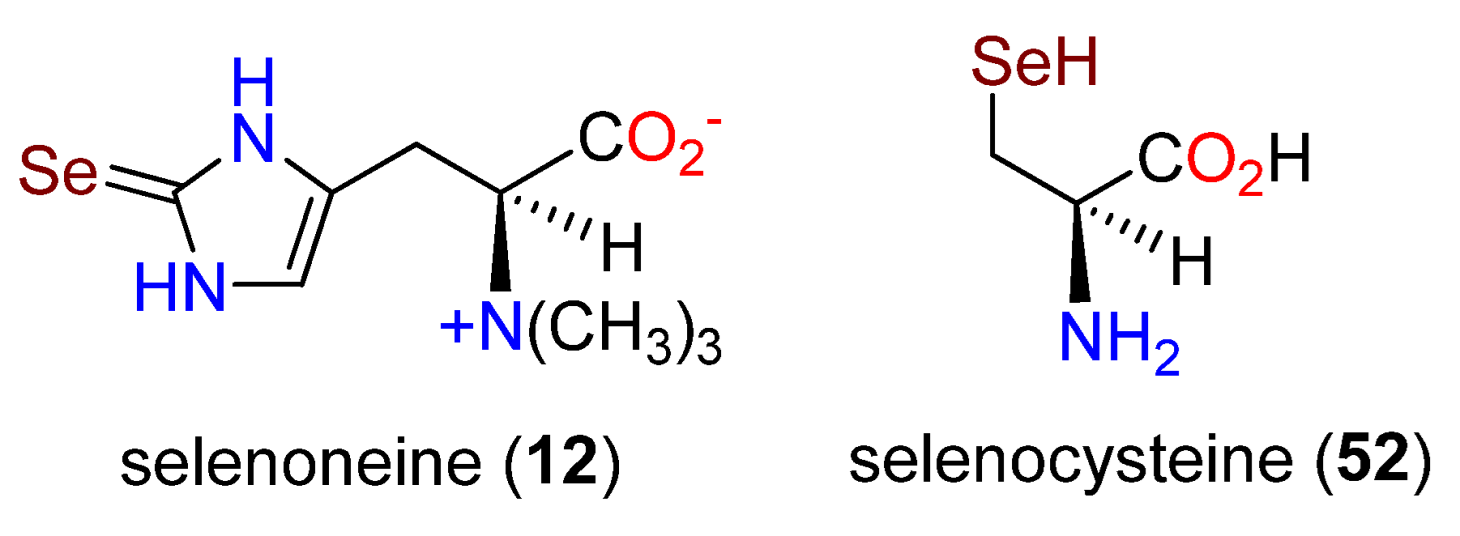
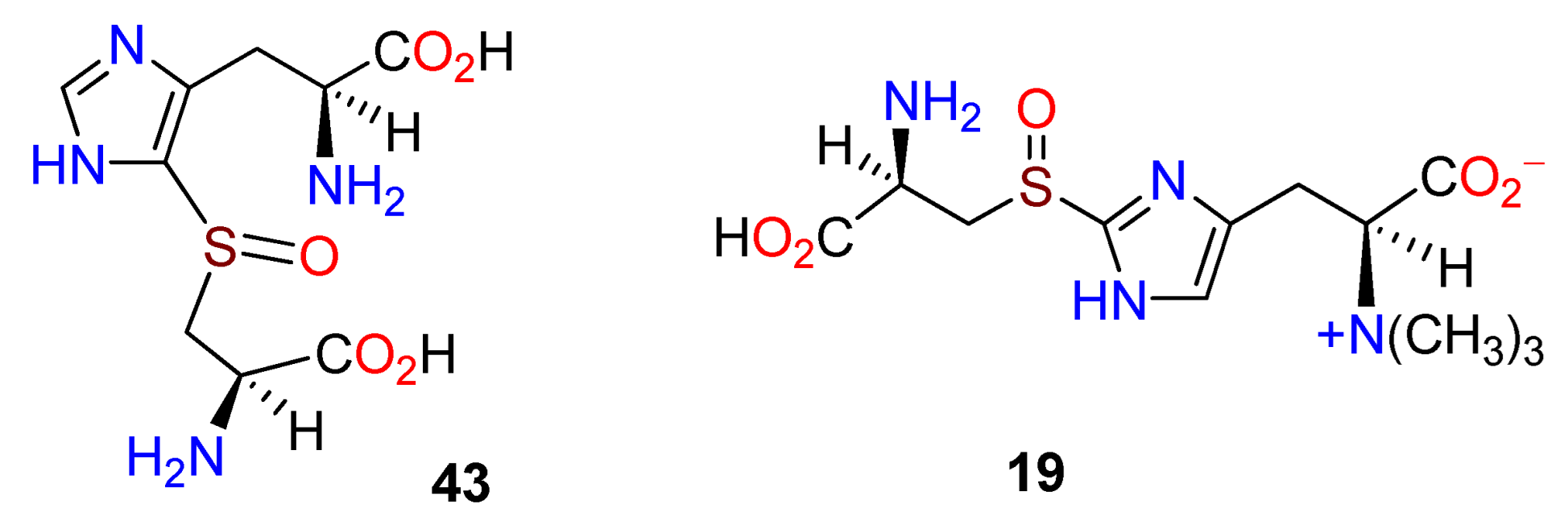
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordell, G.A.; Lamahewage, S.N.S. Ergothioneine, Ovothiol A, and Selenoneine—Histidine-Derived, Biologically Significant, Trace Global Alkaloids. Molecules 2022, 27, 2673. https://doi.org/10.3390/molecules27092673
Cordell GA, Lamahewage SNS. Ergothioneine, Ovothiol A, and Selenoneine—Histidine-Derived, Biologically Significant, Trace Global Alkaloids. Molecules. 2022; 27(9):2673. https://doi.org/10.3390/molecules27092673
Chicago/Turabian StyleCordell, Geoffrey A., and Sujeewa N. S. Lamahewage. 2022. "Ergothioneine, Ovothiol A, and Selenoneine—Histidine-Derived, Biologically Significant, Trace Global Alkaloids" Molecules 27, no. 9: 2673. https://doi.org/10.3390/molecules27092673
APA StyleCordell, G. A., & Lamahewage, S. N. S. (2022). Ergothioneine, Ovothiol A, and Selenoneine—Histidine-Derived, Biologically Significant, Trace Global Alkaloids. Molecules, 27(9), 2673. https://doi.org/10.3390/molecules27092673