1. Introduction
Pandemic coronavirus disease 2019 (COVID-19) is one of the most serious challenges we face [
1]. The struggle against this disease requires the solving of various problems, including the urgent development of antiviral drugs with direct action on the therapeutic target proteins of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that caused the pandemic [
2]. By the beginning of the pandemic, deep knowledge of the mechanisms of coronavirus replication at the molecular level had been gained thanks to studies of earlier diseases such as SARS [
3] and MERS [
4]. Currently, several therapeutic target proteins of this coronavirus are well established [
5,
6,
7,
8,
9]. In this work, we focus our study on the SARS-CoV-2 2′-
O-methyltransferase (2′-
O-MTase; EC 2.1.1.296), which is referred to henceforth as nsp16 [
10]. Non-Structural Protein 16 (nsp16), a viral RNA 2′-
O-MTase, is one of the highly viable targets for the design of agents against SARS-CoV-2. Nsp16, with its cofactor nsp10, forms a heterodimer and stimulates 2′-
O-methyltransferase activity. 2’-
O-methylation is essential for formation of the cap structure in viral RNAs. The presence of the cap part on RNA of SARS-CoV-2 allows it to make its genetic information readable to the host translation machinery. It has been shown that this 2′O-MTase is indispensable for replication of coronaviruses in cell cultures [
11].
Given the importance of the RNA capping process for mRNA stability and translation, and as an immune evasion mechanism, RNA-capping machineries are an attractive target for antiviral-drug design [
12,
13]. In the viral RNA capping process, non-structural protein 16 (nsp16), an
S-adenosyl methionine (SAM)-dependent 2′-
O-MTase, methylates the first transcribed nucleotide at the ribose 2′-OH position, to form a cap-1 structure. To perform a methyl transfer, nsp16 utilizes SAM that contains an activated methyl group and represents a common co-substrate for methyl transfer reactions in living organisms. Blockage of the SAM-binding site of nsp16 results in inhibition of the methylation process catalyzed by this enzyme. 2′-
O-methylation is essential for formation of the cap structure in coronavirus (CoV) RNAs, and, therefore, plays an important role in the replication of CoVs. Furthermore, 2′-
O-methylation allows CoVs to escape the recognition of immune sensors and resist the type I interferon-mediated anti-infective immune response. Importantly, 2′-
O-MTase/nsp16 is highly conserved in structure and function among various CoVs, together with its indispensable role in CoV replication and immune escape, making it an attractive and effective target for broad-spectrum antivirals.
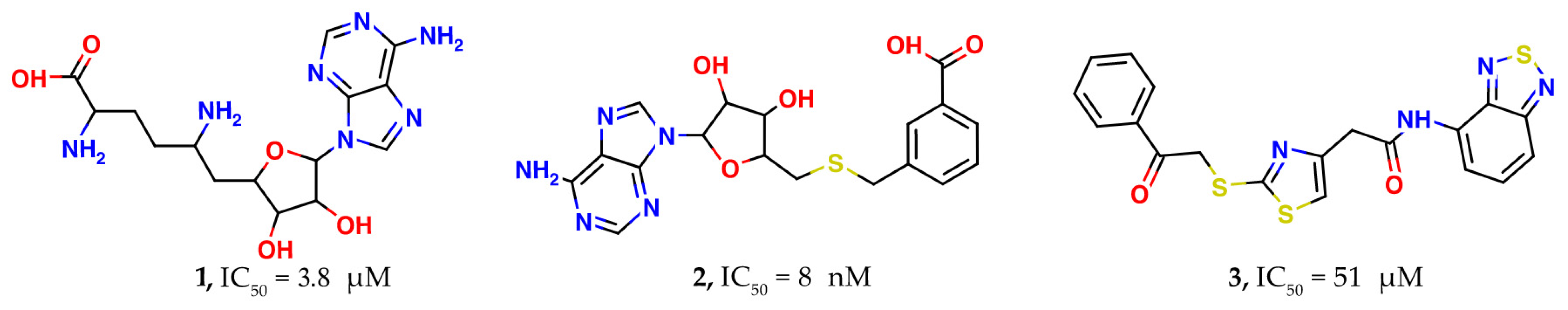
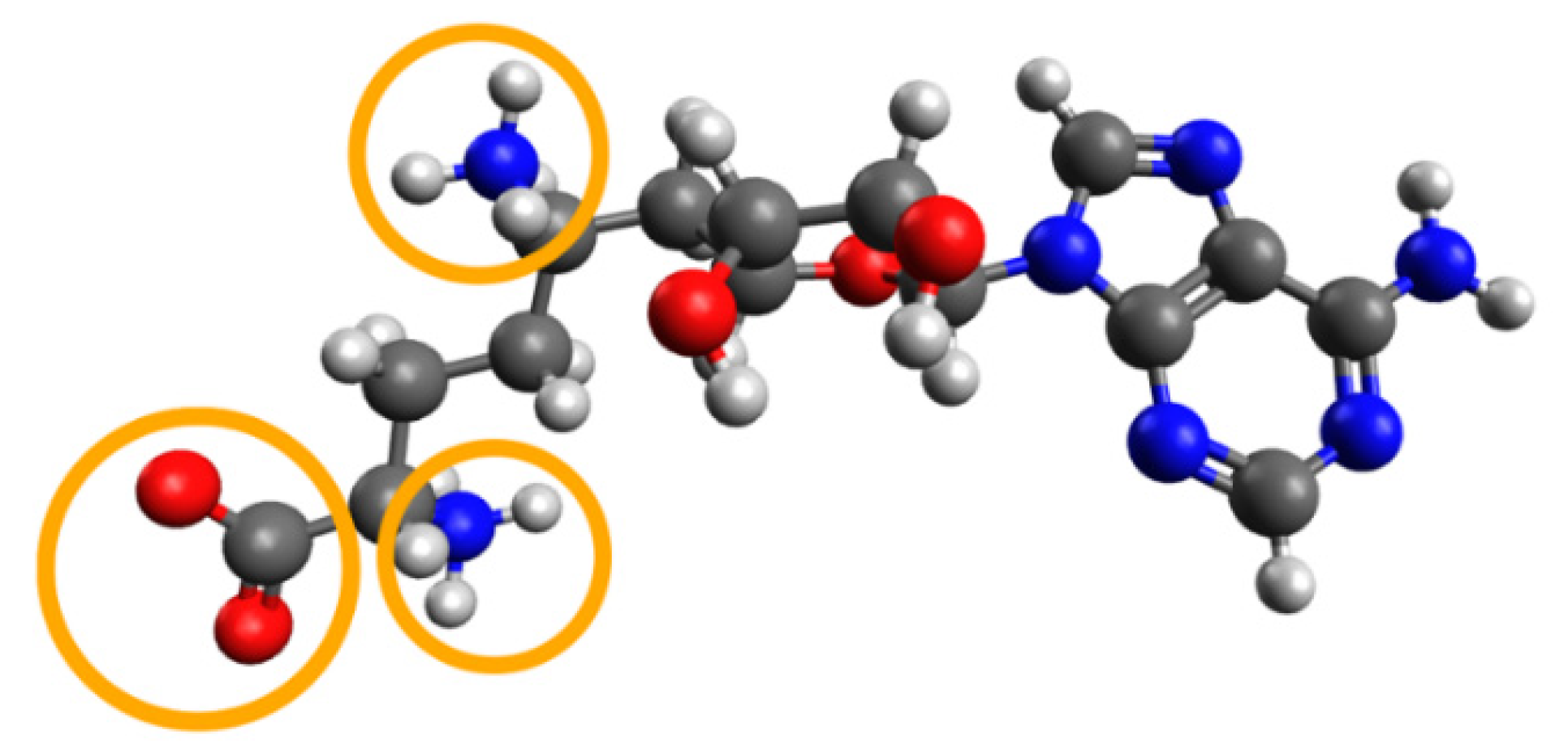
After the first SARS outbreak, the search for nsp16 inhibitors of SARS-CoV-1 led to identification of sinefungin (
1) (see
Figure 1) as a potent suppressor of the methyltransferase activity of nsp16 [
14]. Sinefungin is similar to
S-adenosylmethionine, an endogenous cofactor of nsp16, and binds to the same pocket. Recently, the ability of sinefungin to block the methyltransferase activity of nsp16 in SARS-CoV-2 was confirmed by X-ray crystallography [
11].
In the study by [
15], several nsp16 inhibitors were designed using bioisosteric replacements of the sulfonium and amino acid substructures of the
S-adenosylmethionine. Virtual close analogs of
S-adenosylmethionine were docked into active site of nsp16. Activity testing of the synthesized prioritized compounds showed nanomolar to sub-micromolar IC
50 values for five compounds. The best IC
50 was 8 nM for methylbenzoic acid derivative (
2). However, selectivity testing on human glycine
N-methyltransferase revealed that all inhibitors are non-selective and also inhibit this methyltransferase. The main reason for the non-selectivity can be attributed to the very high similarity between the found nsp16 inhibitors and
S-adenosylmethionine, a co-factor used by most methyltransferases.
Since nsp14 is another viral methyltransferase with a similar active site architecture, some scientific groups initiated a combined search for both nsp16 and nsp14 inhibitors. In the study of [
16], docking-based high-throughput virtual screening of 7 million commercially-available drug-like compounds and
S-adenosylmethionine analogs was performed. As a result, 80 virtual screening hits (39 against nsp14 and 41 against nsp16) were identified, which were purchased and tested using an enzymatic homogeneous time-resolved fluorescent energy transfer assay. Of these, nine compounds showed a micromolar inhibition activity with an IC
50 less than 200 μM. The best inhibitor (
3) was based on a sulfanyl-thiazole core and possessed an IC
50 against nsp16 of 51 µM. Similarly to the above-mentioned study, most of the compounds showed poor selectivity against human glycine
N-methyltransferase.
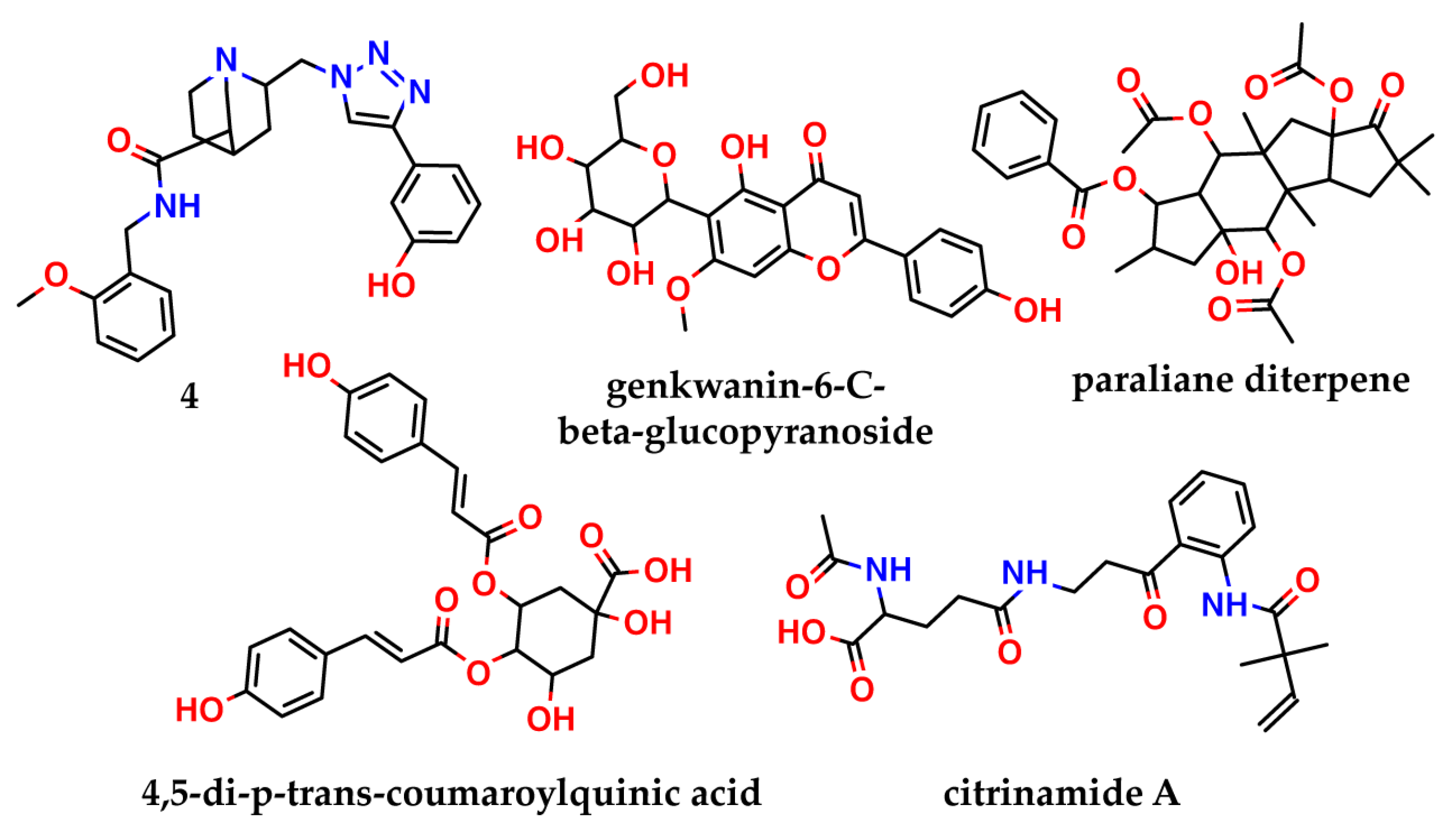
Pharmacophore-based screening for 48 million drug-like compounds of the Zinc database was used to identify nsp16 inhibitors in [
17]. A 3D pharmacophore model was constructed based on the complex of nsp16 with sinefungin (PDB ID: 6WKQ). The 24 best-scoring ligands were then docked in the SAM-binding pocket. Finally, molecular dynamics (MD) simulation experiments for the three best compounds were carried out, as a refinement step. These simulations revealed one compound (
4,
Figure 2) with a triazine scaffold as the potential nsp16 inhibitor. Testing of its activity has not yet been performed.
In the study by [
10], the authors applied an virtual drug repurposing approach to identify nsp16 inhibitors binding to active site of the enzyme. Shape-based screening among FDA approved drugs, followed by molecular dynamics (MD) simulation, revealed that raltegravir and maraviroc can bind tightly to the active site of the protein, compared to sinefungin, and could be potential candidates for inhibition of nsp16. No in vitro confirmation of their activity has been completed to date.
Another strategy to target nsp16, is disrupting the nsp16–nsp10 complex, without affecting the
S-adenosylmethionine-binding site. Such an approach was used, for example, in [
18]. The authors virtually screened the North African Natural Products database for compounds that can interact with the nsp10 interface and disturb the nsp10–nsp16 complex formation. They identified four compounds (genkwanin-6-
C-beta-glucopyranoside, paraliane diterpene, 4,5-di-
p-
trans-coumaroylquinic acid, and citrinamide A—
Figure 2) that showed the best binding affinity, predicted by AutoDock Vina. Of the four compounds, genkwanin-6-
C-beta-glucopyranoside showed the most stable complex with nsp10 in a molecular dynamics simulation, but it was not tested in vitro.
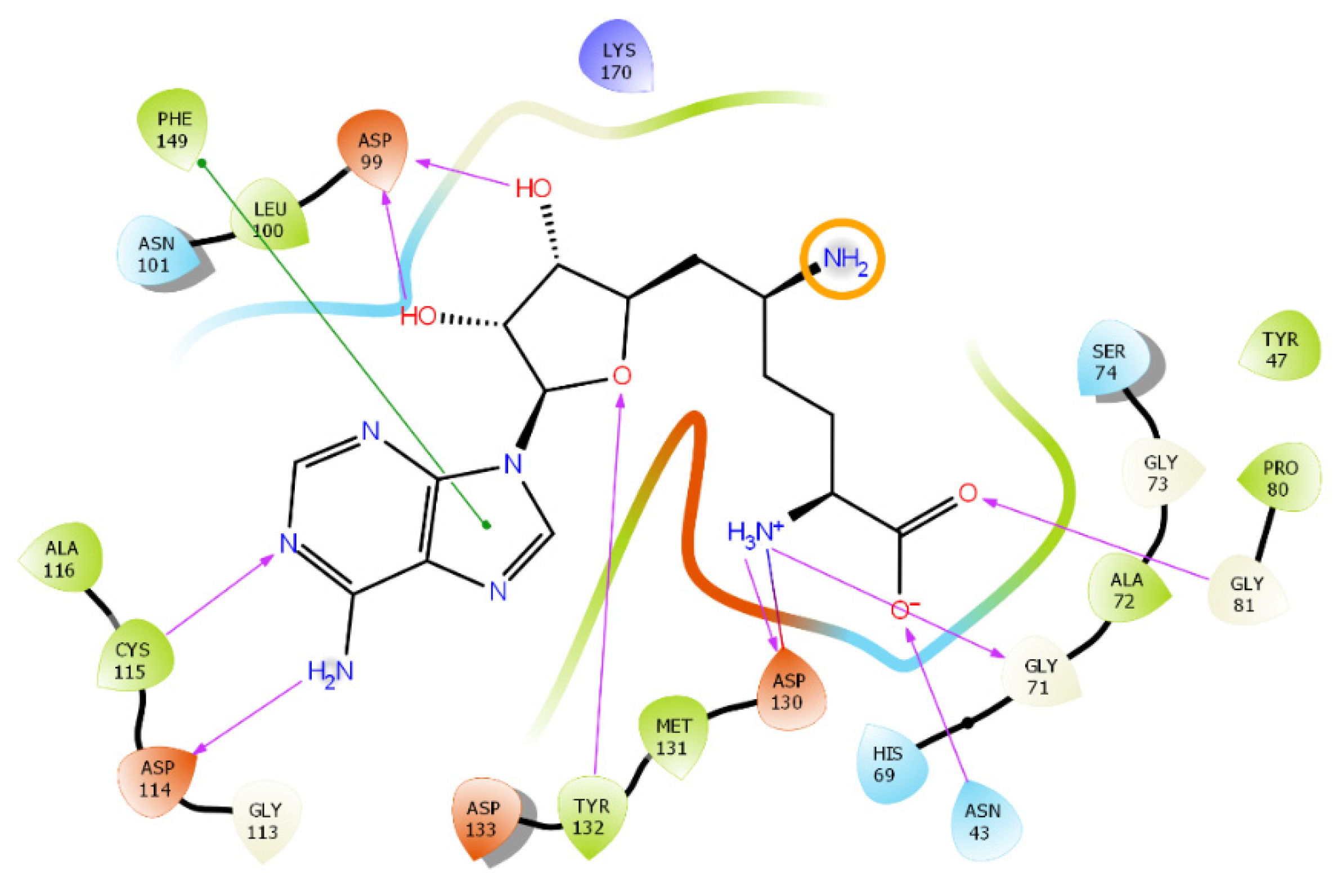
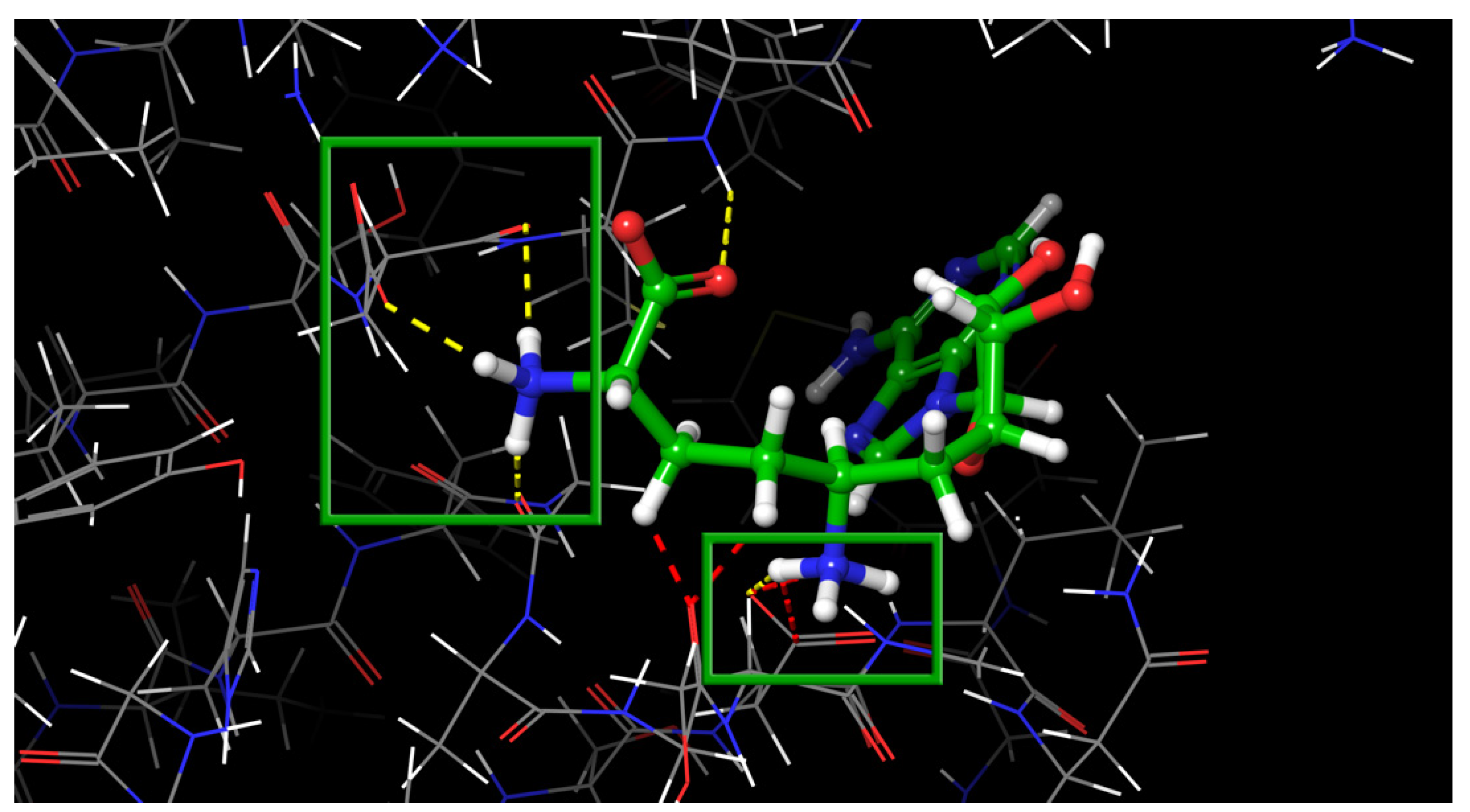
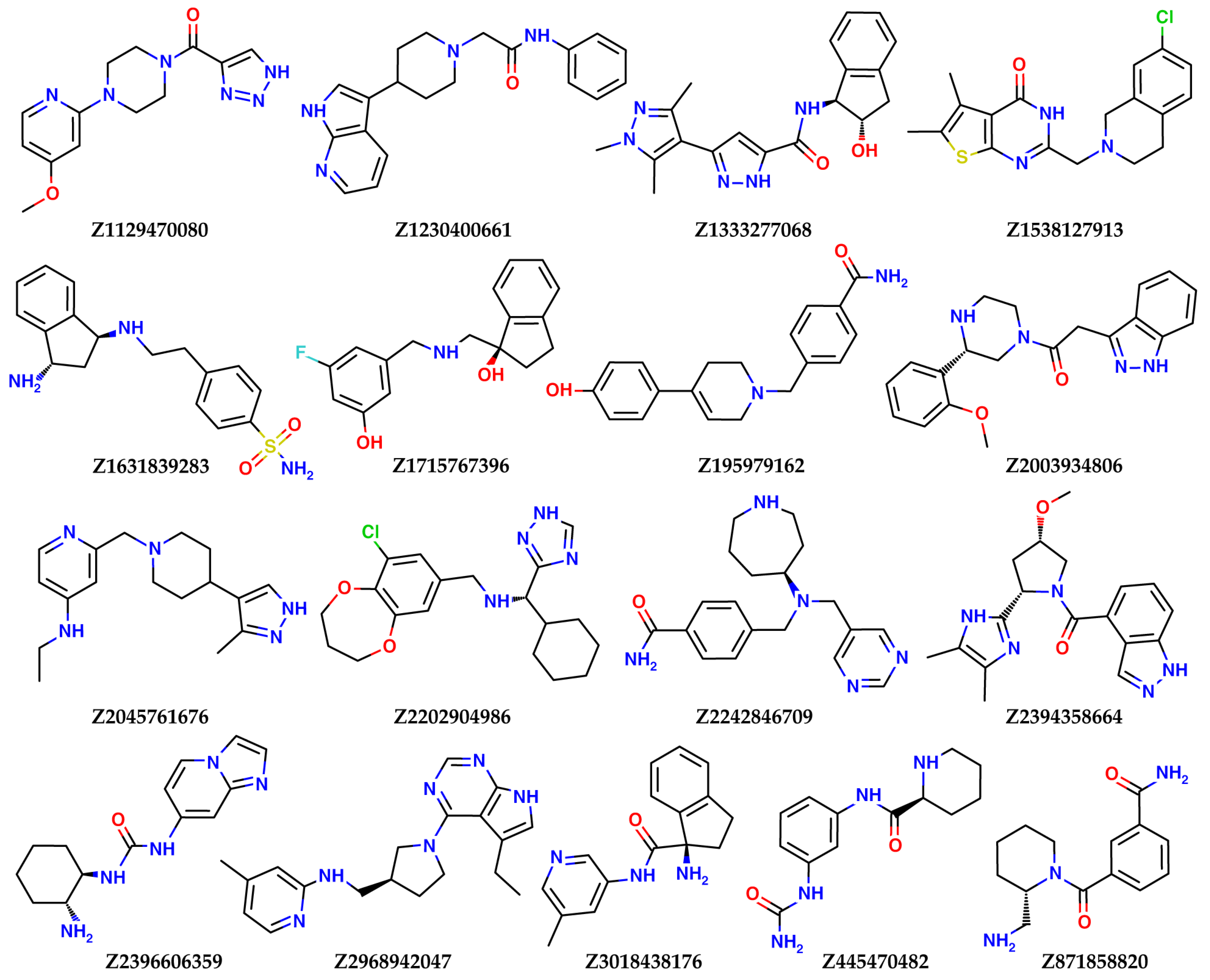
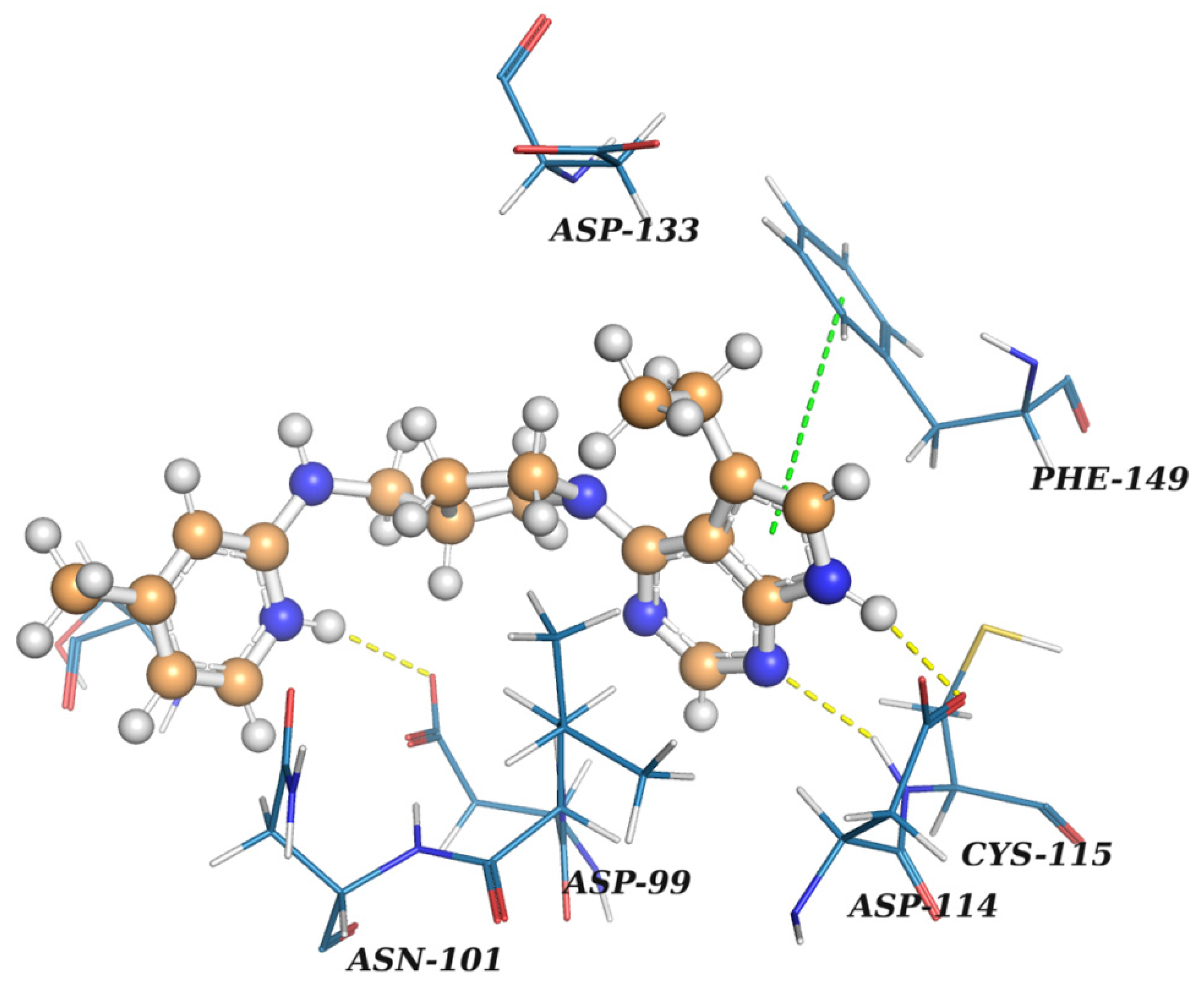
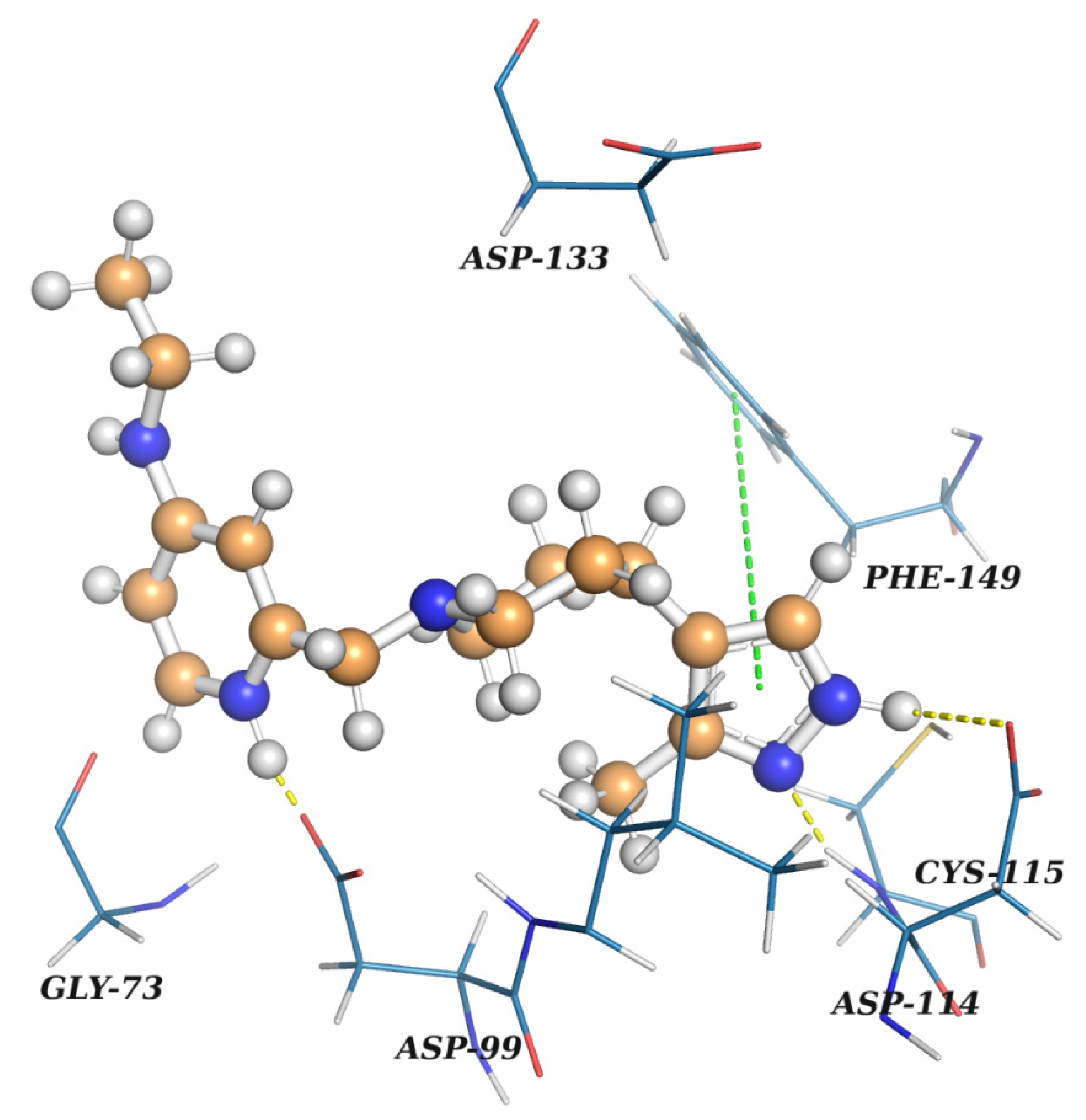
In this study, virtual screening of the databases of on-the-shelf low molecular weight compounds using docking was carried out, followed by quantum-chemical calculations of the protein–ligand binding enthalpy, with the goal of finding inhibitors of nsp16. The inhibition strategy was to target the SAM-binding site of the enzyme. As a result, twenty one compounds were selected for experimental testing of their inhibition of the nsp16 target protein.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}