
Synthesis of (Hyper)Branched Monohydroxyl Alkoxysilane Oligomers toward Silanized Urethane Prepolymers


Abstract
:1. Introduction
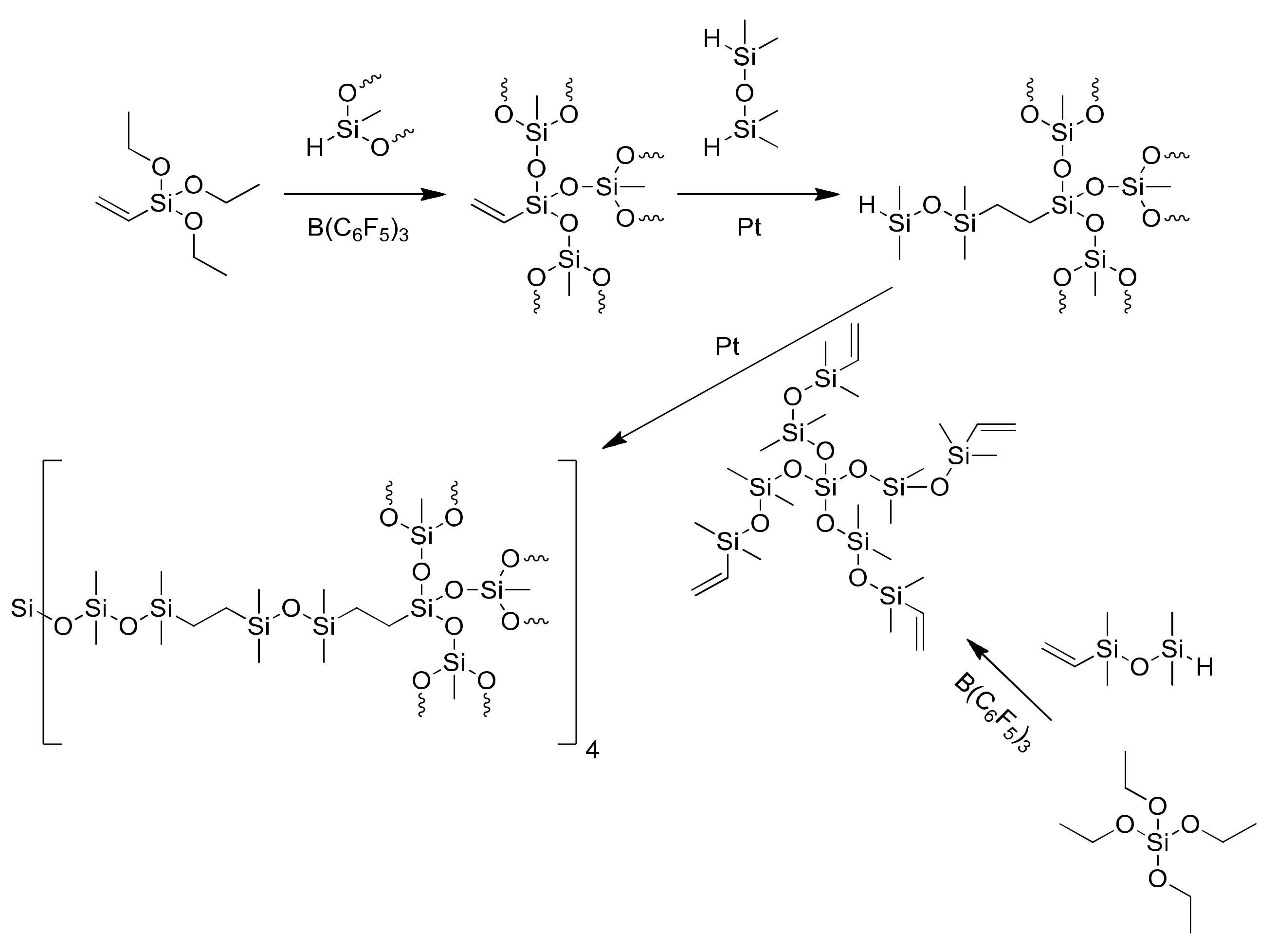
2. Results and Discussion
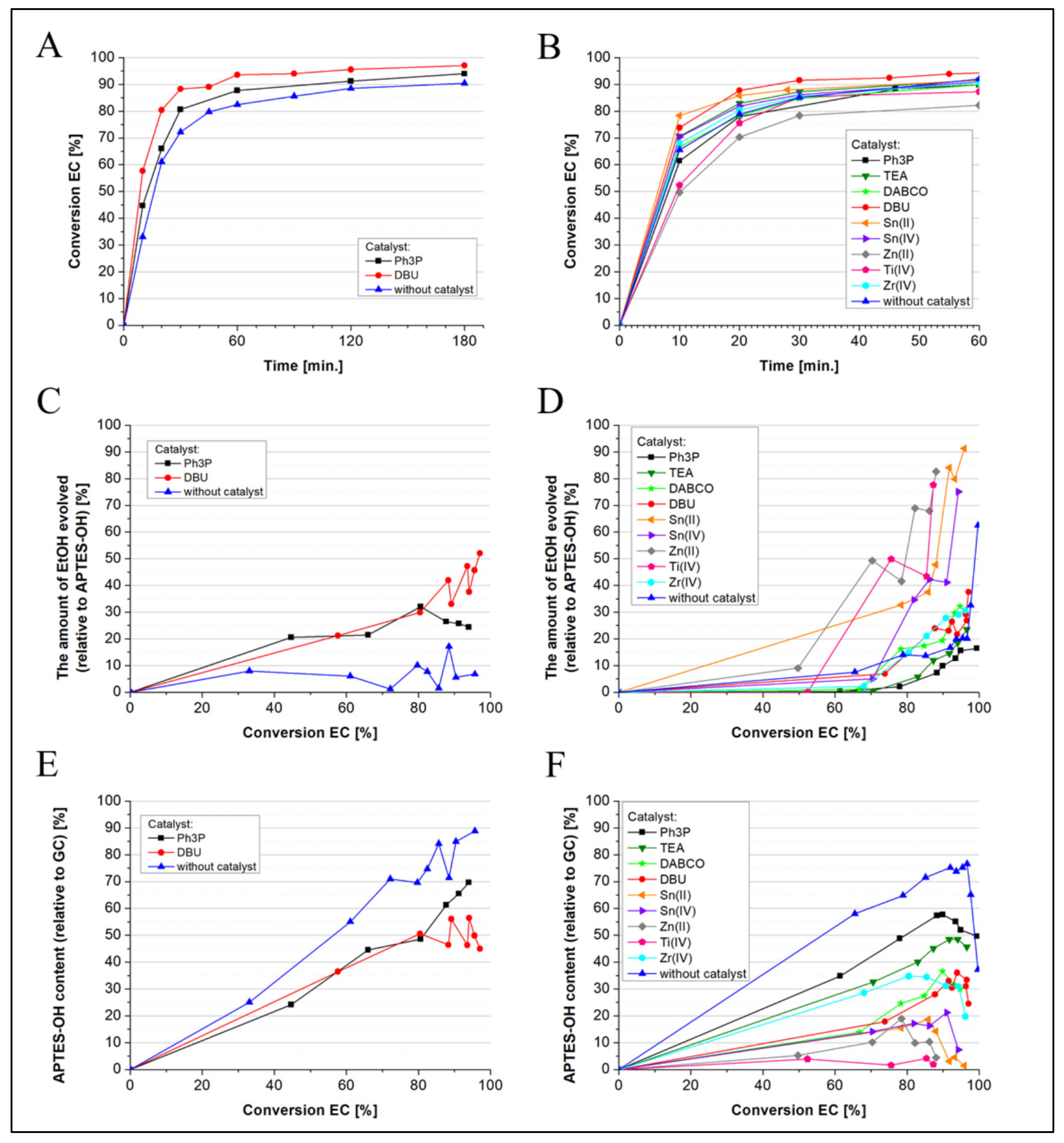
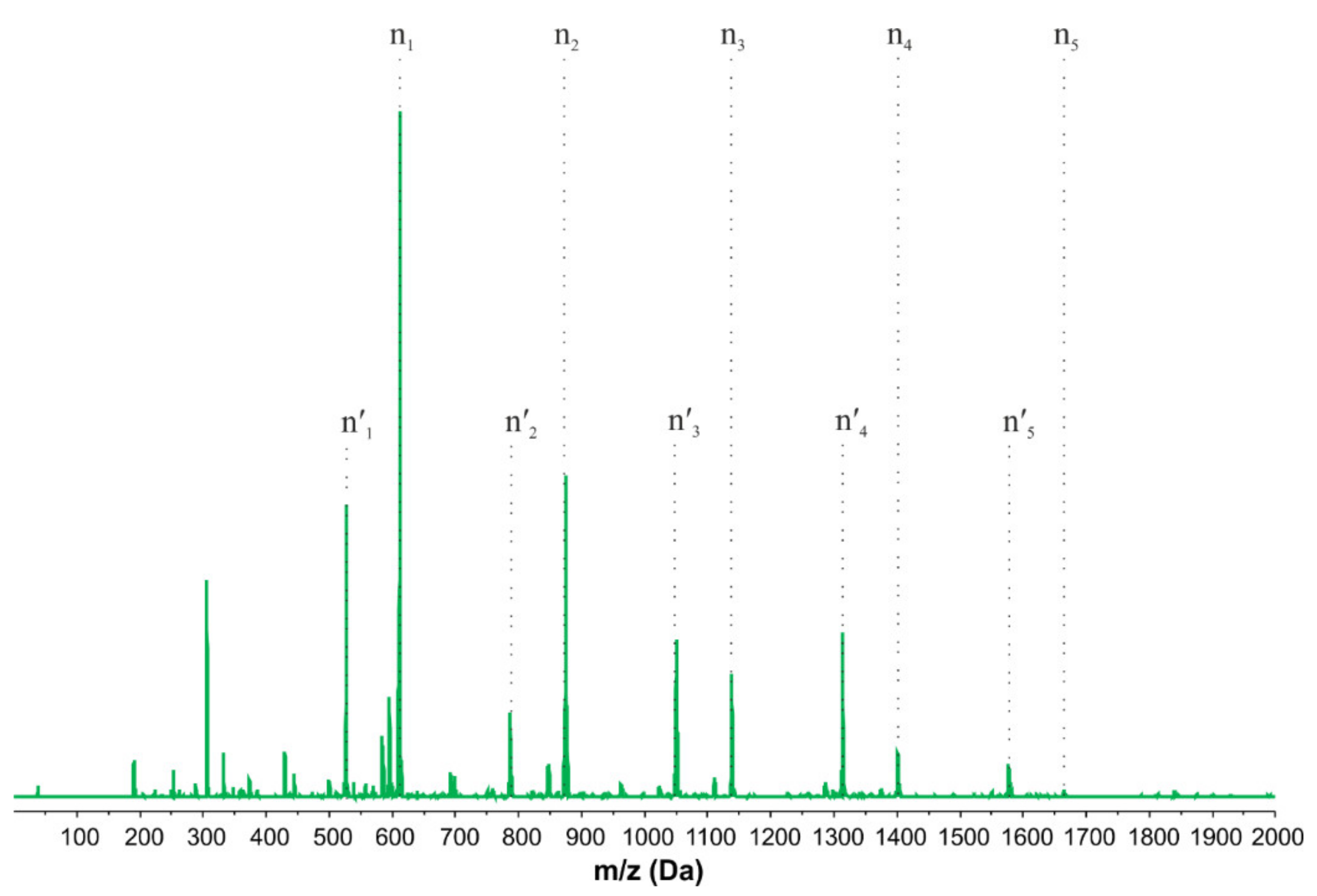
2.1. Influence of Temperature and Catalysts on the Degree of Polymerization of Hydroxyl-Terminated Alkoxysilane Oligomers
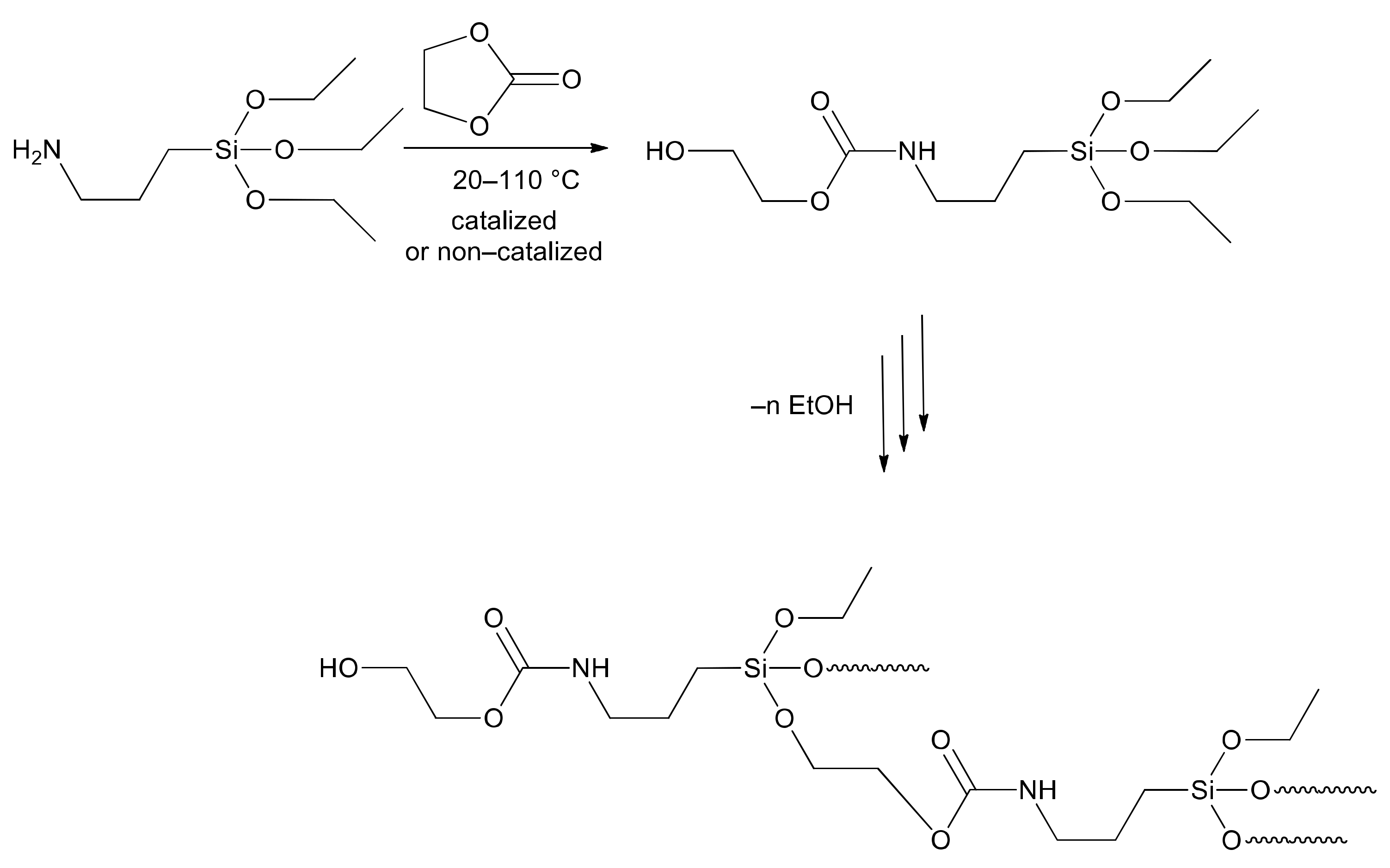
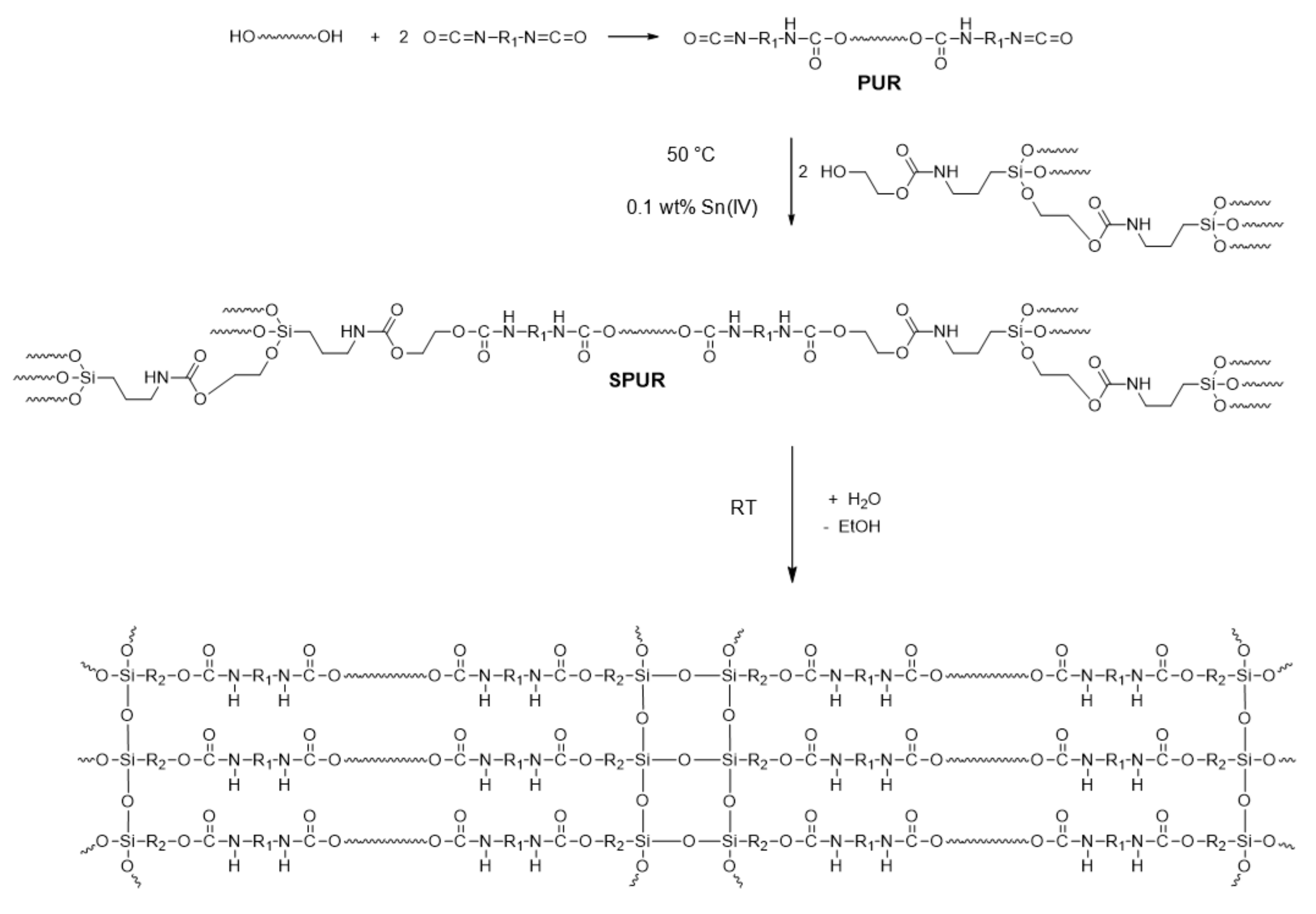
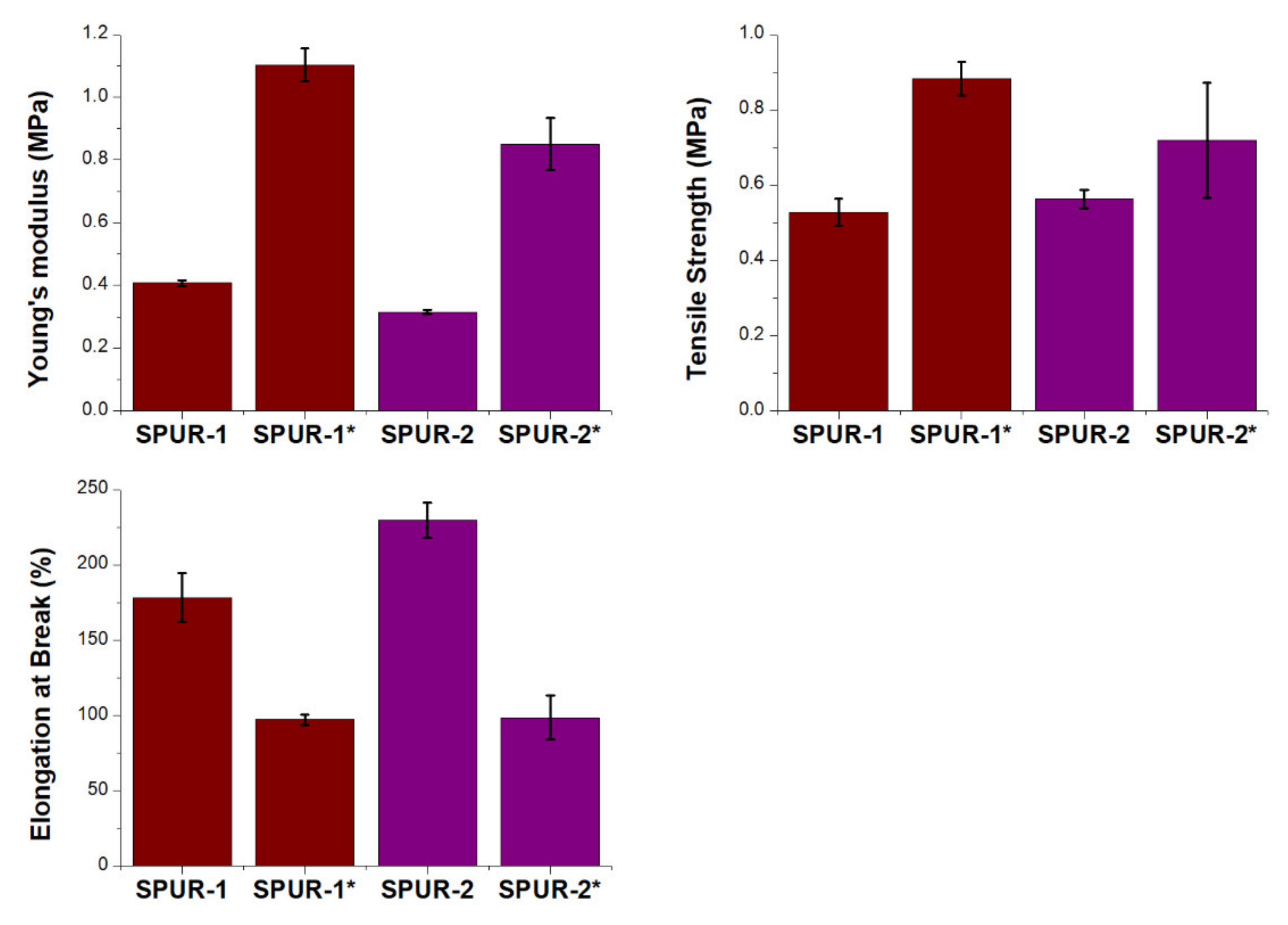
2.2. Synthesis of SPUR
3. Materials and Methods
3.1. Materials
3.2. Synthesis Methods
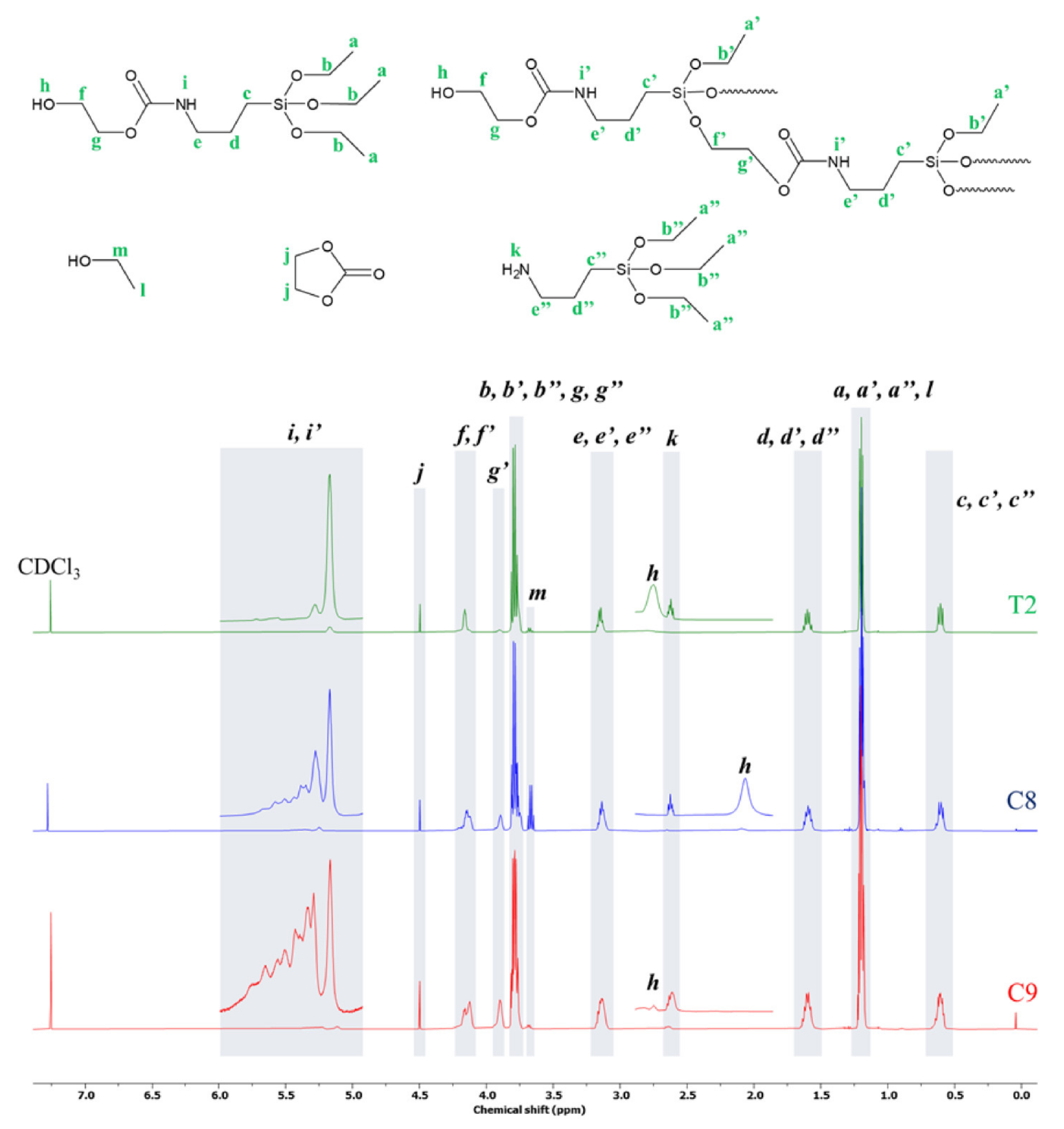
3.2.1. Synthesis of Hyperbranched Polymers Terminated with a Hydroxyl Group
3.2.2. Esterification of Oleic Acid with APTES-OH Blocking of Monomer Stability in MALDI-ToF Analysis
3.2.3. Synthesis of Silane-Modified Polyurethane (SPUR)
3.3. Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Olejnik, A.; Sztorch, B.; Brząkalski, D.; Przekop, R.E. Silsesquioxanes in the Cosmetics Industry—Applications and Perspectives. Materials 2022, 15, 1126. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J. Polymers for Personal Care and Cosmetics; The Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar]
- Zhang, Y. Preparation of High-temperature Silicone Rubber Mold and its Application in Food Industry. Adv. J. Food Sci. Technol. 2016, 12, 145–149. [Google Scholar] [CrossRef]
- Boudot, C.; Kühn, M.; Kühn-Kauffeldt, M.; Schein, J. Vacuum arc plasma deposition of thin titanium dioxide films on silicone elastomer as a functional coating for medical applications. Mater. Sci. Eng. C 2017, 74, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Arango-Ospina, M.; Xie, F.; Gonzalo-Juan, I.; Riedel, R.; Ionescu, E.; Boccaccini, A.R. Review: Silicon oxycarbide based materials for biomedical applications. Appl. Mater. Today 2020, 18, 100482. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, X.; Kundu, S.; Nag, A.; Afsarimanesh, N.; Sapra, S.; Mukhopadhyay, S.C.; Han, T. Silicon-Based Sensors for Biomedical Applications: A Review. Sensors 2019, 19, 2908. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Liu, Y.; Xu, Y. Silicon-based materials as high capacity anodes for next generation lithium ion batteries. J. Power Sources 2014, 267, 469–490. [Google Scholar] [CrossRef]
- Zuoa, X.; Zhua, J.; Müller-Busch, P. Silicon based lithium-ion battery anodes: A chronicle perspective review. Nano Energy 2017, 31, 113–143. [Google Scholar] [CrossRef]
- Namithaa, L.K.; Sebastian, M.T. High permittivity ceramics loaded silicone elastomer composites for flexible electronics applications. Ceram. Int. 2017, 43, 2994–3003. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, M.G.; Kramer, R. All-Printed Flexible and Stretchable Electronics. Adv. Mater. 2017, 29, 1604965. [Google Scholar] [CrossRef]
- Barroso, G.; Li, Q.; Bordiab, R.K.; Motz, G. Polymeric and ceramic silicon-based coatings—A review. J. Mater. Chem. A 2019, 7, 1936. [Google Scholar] [CrossRef]
- Giese-Hinz, J.; Kothe, C.; Louter, C.; Weller, B. Mechanical and chemical analysis of structural silicone adhesives with the influence of artificial aging. Int. J. Adhes. Adhes. 2021, 103019. [Google Scholar] [CrossRef]
- Antosik, A.K.; Mozelewska, K.; Piątek-Hnat, M.; Czech, Z.; Bartkowiak, M. Silicone pressure-sensitive adhesives with increased thermal resistance. J. Therm. Anal. Calorim. 2021, 1–9. [Google Scholar] [CrossRef]
- Rochow, E.G. Silicon and Silicones; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- O’Lenick, A.J. Basic chemistry and selected applications. J. Surfactants Deterg. 2000, 3, 229–236. [Google Scholar] [CrossRef]
- Kalchauer, W.; Pachaly, B. 12.6 Müller–Rochow Synthesis: The Direct Process to Methylchlorosilanes, Part 12. Inorganic Reactions. In Handbook of Heterogenous Catalysis; Wiley: Weinheim, Germany, 2008; pp. 2635–2646. [Google Scholar]
- Pachaly, B.; Weis, J. Chapter 79: The Direct Process to Methylchlorosilanes: Reflections on Chemistry and Process Technology. In Organosilicon Chemistry III: From Molecules to Materials; Wiley: Weinheim, Germany, 1998; pp. 478–484. [Google Scholar]
- Brook, M.A. Silicon in Organic, Organometallic and Polymer Chemistry; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Fawcett, A.S.; Brook, M.A. Thermoplastic Silicone Elastomers through Self-Association of Pendant Coumarin Groups. Macromolecules 2014, 47, 1656–1663. [Google Scholar] [CrossRef]
- Nowacka, M.; Herc, A.S.; Kowalewska, A. Thiol-ene addition of mercaptoalcohols to poly(vinylsiloxanes) under visible light photocatalysis—An approach towards cross-linkable hydrophilic silicones. Polyhedron 2020, 185, 114588. [Google Scholar] [CrossRef]
- Li, X.; Yu, R.; Zhao, T.; Zhang, Y.; Yang, X.; Zhao, X.; Huang, W. A self-healing polysiloxane elastomer based on siloxane equilibration synthesized through amino-ene Michael addition reaction. Eur. Polym. J. 2018, 108, 399–405. [Google Scholar] [CrossRef]
- Dvornic, P.R.; Owen, M.J. Silicon-Containing Dendritic Polymers; Springer: Dordrecht, Netherlands, 2009. [Google Scholar]
- Malmström, E.; Hult, A. Hyperbranched Polymers. J. Macromol. Science. Part C Polym. Rev. 1997, 37, 555–579. [Google Scholar] [CrossRef]
- Frey, H.; Schlenk, C. Silicon-Based Dendrimers. In Dendrimers II. Topics in Current Chemistry; Vögtle, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; Volume 210. [Google Scholar]
- Chojnowski, J.; Rubinsztajn, S.; Cella, J.A.; Fortuniak, W.; Cypryk, M.; Kurjata, J.; Kaźmierski, K. Mechanism of the B(C6F5)3-Catalyzed Reaction of Silyl Hydrides with Alkoxysilanes. Kinetic and Spectroscopic Studies. Organometallics 2005, 24, 6077–6084. [Google Scholar] [CrossRef]
- Piers, W.E. The chemistry of perfluoroaryl boranes. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2005; Volume 52, pp. 1–76. [Google Scholar]
- Rubinsztajn, S.; Cella, J.A. A New Polycondensation Process for the Preparation of Polysiloxane Copolymers. Macromolecules 2005, 38, 1061–1063. [Google Scholar] [CrossRef]
- Morgan, J.; Chen, T.; Hayes, R.; Dickie, T.; Urlich, T.; Brook, M.A. Facile synthesis of dendron-branched silicone polymers. Polym. Chem. 2017, 8, 2743–2746. [Google Scholar] [CrossRef]
- Grande, J.B.; Urlich, T.; Dickie, T.; Brook, M.A. Silicone dendrons and dendrimers from orthogonal SiH coupling reactions. Polym. Chem. 2014, 5, 6728–6739. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis using SiH Chemistry: The Piers–Rubinsztajn Reaction. Chem. Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Yi, M.; Chen, X.; Wu, S.; Ge, J.; Zhou, X.; Yin, G. Fabrication of Reactive Poly(Phenyl-Substituted Siloxanes/Silsesquioxanes) with Si-H and Alkoxy Functional Groups via the Piers–Rubinsztajn Reaction. Polymers 2018, 10, 1006. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztajn, S.; Cypryk, M.; Chojnowski, J.; Fortuniak, W.; Mizerska, U.; Pospiech, P. Reaction of Silyl Hydrides with Tetrabutoxygermanium in the Presence of B(C6F5)3: Difference between Silicon and Germanium Chemistries and Easy Route to GeH4. Organometallics 2018, 37, 1585–1590. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Mizerska, U.; Fortuniak, W.; Bak-Sypien, I.I. Kinetic and mechanistic studies of the transformation of the catalyst, tris(pentafluorophenyl)borane, in the presence of silyl and germyl hydrides. J. Catal. 2019, 379, 90–99. [Google Scholar] [CrossRef]
- Drohmann, C.; Möller, M.; Gorbatsevich, O.B.; Muzafarov, A.M. Hyperbranched polyalkenylsilanes by hydrosilylation with platinum catalysts. I. Polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 741–751. [Google Scholar] [CrossRef]
- Cai, G.; Weber, W.P. Synthesis of terminal Si–H irregular tetra-branched star polysiloxanes. Pt-catalyzed hydrosilylation with unsaturated epoxides. Polysiloxane films by photo-acid catalyzed crosslinking. Polymer 2004, 45, 2941–2948. [Google Scholar] [CrossRef]
- Zhan, X.; Liu, H.; Zhang, J. Two Branched Silicone Resins with Different Reactive Groups: A Comparative Evaluation. Ind. Eng. Chem. Res. 2018, 57, 5606–5615. [Google Scholar] [CrossRef]
- Wang, X.; Ervithayasuporn, V.; Zhang, Y.; Kawakami, Y. Reversible self-assembly of dendrimer based on polyhedral oligomeric silsesquioxanes (POSS). Chem. Commun. 2011, 47, 1282–1284. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Gao, P.; Li, D.; Yang, F.; Shen, H.; Guo, H.; Xu, F.; Wu, D. POSS dendrimers constructed from a 1–7 branching monomer. Chem. Commun. 2014, 50, 6126. [Google Scholar] [CrossRef]
- Michalczyk, M.; Piec, K.; Zierkiewicz, W.; Ejfler, J.; John, Ł. Possible coordination modes of copper(II) atom in model silsesquioxanes complexes at various pH conditions: DFT study. Chem. Phys. Lett. 2021, 778, 138739. [Google Scholar] [CrossRef]
- Li, D.; Niu, Y.; Yang, Y.; Wang, X.; Yang, F.; Shen, H.; Wu, D. Synthesis and self-assembly behavior of POSS-embedded hyperbranched polymers. Chem. Commun. 2015, 51, 8296–8299. [Google Scholar] [CrossRef] [PubMed]
- Piec, K.; Wątły, J.; Jerzykiewicz, M.; Kłak, J.; Plichta, A.; John, Ł. Mono-substituted cage-like silsesquioxanes bound by trifunctional acyl chloride as a multi-donor N,O-type ligand in copper(II) coordination chemistry: Synthesis and structural properties. New J. Chem. 2021, 45, 4156–4163. [Google Scholar] [CrossRef]
- Zhang, W.; Müller, A.H.E. Architecture, self-assembly and properties of well-defined hybrid polymers based on polyhedral oligomericsilsequioxane (POSS). Prog. Polym. Sci. 2013, 38, 1121–1162. [Google Scholar] [CrossRef]
- SciFinder. Available online: https://scifinder.cas.org (accessed on 16 December 2021).
- Sun, S.-K.; Wang, H.-F.; Yan, X.-P. Engineering Persistent Luminescence Nanoparticles for Biological Applications: From Biosensing/Bioimaging to Theranostics. Acc. Chem. Res. 2018, 51, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, H.; Wu, J.; He, Q.; Li, Y.; Yang, W.; Zhou, Y. Preparation and characterization of APTES modified magnetic MMT capable of using as anisotropic nanoparticles. Appl. Surf. Sci. 2018, 447, 393–400. [Google Scholar] [CrossRef]
- Bouazizi, N.; Vieillard, J.; Bargougui, R.; Couvrat, N.; Thoumire, O.; Morin, S.; Ladam, G.; Mofaddel, N.; Brun, N.; Azzouz, A.; et al. Entrapment and stabilization of iron nanoparticles within APTES modified graphene oxide sheets for catalytic activity improvement. J. Alloys Compd. 2019, 771, 1090–1102. [Google Scholar] [CrossRef]
- Gončuková, Z.; Řezanka, M.; Dolina, J.; Dvořák, L. Sulfonated polyethersulfone membrane doped with ZnO-APTES nanoparticles with antimicrobial properties. React. Funct. Polym. 2021, 162, 104872. [Google Scholar] [CrossRef]
- Tabatabaee, F.; Khorasani, M.; Ebrahimi, M.; González, A.; Irusta, L.; Sardon, H. Synthesis and comprehensive study on industrially relevant flame retardant waterborne polyurethanes based on phosphorus chemistry. Prog. Org. Coat. 2019, 131, 397–406. [Google Scholar] [CrossRef]
- Tachibana, Y.; Shi, X.; Graiver, D.; Narayan, R. Hydroxyl Terminated Hydrophilic Silanes. Silicon 2012, 4, 167–174. [Google Scholar] [CrossRef]
- Shi, X.; Graiver, D.; Narayan, R. Hydrolysis and Condensation of Hydrophilic Alkoxysilanes Under Acidic Conditions. Silicon 2012, 4, 109–119. [Google Scholar] [CrossRef]
- Rokicki, G.; Piotrowska, A. A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer 2002, 43, 2927–2935. [Google Scholar] [CrossRef]
- Rokicki, G.; Parzuchowski, P.G.; Mazurek, M. Non-isocyanate polyurethanes: Synthesis, properties, and applications. Polym. Adv. Technol. 2015, 26, 707–761. [Google Scholar] [CrossRef]
- Florjańczyk, Z.; Rokicki, G.; Parzuchowski, P.G.; Mazurek-Budzyńska, M.; Dębowski, M. Polymeric Materials Based on Carbon Dioxide: A Brief Review of Studies Carried Out at the Faculty of Chemistry, Warsaw University of Technology. Polymers 2022, 14, 718. [Google Scholar] [CrossRef]
- Katayama, S.; Yoshinaga, I.; Kubo, Y.; Yamada, N. Catalytic Effect of Chemically Modified Metal Alkoxides on Hydrolysis and Condensation Reactions of Methyl- and Phenyl-Ethoxysilanes. J. Ceram. Soc. Jpn. 2003, 111, 743–748. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Temperature [°C] | Catalyst | Time [h] |
|---|---|---|---|
| T1 | 20 | - | 24 |
| T2 | 25 | - | 23 |
| T3 | 30 | - | 23 |
| T4 | 50 | - | 22 |
| T5 | 70 | - | 25 |
| T6 | 90 | - | 23 |
| T7 | 110 | - | 25 |
| C1 | 30 | DBU | 4 |
| C2 | 30 | Ph3P | 3 |
| C3 | 50 | DBU | 3 |
| C4 | 50 | Ph3P | 3 |
| C5 | 50 | Sn(II) | 3 |
| C6 | 50 | Sn(IV) | 3.5 |
| C7 | 50 | DABCO | 3 |
| C8 | 50 | Ti(IV) | 3 |
| C9 a | 50 | Ti(IV) | 3 + 3.5 b |
| C10 | 50 | Zn(II) | 4 |
| C11 | 50 | Zr(IV) | 4.5 |
| C12 | 50 | TEA | 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, S.; Dębowski, M.; Iuliano, A.; Brzeski, S.; Plichta, A. Synthesis of (Hyper)Branched Monohydroxyl Alkoxysilane Oligomers toward Silanized Urethane Prepolymers. Molecules 2022, 27, 2790. https://doi.org/10.3390/molecules27092790
Kowalczyk S, Dębowski M, Iuliano A, Brzeski S, Plichta A. Synthesis of (Hyper)Branched Monohydroxyl Alkoxysilane Oligomers toward Silanized Urethane Prepolymers. Molecules. 2022; 27(9):2790. https://doi.org/10.3390/molecules27092790
Chicago/Turabian StyleKowalczyk, Sebastian, Maciej Dębowski, Anna Iuliano, Sebastian Brzeski, and Andrzej Plichta. 2022. "Synthesis of (Hyper)Branched Monohydroxyl Alkoxysilane Oligomers toward Silanized Urethane Prepolymers" Molecules 27, no. 9: 2790. https://doi.org/10.3390/molecules27092790