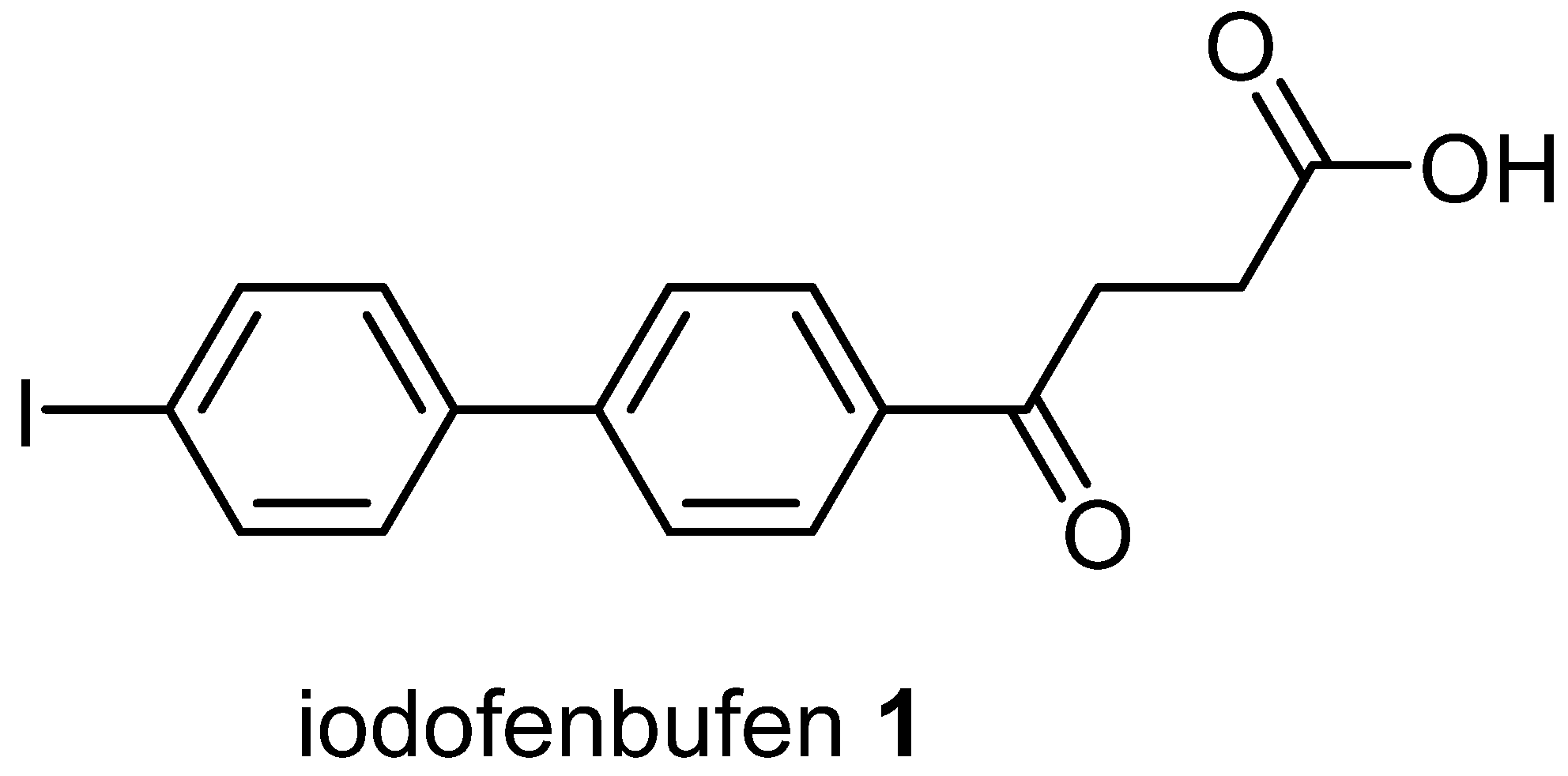
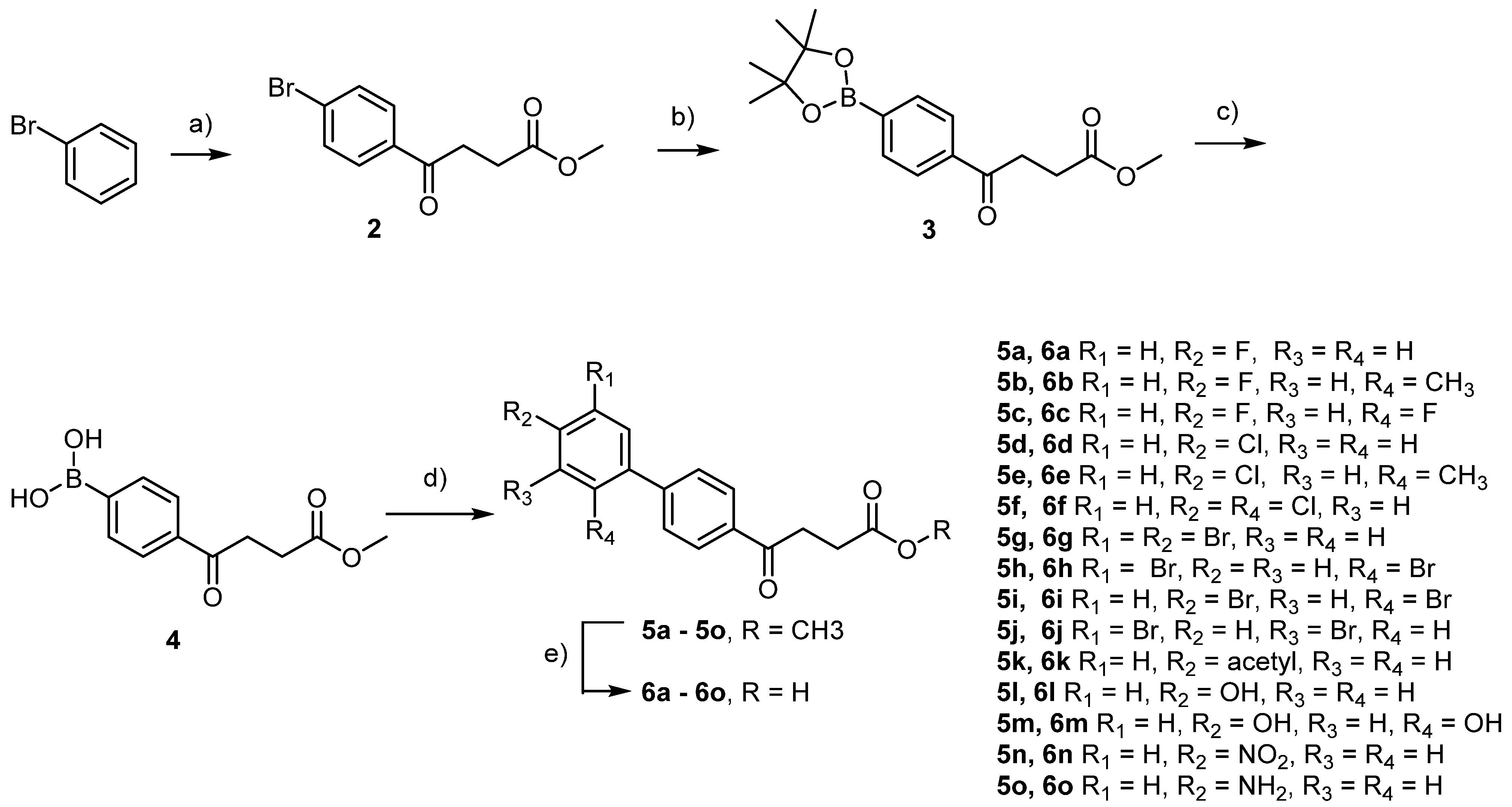
4.2. Chemical Preparation
para-Bromofenbufen methyl ester 2
![Molecules 27 02850 i001]()
To a three-neck round bottomed flask was charged a mixture of succinic anhydride (31 g, 0.31 mol, 1.2 eq). CH
2Cl
2 (50 mL) was added and the mixture was stirred until dissolution. The mixture was cooled down by an ice bath followed by adding AlCl
3 (104 g, 0.78 mol, 3 eq). The bath was removed and bromobenzene (27 mL, 0.26 mol) was added. CH
2Cl
2 (50 mL) was added and the sticky solid was agitated using a spatula. The mixture was poured into a mixture containing ice (120 g) and HCl (12 N, 65 mL) followed by Büchner filtration through suction. The white residue was further concentrated under reduced pressure. While attempting to recrystallize by using CH
3OH for dissolution, a significant amount of methylated product was obtained but not to completeness. The mixture (35.5 g) after concentration under reduced pressure was submitted to an acid protection procedure as follows. The mixture was dried by distilling with toluene (10 mL) and CH
2Cl
2 (10 mL). CH
3OH (100 mL) and H
2SO
4 (7.5 mL) was added. Stirring was allowed for 40 min followed by extraction using EtOAc (200 mL), Na
2CO
3 (satd., 50 mL × 2). The organic layers were collected and dried over Na
2SO
4 followed by gravitational filtration. The filtrate was concentrated under reduced pressure to give 49% yield of the white solid (35 g, 0.13 mol) over two steps (m.p. 48–50 °C (lit.[
46] m.p. 51.5 °C).
1H-NMR (500 MHz, CDCl3) δ 2.70 (t, J = 6.5 Hz, 2H, HAliphatic), 3.21 (t, J = 6.7 Hz, 2H, HAliphatic), 3.64 (s, 3H, HOCH3), 7.54 (d, J = 8.0 Hz, 2H, HAr), 7.78 (d, J = 8.5 Hz, 2H, HAr); 13C-NMR (125 MHz, CDCl3).
δ 27.92 (aliphatic, CH2), 33.33 (aliphatic, CH2), 51.88 (OCH3, CH3), 128.43 (Ar, C), 129.55 (Ar, CH), 131.95 (Ar, CH), 135.24 (Ar, C), 173.22 (CO-OCH3, C), 197.06 (Ar-CO, C).
methyl 4-oxo-4-(p-boronopinacoaryl)butanoate
3 [19] ![Molecules 27 02850 i002]()
A two-necked round-bottomed flask (50 mL) encompassing KOAC (7.3 g, 73.8 mmol, 4.0 eq) was dried in an oven at 120 °C for over 96 h. A second flask (50 mL) charging diboronopinacol (9.4 g, 36.9 mmol, 2.0 eq) was dried by distillation with toluene at 50 °C under reduced pressure. Bromo compound 2 (5.0 g, 18.5 mmol, 1 eq) was added and co-distilled with toluene. A spin-like flask (50 mL) containing a complex of dichloro-[1,1′-bis(diphenylphosphino)ferrocene] palladium (II) [PdCl2(dppf)] (500 mg, 0.68 mmol, 10.0% wt) was dried twice with toluene by co-distillation at 55 °C under reduced pressure. A flask containing dried DMSO (over 4 Å MS for 48 h) was bubbled with N2 for 15 min. To the two-necked round bottomed flask containing KOAc, a mixture of bromo compound and diboronpinacol in DMSO (30 mL) and a solution of PdCl2(dppf) in DMSO (10 mL) were added sequentially under sufficient stirring. The mixture was moved to an oil bath that had been preheated to 90 °C and the stirring was continued through bubbling. The mixture turned from light orange to dark brown after 10 min. TLC (EtOAc/n-hexane 2:8) indicated the consumption of the starting material 2 (Rf = 0.52) and formation of the product 3 (Rf = 0.52) with intense blue after staining. The reaction was terminated at 110 min post reaction by partitioning between CH2Cl2 (40 mL) and HCl (aq. 0.2 N, 20 mL). The organic layer was dried over Mg2SO4 followed by filtration using a celite pad and concentration under reduced pressure. The black residue (25 g) contained a significant amount of DMSO that can be further reduced by a second extraction or purified through flash chromatography using the gradient mode of EtOAc/n-hexane 1:9 → 2:8 to give a pleasant Hinoki-essential oil-odor pale yellow viscous gum in a 78% yield (4.6 g).
1H-NMR (500 MHz, CDCl3) δ 1.33 (s, 12H, 4 × CH3), 2.75 (t, J = 6.5 Hz, 2H, -CH2), 3.31 (t, J = 6.5 Hz, 2H, -CH2), 3.68 (s, 3H, -OCH3), 7.87 (d, J = 8.0 Hz, 2H, aromatic), 7.93 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 24.86 (CH3, Bpin) 28.01 (CH2), 33.60 (CH2), 51.81 (CH3, O-CH3), 84.21 (C, Bpin), 126.99 (CH, aromatic), 134.94 (CH, aromatic), 138.40 (C, aromatic), 173.33 (C, COO), 198.31 (C, CO).
4-(4-methoxy-4-oxobutanoyl)phenyl)boronic acid 4
To a flask (100 mL) containing compound 3 (4.60 g, 14.4 mmol, 1.0 eq) was added THF (30 mL), H2O (7.5 mL) and NaIO4 (9.20 g, 43.2 mmol, 3.0 eq), sequentially. After 10 min, 1N HCl (1.5 mL, 1.5 mmol, 0.1 eq) was added. The white precipitate was stirred for 2 h to show a complete consumption of starting material (Rf = 0.88) and formation of product (Rf = 0.40) from TLC (acetone/n-hexane = 5:5). The mixture was partitioned between EtOAc (30 mL) and Na2CO3 (10 mL), followed by washing with sat. NaCl(aq) (25 mL). The aqueous layers were further extracted with EtOAc (15 mL) × 3. The organic layers combined were dried over MgSO4 and filtered followed by concentration under reduced pressure to provide a white solid 4 (2.63 g, 77%). 148–153 °C.
1H-NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.71 (t, J = 6.5 Hz, 2H, -CH2), 3.33 (t, J = 6.5 Hz, 2H, -CH2), 3.66 (s, 3H, -OCH3), 7.82 (d, 2H, aromatic), 7.92 (bs, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.79 (CH2), 33.38 (CH2), 52.20 (CH3, O-CH3), 127.82 (CH, aromatic), 134.92 (CH, aromatic), 138.79 (C, aromatic), 140.47 (C, bs, Ar-B(OH)2), 175.28 (C, COO), 200.54 (C, CO); analysis for C11H13BO5, calculated [M + Na]+ (m/z) = 259.0748 (100.0%), 258.0785 (24.8%), 260.0782 (9.7%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 259.0749 (46.6%), 258.0780 (11.2%), 260.0781 (5.8%), δ [ppm] = 0.4.
4-Boronopinacol phenylmethanol
Reagents of commercial 4-bromophenyl methanol (2.0 g, 10.7 mmol, 1 eq), bis(pinacolato)diboron (5.4 g, 21.4 mmol, 2 eq), [PdCl
2(dppf)] (200 mg, 10.0% wt) and KOAc (4.2 g, 42.8 mmol, 4 eq) as well as solvent DMF (20 mL) were used. Following the same procedure as that described for
3, the reaction was performed under reflux for 4 h. The mixture was filtered followed by concentration under reduced pressure using an oil pump. The residue was extracted using EtOAc (30 mL) and saline (15 mL × 3). The organic layer was collected, dried over Na
2SO
4 and filtered through celite. After concentrating the filtrate under reduced pressure, the crude product (2.3 g) was chromatographed using gradient mode of EtOAc/
n-hexane 2:8 → 4:6 to give a pale yellow solid in 81% yield (2 g). analysis for C
13H
19BO
3 m.p. 63–65 °C (lit.[
47] m.p. 62–64 °C white powder, lit.[
48] oil).
1H-NMR (500 MHz, CDCl3) δ 1.35 (s, 12H, HMethyl), 4.72 (s, 2H, CH2), 7.37 (d, J = 7.5 Hz, 2H, HAr), 7.80 (d, J = 8.0 Hz, 2H, HAr), 13C-NMR (125 MHz, CDCl3) δ 24.85 (methyl, CH3), 65.29 (OH-CH2, CH2), 83.80 (BPin, C), 126.06 (Ar, CH), 135.05 (Ar, CH), 143.95 (Ar, C).
Methyl 4-(4′-chloro-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5d
![Molecules 27 02850 i005]()
A flask charging K2CO3 (352 mg, 0.70 mmol, 3.1 eq) was dried at 120 °C for 48 h. The Erlenmeyer flask containing toluene (15 mL) and EtOH (15 mL) was bubbled thoroughly by N2 for 15 min. To the cooled flask of K2CO3 was added the bubbled cosolvents (3 mL), the mixture of 4 (200 mg, 0.85 mmol, 1.0 eq) and 1-bromo-4-chlorobenzene (326 mg, 1.70 mmol, 2.0 eq) in cosolvents (5 mL) of EtOH and toluene and PdCl2 (55 mg, 0.31 mmol, 6 mol%), sequentially. The mixture turned from red-brown to gray-black and finally to black in 1 h. The 3-h reaction indicated the consumption of the starting material 4 (Rf = 0.20) and formation of the product (Rf = 0.74) from TLC (acetone/n-hexane = 3/7). The mixture was extracted with 1 N HCl (0.2 mL), sat. NaCl(aq) (1.5 mL) and H2O (5mL), sequentially. The organic layer was washed with CH2Cl2 (6 mL) and sat. NaCl(aq) (1.5 mL). The organic layer along with the two organic layers used to back-extract the aqueous layer were dried over MgSO4 and filtered. After concentration of the filtrate under reduced pressure, the white residue (693.1 mg) was chromatographed using EtOAc/n-hexane in a gradient mode of 1/9 → 2/8 → 3/7 to provide the white solid 5d in 77% yield (198 mg). m.p.: 117–120 °C.
1H NMR (500 MHz, CDCl3) δ 2.77 (t, J = 6.5 Hz, 2H, -CH2), 3.33 (t, J = 6.5 Hz, 2H, -CH2), 3.70 (s, 3H, -OCH3), 7.41 (dd, J = 7.0, 1.5 Hz, 2H, aromatic), 7.53 (dd, J = 7.0, 1.5 Hz, 2H, aromatic), 7.63 (d, J = 8.5 Hz, 2H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.04 (CH2), 33.43 (CH2), 51.82 (CH3, O-CH3), 127.09 (CH, aromatic), 128.48 (CH, aromatic), 128.71 (CH, aromatic), 129.13 (CH, aromatic), 134.48 (C, aromatic), 135.52 (C, aromatic), 138.27 (C, aromatic), 144.58 (C, aromatic), 173.32 (C, COO), 197.50 (C, CO); analysis for C17H15ClO3, calculated [M + Na]+ (m/z) = 325.0602 (100.0%), 327.0572 (32.0%), 326.0635 (18.4%), 328.0606 (5.9%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ (m/z) = 325.0609 (100.0%), 327.0581 (30.9%), 326.0639 (15.9%), 328.0608 (5.1%), δ [ppm] = 2.1.
methyl 4-(4′-fluoro-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5a
Reagents of 4 (52 mg, 0.21 mmol, 1.0 eq), bromo-4-fluorobenzene (73.5 mg, 0.42 mmol, 2.0 eq), K2CO3 (87 mg, 0.63 mmol, 3 eq) and PdCl2 (14.5 mg, 0.08 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (3 mL) were used. Followed the procedure for that of 5d for 3 h. TLC (acetone/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.18) and formation of the product (Rf = 0.52). The white solid of 5a was obtained in 82% yield (50 mg, 0.17 mmol). m.p.: 127–130 °C.
1H NMR (500 MHz, CDCl3) δ 2.77 (t, J = 6.5 Hz, 2H, -CH2), 3.32 (t, J = 6.5 Hz, 2H, -CH2), 3.70 (s, 3H,-OCH3), 7.13 (ddd, J = 11.0, J = 5.0, J = 2.5 Hz, 2H, aromatic), 7.56 (ddd, J = 11.0, J = 5.0, J = 2.5 Hz, 2H, aromatic), 7.61 (d, J = 8.5 Hz, 2H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ28.03 (CH2), 33.34 (CH2), 51.80 (CH3, O-CH3), 115.88 (d, 2JCF = 21.4 Hz, CH, aromatic), 127.07 (CH, aromatic), 128.66 (CH, aromatic), 128.89 (d, 3JCF = 8.1 Hz, CH, aromatic), 135.24 (C, aromatic), 135.95 (d, 4JCF = 2.8 Hz, C, aromatic), 144.83 (C, aromatic), 162.98 (d, 1JCF = 248.4 Hz, C, aromatic) 173.33 (C, COO), 197.52 (C, CO); analysis for C17H15FO3, calculated [M + Na]+ (m/z) = 309.0903 (100.0%), 310.0931 (18.4%), 311.0965 (1.6%), [2M + Na]+ (m/z) = 595.1903 (100.0%), 596.1936 (36.8%), 597.1970 (6.6%), 597.1945 (1.2%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 309.0908 (75.0%), 310.0944 (14.2%), 311.0969 (1.22%), δ [ppm] = 1.8 [2M + Na]+ (m/z) = 595.1893 (100.0%), 596.1929 (36.8%), 597.1970 (37.2%), 597.1954 (8.9%), δ [ppm] = −1.7.
methyl 4-(4′-fluoro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5b, ethyl 4-(4′-fluoro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5bbyp
![Molecules 27 02850 i007]()
The reagents included 4 (52 mg, 0.21 mmol, 1.0 eq), 2-bromo-5-fluorotoluene (80 mg, 0.42 mmol, 2.0 eq), K2CO3 (87 mg, 0.63 mmol, 3 eq), PdCl2 (13 mg, 0.082 mmol, 6 mol%) and solvent of toluene (4 mL) and the procedure followed that of 5d. The reaction post 1 h remained pale brown and TLC (acetone/n-hexane = 3:7) indicated no consumption of starting material (Rf = 0.20). Additional EtOH (0.5 mL) was added and it turned gray and clear. After a further reaction at rt for 1.5 h, it was heated to 70–75 °C for 17.5 h. TLC (acetone/n-hexane = 3:7) indicated the formation of the two products (Rf = 0.68, 0.70). After chromatography, a plastic smell and viscous liquid of the mixture of 5b and 5bbyp (40 mg, 62%). The mixture was purified using HPLC with isocratic condition of EtOAc/n-hexane = 1:9 to give the methyl fenbufen analog 5b and ethyl fenbufen analog 5bbyp in a ratio of 1:1. Another batch of experiment was optimized by using toluene/EtOH = 1:1 (3 mL) at 70 °C for 30 min. The viscous transparent liquid 5b was obtained in 75% yield (47 mg). 5b m.p.: 75–77 °C, 5bbyp m.p.: 51–53 °C.
methyl 4-(4′-fluoro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5b
1H NMR (500 MHz, CDCl3) δ 2.23 (s, 3H, Ar-CH3), 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H,-OCH3), 6.93 (t, J = 8.5, J = 2.5 Hz, 1H, aromatic), 6.97 (ddd, J = 9.5, J = 8.5, J = 2.5 Hz, 1H, aromatic), 7.15 (t, J = 8.5, J = 6.0 Hz, 1H, aromatic), 7.37 (d, J = 8.5 Hz, 2H, aromatic), 8.12 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 20.51 (Ar-CH3), 28.05 (CH2), 33.41 (CH2), 51.84 (CH3, O-CH3), 112.77 (d, 2JCF = 21.2 Hz, CH, aromatic), 116.99 (d, 2JCF = 21.2 Hz, CH, aromatic), 128.00 (CH, aromatic), 129.58 (CH, aromatic), 130.97 (d, 3JCF = 8.5 Hz, CH, aromatic), 135.14 (C, aromatic), 136.72 (d, 4JCF = 2.5 Hz, C, aromatic), 137.63 (d, 3JCF = 7.9 Hz, C, aromatic), 146.12 (C, aromatic), 162.31 (d, 1JCF = 245.1 Hz, C, aromatic) 173.37 (C, COO), 197.69 (C, CO); analysis for C18H17FO3, calculated [M + Na]+ (m/z) = 323.1054 (100.0%), 324.1087 (19.5%), 325.1121 (1.8%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 323.1058 (94.8%), 324.1086 (16.6%), 325.1115 (2.4%), δ [ppm] = 1.2.
ethyl 4-(4′-fluoro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5bbyp
1H NMR (500 MHz, CDCl3) δ 1.26 (t, J = 7.0 Hz, 3H, CH3), 2.23 (s, 3H, Ar-CH3), 2.77 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 4.16 (q, J = 7.0 Hz, 2H,-OCH2), 6.93 (dd, JHH = 8.5 Hz, JHF = 2.5 Hz, 1H, aromatic), 6.97 (ddd, J = 9.5, J = 8.5, J = 2.5 Hz, 1H, aromatic), 7.15 (dd, J = 8.5, J = 6.0 Hz, 1H, aromatic), 7.37 (d, J = 8.5 Hz, 2H, aromatic), 8.01 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 14.19 (-CH3, O-CH2CH3), 20.51 (Ar-CH3), 28.33 (CH2), 33.41 (CH2), 60.68 (CH2, O-CH2CH3), 112.76 (d, 2JCF = 20.9 Hz, CH, aromatic), 116.98 (d, 2JCF = 21.3 Hz, CH, aromatic), 128.00 (CH, aromatic), 129.57 (CH, aromatic), 130.97 (d, 3JCF = 8.5 Hz, CH, aromatic), 135.20 (C, aromatic), 136.73 (d, 4JCF = 2.3 Hz, C, aromatic), 137.63 (d, 3JCF = 7.9 Hz, C, aromatic), 146.07 (C, aromatic), 162.30 (d, 1JCF = 245.1 Hz, C, aromatic) 172.92 (C, COO), 197.80 (C, CO); analysis for C19H19FO3, calculated [M + Na]+ (m/z) = 337.1210 (100.0%), 338.1244 (20.5%), 339.1278 (2.0%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 337.1209 (100.0%), 338.1236 (16.8%), 339.1268 (2.3%), δ [ppm] = −0.3.
methyl 4-(2′,4′-difluoro-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5c
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 1-bromo-2, 4-difluorobenzene (328 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure for that of 5d for 3 h. TLC (EtOAc/n-hexane = 4/6) indicated the consumption of starting material (Rf = 0.20) and formation of the product (Rf = 0.72). The white solid of 5c was obtained in 69% yield (176 mg, 0.58 mmol). m.p.: 92–94 °C.
1H NMR (500 MHz, CDCl3) δ 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, OCH3), 6.92 (td, J = 9.0, J = 2.5 Hz, 1H, aromatic), 6.96 (dd, J = 8.5, J = 2.5 Hz, 1H, aromatic), 7.41 (td, J = 8.5, J = 8.5, J = 6.0 Hz, 1H, aromatic), 7.59 (d, J = 8.5, J = 2.0 Hz, 2H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.03 (CH2), 33.44 (CH2), 51.83 (CH3, O-CH3), 104.61 (t, 2JCF = 25.8 Hz, CH, C-3′), 111.85 (dd, 2JCF = 21.0 Hz, 4JCF = 3.8 Hz, CH, C-5′), 124.18 (dd, 2JCF = 13.4 Hz, 4JCF = 3.8 Hz, C, C-1′) 128.27 (CH, aromatic), 129.10 (d, JCF = 2.8 Hz, CH, aromatic), 131.39 (dd, 3JCF = 9.6 Hz, 3JCF = 4.6 Hz, CH, C-6′), 135.63 (C, aromatic), 139.80 (C, aromatic), 159.79 (dd, 1JCF = 250.1 Hz, 3JCF = 12.5 Hz, C-F, C-2′), 162.82 (dd, 1JCF = 249.0 Hz, 3JCF = 12.5 Hz, C-F, C-4′), 173.31 (C, COO), 197.55 (C, CO); analysis for C17H14F2O3, calculated [M + Na]+ (m/z) = 327.0803 (100.0%), 328.0837 (18.4%), 329.0870 (1.6%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 327.0802 (96.2%), 328.0835 (17.0%), 329.0868 (2.3%), δ [ppm] = 1.5.
methyl 4-(4′-chloro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5e
Reagents of 4 (52 mg, 0.21 mmol, 1.0 eq), 2-bromo-5-chlorotoluene (86 mg, 0.42 mmol, 2.0 eq), K2CO3 (96 mg, 0.70 mmol, 3.3 eq) and PdCl2 (13 mg, 0.082 mmol, 6 mol%) and cosolvents of toluene/CH3OH = 1:1 (3 mL) were used. Following the procedure for that of 5d for 3 h. TLC (acetone/n-hexane = 3:7) indicated the consumption of starting material (Rf = 0.20) and formation of the product (Rf = 0.62). The white solid of 5e was obtained in 71% yield (46 mg, 0.15 mmol). m.p.: 77–78 °C.
1H NMR (500 MHz, CDCl3) δ 2.22 (s, 3H, Ar-CH3), 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.30 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, -OCH3), 7.12 (d, J = 8.5 Hz, 1H, aromatic), 7.21 (d, J = 8.5, J = 2.0 Hz, 1H, aromatic), 7.26 (s, 1H, aromatic), 7.37 (d, J = 8.5 Hz, 2H, aromatic), 8.01 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 20.25 (Ar-CH3), 28.02 (CH2), 33.40 (CH2), 51.81 (CH3, O-CH3), 126.02 (CH, aromatic), 128.02 (CH, aromatic), 129.39 (CH, aromatic), 130.32 (CH, aromatic), 130.68 (CH, aromatic), 133.59 (C, aromatic), 135.27 (C-Cl, aromatic), 137.08 (C, aromatic), 139.16 (C, aromatic), 145.83 (C, aromatic) 173.32 (C, COO), 197.63 (C, CO); analysis for C18H17ClO3, calculated [M + H]+ (m/z) = 317.0946 (100.0%), [M + Na]+ (m/z) = 339.0764 (100.0%), 341.0729 (32.0%), 340.0792 (19.5%), ESI-Q-TOF HR-ESI-MS found: [M + H]+ (m/z) = 317.0942 (12.7%), δ [ppm] = −0.8, [M + Na]+ = 339.0766 (100.0%), 341.0743 (38.5%), 340.0786 (25.9%), δ [ppm] = 0.6.
methyl 4-(2′,4′-dichloro-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5f
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 1-bromo-2, 4-dichlorobenzene (384 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure for that of 5d for 27 h. TLC (EtOAc/n-hexane = 4/6) indicated the consumption of starting material (Rf = 0.20) and formation of the product (Rf = 0.76). The white solid of 5f was obtained in 69% yield (197 mg, 0.58 mmol).
1H NMR (500 MHz, CDCl3) δ 2.73 (t, J = 6.5 Hz, 2H, CH2), 3.29 (t, J = 6.5 Hz, 2H, CH2), 3.65 (s, 3H, -OCH3), 7.20 (dd, J = 8.5, 2.0 Hz, 1H, aromatic), 7.26 (dd, J = 8.5, 2.0 Hz, 1H, aromatic), 7.45 (dd, J = 7.0, 2.0 Hz, 3H, aromatic), 7.98 (dd, J = 7.0, 2.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.00 (CH2), 33.46 (CH2), 51.85 (O-CH3), 127.33 (CH, aromatic), 127.94 (CH, aromatic), 129.69 (CH, aromatic), 129.91 (CH, aromatic), 131.81 (CH, aromatic), 133.08 (C-Cl, aromatic), 134.48 (C-Cl, aromatic), 135.80 (C, aromatic), 137.90 (C, aromatic), 143.09 (C, aromatic), 174.04 (C, COO), 198.44 (C, CO); analysis for C17H14Cl2O3, calculated [M + Na]+ (m/z) = 359.0212 (100.0%), 361.0183 (63.9%), 360.0246 (18.4%), 362.0216 (11.8%), 363.0153 (10.2%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 359.0212 (88.1%), 361.0185 (60.0%), 360.0250 (16.6%), 362.0220 (10.8%), 363.0158 (10.3%), δ [ppm] = 0.
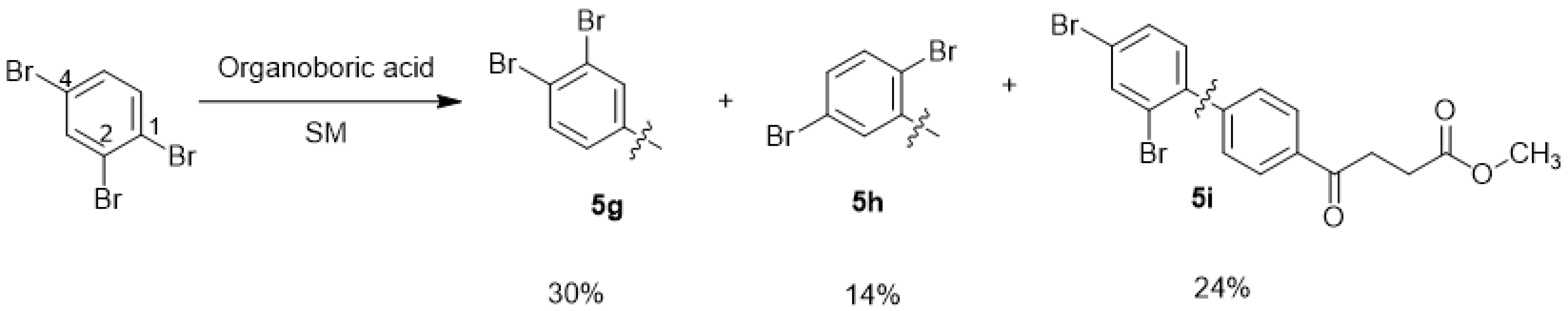
methyl 4-(3′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5g, methyl 4-(2′,5′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5h, methyl 4-(2′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5i
![Molecules 27 02850 i011]()
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 1, 2, 4-tribromobenzene (535 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure that for 5d for 2 h. TLC (EtOAc/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.20) and formation of the product (Rf = 0.54–0.64). The white solid was obtained as a mixture in 68% yield (246 mg). A portion of the sample (50 mg) was analyzed using normal phase HPLC per isocratic mode with eluents of EtOAc/n-hexane = 1/10 to give white solid 5g, viscous liquid 5h and white solid 5i in 30% (20 mg), 14% (9 mg) and 24% (16 mg) yield, respectively. Due to the limited amount of 5h, spectroscopic measurement was not taken. It was directly deprotected for subsequent spectroscopic analysis and biological assay.
methyl 4-(3′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5g
1H NMR (500 MHz, CDCl3) δ 2.77 (t, J = 6.5 Hz, 2H, CH2), 3.32 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, -OCH3), 7.38 (d, J5′,6′ = 8.5 Hz, J2′,5′ = 2.0 Hz, 1H, aromatic), 7.61 (d, J = 8.5 Hz, 2H, aromatic), 7.68 (d, J = 8.5 Hz, 2H, aromatic), 7.84 (s, J2′,5′ = 2.0 Hz, 1H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.00 (CH2), 33.47 (CH2), 51.83 (CH3, O-CH3), 124.78 (C-Br, aromatic), 125.47 (C-Br, aromatic), 127.10 (CH, aromatic), 127.21 (CH, aromatic), 128.78 (CH, aromatic), 132.23 (CH, aromatic), 134.07 (CH, aromatic), 135.97 (C, aromatic), 140.55 (C, aromatic), 143.18 (C, aromatic), 173.28 (C, COO), 197.42 (C, CO); analysis for C17H14Br2O3, calculated [M + Na]+ (m/z) = 448.9181 (100.0%), 446.9207 (51.4%), 450.9161 (48.6%), 449.9215 (9.7%), 451.9195 (8.9%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 448.9183 (100.0%), 446.9204 (78.7%), 450.9177 (81.2%), 449.9230 (32.8%), 451.9214 (1.3%), δ [ppm] = 0.4.
methyl 4-(2′,5′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5h
1H NMR (500 MHz, CDCl3) δ 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H,-OCH3), 7.35 (d, J5′,6′ = 8.5 Hz, J3′,5′ = 2.5 Hz, 1H, aromatic), 7.44 (s, J3′,5′ = 2.5 Hz, 1H, aromatic), 7.47 (d, J = 8.5 Hz, 2H, aromatic), 7.52 (d, J = 8.5 Hz, 2H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.01 (CH2), 33.50 (CH2), 51.85 (CH3, O-CH3), 120.94 (C-Br, aromatic), 121.32 (C-Br, aromatic), 127.93 (CH, aromatic), 129.60 (CH, aromatic), 132.30 (CH, aromatic), 133.66 (CH, aromatic), 134.61 (CH, aromatic), 135.99 (CH, aromatic), 143.25 (C, aromatic), 144.48 (C, aromatic), 173.30 (C, COO), 197.55 (C, CO); analysis for C17H14Br2O3, calculated [M + Na]+ (m/z) = 448.9181 (100.0%), 446.9207 (51.4%), 450.9161 (48.6%), 449.9215 (9.7%), 451.9195 (8.9%), 447.9235 (5.0%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 448.9187 (100.0%), 446.9207 (57.1%), 450.9177 (54.2%), 449.9205 (27.7%), 451.9360 (2.1%), 447.9314 (3.4%), δ [ppm] = 1.3.
methyl 4-(2′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5i
1H NMR (500 MHz, CDCl3) δ 2.78 (t, J = 6.5 Hz, 2H, -CH2), 3.38 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, OCH3), 7.17 (d, J = 8.0 Hz, 1H, aromatic), 7.46 (d, J = 8.0 Hz, 2H, aromatic) 7.50 (d, J3′,4′ = 8.0 Hz, J4′,6′ = 2.0 Hz, 1H, aromatic), 7.84 (s, J4′,6′ = 2.0 Hz, 1H, aromatic), 8.03 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.02 (CH2), 33.48 (CH2), 51.84 (CH3, O-CH3), 122.32 (C-Br, aromatic), 122.89 (C-Br, aromatic), 127.91 (CH, aromatic), 129.59 (CH, aromatic), 130.73 (CH, aromatic), 131.94 (CH, aromatic), 135.60 (CH, aromatic), 135.86 (CH, aromatic), 140.46 (C, aromatic), 144.78 (C, aromatic), 173.29 (C, COO), 197.56 (C, CO).
methyl 4-(3′,5′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5j
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 1-bromo-2, 4-difluorobenzene (538 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure for that of 5d for 3 h. TLC (EtOAc/n-hexane = 4/6) indicated the consumption of starting material (Rf = 0.40) and formation of the product (Rf = 0.72). The white solid of 5j was obtained in 36% yield (141 mg, 0.31 mmol). m.p.: 107–108 °C.
1H NMR (500 MHz, CDCl3) δ 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H,-OCH3), 7.60 (d, J = 8.5 Hz, 2H, aromatic), 7.66 (dd, J = 9.0, 1.0 Hz, 3H, aromatic), 8.04 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.00 (CH2), 33.50 (CH2), 51.86 (CH3, O-CH3), 123.47 (C-Br, aromatic), 127.33 (CH, aromatic), 128.78 (CH, aromatic), 129.09 (CH, aromatic), 133.52 (CH, aromatic), 136.24 (C, aromatic), 142.80 (C, aromatic), 143.39 (C, aromatic), 173.28 (C, COO), 197.41 (C, CO); analysis for C17H14Br2O3, calculated [M + Na]+ (m/z) = 448.9181 (100.0%), 446.9202 (51.4%), 450.9161 (48.6%), 449.9215 (9.7%), 451.9195 (8.9%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 448.9192 (25.8%), 446.9194 (13.7%), 450.9177 (12.8%), 449.9228 (4.8%), 451.9183 (2.8%), δ [ppm] = −2.4.
methyl 4-(4′-acetyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5k
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 4-bromoacetophenone (338.4 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure for that of 5d for 1 h. TLC (acetone/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.18) and formation of the product (Rf = 0.40). The mixture was chromatographed using eluents of CH3OH/CH2Cl2 = 1/99 to provide the white solid 5k in 81% yield (215 mg, 0.69 mmol). 151–158 °C.
1H NMR (500 MHz, CDCl3) δ 2.62 (s, 3H, Ar-COCH3), 2.78 (t, J = 6.5 Hz, 2H, Ar–COCH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, COOCH3), 7.69 (d, J = 8.0 Hz, 2H, aromatic), 7.70 (d, J = 8.5 Hz, 2H, aromatic), 8.03 (d, J = 8.0 Hz, 2H, aromatic), 8.06 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 26.63 (CH3, COCH3), 27.99 (CH2), 33.45 (CH2), 51.81 (CH3, O-CH3), 127.40 (CH, aromatic), 127.44 (CH, aromatic), 128.70 (CH, aromatic), 128.96 (CH, aromatic), 135.96 (C, aromatic), 136.57 (C, aromatic), 144.26 (C, aromatic), 144.23 (C, aromatic), 173.28 (C, COO), 197.51 (C, CO); analysis for C19H18O4, calculated [M + Na]+ (m/z) = 333.1103 (100.0%), 334.1131 (20.5%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 333.1104 (100.0%), 334.1142 (22.46%), δ [ppm] = 0.4.
methyl 4-(4′-hydroxy-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5l
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), 4-bromophenol (294 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (6 mL) were used. Following the procedure for that of 5d for 5 h. TLC (acetone/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.14) and formation of the product (Rf = 0.24). The mixture was chromatographed using eluents of CH3OH/CH2Cl2 in a gradient mode of 1/99 → 1/49 → 1/19 to provide the white solid 5l in 90% yield (214 mg, 0.75 mmol). m.p.: 179–182 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.32 (t, J = 6.5 Hz, 2H, CH2), 3.67 (s, 3H,-OCH3), 6.87 (d, J = 8.5 Hz, 2H, aromatic), 7.47 (d, J = 8.5 Hz, 2H, aromatic), 7.61 (d, J = 8.5 Hz, 2H, aromatic), 7.97 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.61 (CH2), 33.86 (CH2), 52.18 (CH3, O-CH3), 116.40 (CH, aromatic), 126.99 (CH, aromatic), 128.92 (CH, aromatic), 129.20 (CH, aromatic), 131.61 (C, aromatic), 134.90 (C, aromatic), 146.70 (C, aromatic), 158.26 (C-OH, aromatic), 174.62 (C, COO), 199.26 (C, CO); analysis for C17H16O4, calculated [M + Na]+ (m/z) = 307.0946 (100.0%), 308.0974 (18.4%), 309.1008 (1.6%), [2M + Na]+ (m/z) = 591.1989 (100.0%), 592.2023 (36.8%), 593.2056 (6.6%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 307.0947 (94.6%), 308.0987 (31.3%), 309.1035 (2.2%), δ [ppm] = 0.1, [2M + Na]+ (m/z) = 591.1971 (100.0%), 592.2017 (65.4%), 593.2035 (23.8%), δ [ppm] = −3.0.
methyl 4-(2′,4′-dihydroxy-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5m
Reagents of 4 (400 mg, 1.69 mmol, 1 eq), 4-bromoresorcinol (642 mg, 3.40 mmol, 2 eq), K2CO3 (705 mg, 5.10 mmol, 3 eq) and PdCl2 (110 mg, 0.62 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (4.5 mL) were used. Following the procedure for that of 5d for 19 h. TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of starting material (Rf = 0.66) and formation of the product (Rf = 0.50). The mixture was chromatographed using eluents of CH3OH/CH2Cl2 = 1/29 to provide the impure brown solid 5m in 63% crude yield (321 mg, 1.07 mmol). A portion (110 mg) was purified using HPLC with eluents of CH3OH/CH2Cl2 = 1/49 to afford a white solid of 5m in 31% yield (55 mg). m.p.: 82–86 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2),3.67 (s, 3H, -OCH3), 6.40 (d, J = 7.5 Hz, 2H, aromatic), 7.12 (d, J = 8.5, 1H, aromatic), 7.65 (d, J = 8.5 Hz, 2H, aromatic), 7.94 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.70 (CH2), 33.91 (CH2), 52.17 (O-CH3), 103.75 (CH, aromatic), 108.14 (CH, aromatic), 119.84 (C, aromatic), 128.45 (CH, aromatic), 129.85 (CH, aromatic), 131.76 (CH, aromatic), 134.52 (C, aromatic), 145.45 (C, aromatic), 156.11 (C-OH, aromatic), 158.97 (C-OH, aromatic), 174.76 (C, COO), 199.62 (C, CO); analysis for C17H16O5, calculated [M + H]+ (m/z) = 301.1071 (100.0%), 302.1104 (18.4%), [M + Na]+ (m/z) = 323.0890 (100.0%), 324.0923 (18.4%), 325.0957 (1.6%), ESI-Q-TOF HR-ESI-MS found: [M + H]+ = 301.1060 (11.3%), 302.1004 (4.0%), δ [ppm] = 3.6, [M + Na]+ = 323.0894 (100.0%), 324.0884 (18.7%), 325.1078 (3.6%), δ [ppm] = 1.2.
methyl 4-(4′-nitro-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5n
Reagents of 4 (200 mg, 0.85 mmol, 1 eq), bromo-4-nitrobenzene (343.4 mg, 1.70 mmol, 2 eq), K2CO3 (352 mg, 2.55 mmol, 3 eq) and PdCl2 (55 mg, 0.31 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (7.5 mL) were used. Following the procedure for that of 5d for 30 min. TLC (acetone/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.20) and formation of the product (Rf = 0.50). The white solid of 5n was obtained in 93% yield (248 mg, 0.79 mmol). m.p.: 146–150 °C.
1H NMR (500 MHz, CDCl3) δ 2.79 (t, J = 6.5 Hz, 2H, -CH2), 3.34 (t, J = 6.5 Hz, 2H, -CH2), 3.70 (s, 3H,-OCH3), 7.70 (d, J = 8.5 Hz, 2H, aromatic), 7.75 (d, J = 8.5 Hz, 2H, aromatic), 8.08 (d, J = 8.5 Hz, 2H, aromatic), 8.30 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 27.97 (CH2), 33.51 (CH2), 51.86 (CH3, O-CH3), 124.20 (CH, aromatic), 127.64 (CH, aromatic), 128.07 (CH, aromatic), 128.85 (CH, aromatic), 136.52 (C, aromatic), 143.22 (C, aromatic), 146.16 (C, aromatic), 147.65 (C-NO2, aromatic), 173.26 (C, COO), 197.43 (C, CO); analysis for C17H15NO5, calculated [M + Na]+ (m/z) = 336.0848 (100.0%), 337.0876 (18.4%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 336.0846 (100.0%), 337.0880 (20.7%), δ [ppm] = −0.5.
methyl 4-(4′-amino-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 5o
Reagents of 4 (400 mg, 1.69 mmol, 1 eq), 4-bromoaniline (581 mg, 3.38 mmol, 2 eq), K2CO3 (796 mg, 5.04 mmol, 3 eq) and PdCl2 (110 mg, 0.62 mmol, 6 mol%) and cosolvents of toluene/EtOH = 1:1 (6 mL) were used. Following the procedure for that of 5d for 6 h. TLC (acetone/n-hexane = 3/7) indicated the consumption of starting material (Rf = 0.22) and formation of the product (Rf = 0.26). The mixture was chromatographed using eluents of acetone/n-hexane/Et3N = 2/8/0.3. However, the crude mixture was not dissolved and precipitated in column. After a rough elution of most 4-bromoaniline and a small part of the product, the eluents were changed as EtOAc/Et3N = 10/0.3 to dissolve the precipitates. After concentration under reduced pressure, a yellow impure solid was obtained in a quantitative yield (454 mg). m.p.: 94–100 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 19:1) δ 2.71 (t, J = 6.5 Hz, 2H, CH2), 3.27 (t, J = 6.5 Hz, 2H, CH2), 3.64 (s, 3H,-OCH3), 6.86 (d, J = 8.5 Hz, 2H, aromatic), 7.44 (d, J = 8.5 Hz, 2H, aromatic), 7.56 (d, J = 8.5 Hz, 2H, aromatic), 7.94 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 19:1) δ 27.96 (CH2), 33.21 (CH2), 51.74 (CH3, O-CH3), 116.99 (CH, aromatic), 126.25 (CH, aromatic), 128.16 (CH, aromatic), 128.55 (CH, aromatic), 134.28 (C, aromatic), 145.52 (C-NH2, aromatic), 173.63 (C, COO), 197.95 (C, CO); analysis for C17H17NO3, calculated [M + H]+ (m/z) = 284.1287 (100.0%), 285.1315 (18.4%), [M + Na]+ (m/z) = 306.1106 (100.0%), 307.1134 (18.4%), [2M + Na]+ (m/z) = 589.2309 (100.0%), 590.2343 (36.8%), 591.2376 (3.9%), ESI-Q-TOF HR-ESI-MS found: [M + H]+ (m/z) = 284.1292 (51.6%), 285.1350, (14.9%) δ [ppm] = 2.0, [M + Na]+ = 306.1106 (66.8%), 307.1165 (17.2%), δ [ppm] = −0.2, [2M + Na]+ (m/z) = 589.2288 (100.0%), 590.2331 (40.9%), 591.2340 (11.4%), δ [ppm] = −3.6.
4-(4′-fluoro-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid
6a [29]To a two-neck round bottomed flask was added 5a (15 mg, 0.052 mmol), TFA (1.5 mL) and H2O (0.5 mL), sequentially. It was then stirred at 110 °C for 3 h. TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of 5a (Rf = 0.94) and formation of the product 6a (Rf = 0.34). After concentration under reduced pressure, the residue was chromatographed using eluents of CH3OH/CH2Cl2 in a gradient mode of 1/49 → 1/19 to give a white solid 6a in 63% yield (9 mg, 0.033 mmol). m.p.: 133–135 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 10:1) δ 2.70 (t, J = 6.5 Hz, 2H, CH2), 3.27 (t, J = 6.5 Hz, 2H, CH2), 7.09 (t, J2′,3′ = 8.5 Hz, 2H, aromatic, H-2′), 7.52 (t, J2′,3′ = 8.5, JHF = 3.5 Hz, 2H, aromatic, H-3′), 7.57 (d, J = 8.5 Hz, 2H, aromatic), 7.97 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/ CD3OD = 10:1) δ 27.97 (CH2), 33.42 (CH2), 115.80 (d, 2JCF = 21.5 Hz, CH, aromatic), 127.02 (CH, aromatic), 128.61 (CH, aromatic), 128.84 (d, 3JCF = 8.1 Hz, CH, aromatic), 135.16 (C, aromatic), 135.87 (d, 4JCF = 2.6 Hz, C, aromatic), 144.88 (C, aromatic), 162.95 (d, 1JCF = 246.5 Hz, C, aromatic) 175.21 (C, COO), 198.35 (C, CO); analysis for C16H13FO3, calculated [M − H]− (m/z) = 271.0771 (100.0%), 272.0810 (17.3%), 273.0843 (1.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 271.0770 (100.0%), 272.0805 (17.3%), 273.0833 (2.1%), δ [ppm] = −0.15.
4-(4′-fluoro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6b
Reagents of 5b (45 mg, 0.15 mmol), TFA (1.5 mL) and H2O (1.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.94) and formation of the product 6b (Rf = 0.42). After column chromatography, a white solid 6b was obtained in 73% yield (30 mg, 0.11 mmol). 135–138 °C.
1H NMR (500 MHz, CD3OD/CDCl3 = 1:1) δ 2.24 (s, 3H, Ar-CH3), 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.49 (t, J = 6.5 Hz, 2H, CH2), 6.97 (td, J = 8.5, JHF = 2.5 Hz, 1H, aromatic), 7.04 (dd, J = 9.5, J = 2.5 Hz, 1H, aromatic), 7.21 (dd, J = 8.0, JHF = 6.0 Hz, 1H, aromatic), 7.42 (d, J = 8.0 Hz, 2H, aromatic), 8.06 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 20.60 (Ar-CH3), 29.05 (CH2), 34.52 (CH2), 51.84 (CH3, O-CH3), 113.65 (d, 1JCF = 21.4 Hz, CH, aromatic), 117.85 (d, 2JCF = 21.4 Hz, CH, aromatic), 129.12 (CH, aromatic), 130.70 (CH, aromatic), 132.20 (d, 3JCF = 8.4 Hz, CH, aromatic), 136.75 (C, aromatic), 138.50 (d, 4JCF = 2.5 Hz, C, aromatic), 139.09 (d, 3JCF = 7.9 Hz, C, aromatic), 147.44 (C, aromatic), 163.76 (d, 1JCF = 243.6 Hz, C, aromatic) 176.72 (C, COOH), 200.19 (C, Ar-CO); analysis for C17H15FO3, calculated [M − H]− (m/z) = 285.0927 (100.0%), 286.0966 (18.4%), 287.1000 (1.6%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 285.0922 (100.0%), 286.0957 (18.1%), 287.0978 (2.1%), δ [ppm] = −1.7.
4-(2′,4′-difluoro-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6c
Reagents of 5c (96 mg, 0.32 mmol), TFA (1 mL) and H2O (1 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6c (Rf = 0.32). After column chromatography, a white solid 6c was obtained in 93% yield (86 mg, 0.30 mmol). 112–115 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.74 (t, J = 6.5 Hz, 2H, CH2), 3.35 (t, J = 6.5 Hz, 2H, CH2), 6.97 (m, 2H, aromatic), 7.46 (q, J = 8.5 Hz, 1H, aromatic), 7.59 (m, 2H, aromatic), 8.02 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 27.23 (CH2), 32.69 (CH2), 51.83 (CH3, O-CH3), 103.69 (t, 2JCF = 26.0 Hz, CH, C-3′), 111.14 (dd, 2JCF = 21.3 Hz, 4JCF = 3.5 Hz, CH, C-5′), 123.52 (dd, 2JCF = 13.1 Hz, 4JCF = 3.5 Hz, C, C-1′) 127.56 (CH, aromatic), 128.44 (CH, aromatic), 130.93 (dd, 3JCF = 9.5 Hz, 3JCF = 4.4 Hz, CH, C-6′), 134.97 (C, aromatic), 139.35 (C, aromatic), 159.45 (dd, 1JCF = 250.0, 3JCF = 11.9 Hz, C-F, C-2′), 162.32 (dd, 1JCF = 248.4, 3JCF = 11.9 Hz, C-F, C-4′), 173.33 (C, COO), 197.96 (C, CO); analysis for C16H11F2O3, calculated [M − H]− (m/z) = 289.0682 (100.0%), 290.0715 (17.3%), 291.0749 (1.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 289.0662 (100.0%), 290.0697 (17.1%), 291.0716 (2.0%), δ [ppm] = −6.9.
4-(4′-chloro-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid
6d [49]Reagents of 5d (100 mg, 0.33 mmol), TFA (1 mL) and H2O (1 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1:19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6c (Rf = 0.32). After column chromatography, a white solid 6d was obtained in 85% yield (80 mg, 0.28 mmol). m.p.: 160–163 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.32 (t, J = 6.5 Hz, 2H, CH2), 7.40 (dd, J = 8.5, 2.0 Hz, 2H, aromatic), 7.55 (dd, J = 8.5, 2.0 Hz, 2H, aromatic), 7.65 (d, J = 8.5 Hz, 2H, aromatic), 8.02 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.56 (CH2), 34.06 (CH2), 127.64 (CH, aromatic), 129.08 (CH, aromatic), 129.29 (CH, aromatic), 129.64 (CH, aromatic), 135.02 (C-Cl, aromatic), 136.17 (C, aromatic), 138.91 (C, aromatic), 145.29 (C, aromatic), 175.87 (C, COOH), 199.37 (C, Ar-CO); analysis for C16H13ClO3, calculated [M − H]− (m/z) = 287.0475 (100.0%), 289.0451 (32.0%), 288.0514 (17.3%), 290.0485 (5.5%) ESI-Q-TOF HR-ESI-MS found: [M − H]− = 287.0475 (51.7%), 289.0459 (22.32%), 288.0507 (16.3%), 290.0474 (1.9%) δ [ppm] = 0.07.
4-(4′-chloro-2′-methyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6e
Reagents of 5e (30 mg, 0.095 mmol), TFA (1.5 mL) and H2O (0.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 1.5 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6e (Rf = 0.46). After column chromatography, a white solid 6e was obtained in 77% yield (22 mg, 0.073 mmol).
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.21 (s, 3H, Ar-CH3), 2.73 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 7.12 (d, J = 8.5 Hz, 1H, aromatic), 7.20 (dd, J = 8.5, 2.0 Hz, 1H, aromatic), 7.25 (d, J = 2.0 Hz, 1H, aromatic), 7.38 (dd, J = 8.5, 2.0 Hz, 2H, aromatic), 8.02 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 20.47 (Ar-CH3), 28.62 (CH2), 34.05 (CH2), 126.63 (CH, aromatic), 128.72 (CH, aromatic), 130.13 (CH, aromatic), 130.89 (CH, aromatic), 131.35 (CH, aromatic), 134.27 (C, aromatic), 135.97 (C-Cl, aromatic), 137.86 (C, aromatic), 139.98 (C, aromatic), 146.81 (C, aromatic), 174.68 (C, COO), 199.40 (C, CO); analysis for C17H15ClO3, calculated [M − H]− (m/z) = 301.0632 (100.0%), 303.0607 (32.0%), 302.0671 (18.4%), 304.0641 (5.9%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 301.0631 (100.0%), 303.0609 (33.7%), 302.0669 (19.7%), 304.0640 (7.2%), δ [ppm] = −0.06.
4-(2′,4′-dichloro-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6f
Reagents of 5f (105 mg, 0.31 mmol), TFA (1 mL) and H2O (1 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.92) and formation of the product 6f (Rf = 0.36). After column chromatography, a white solid 6f was obtained in 84% yield (84 mg, 0.26 mmol). m.p.: 135–137 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.75 (t, J = 6.5 Hz, 2H, CH2), 3.36 (t, J = 6.5 Hz, 2H, CH2), 7.29 (d, J = 8.0, Hz, 1H, aromatic), 7.33 (dd, J = 8.0, 2.0 Hz, 1H, aromatic), 7.50 (dt, J = 7.0, 2.0 Hz, 3H, aromatic), 8.02 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 27.22 (CH2), 32.73 (CH2), 126.72 (CH, aromatic), 127.24 (CH, aromatic), 129.02 (CH, aromatic), 129.05 (CH, aromatic), 131.31 (CH, aromatic), 132.33 (C-Cl, aromatic), 133.82 (C-Cl, aromatic), 135.15 (C, aromatic), 137.37 (C, aromatic), 142.67 (C, aromatic), 173.31 (C, COO), 197.96 (C, CO); analysis for C16H12Cl2O3, calculated [M − H]− (m/z) = 321.0085 (100.0%), 323.0061 (63.9%), 322.0124 (17.3%), 324.0095 (11.1%), 325.0032 (10.2%), 326.0065 (1.8%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 321.0069 (100.0%), 323.0041 (60.2%), 322.0102 (17.3%), 324.0072 (10.7%), 325.0017 (10.6%), 326.0036 (2.3%), δ [ppm] = −1.3.
4-(3′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6g
Reagents of 5g (62 mg, 0.15 mmol), TFA (1.5 mL) and H2O (0.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 2 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6e (Rf = 0.40). After column chromatography, a white solid 6g was obtained in 80% yield (50 mg, 0.12 mmol). 137–140 °C.
1H NMR (500 MHz, CDCl3/CD3OD) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.31 (t, J = 6.5 Hz, 2H, CH2), 7.43 (d, J = 8.5 Hz, J = 2.0 Hz, 1H, aromatic), 7.64 (d, J = 8.5 Hz, 2H, aromatic), 7.68 (d, J = 8.5 Hz, 2H, aromatic), 7.86 (s, J = 2.0 Hz, 1H, aromatic), 8.03 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.61 (CH2), 34.15 (CH2), 125.22 (C-Br, aromatic), 125.89 (C-Br, aromatic), 127.71 (CH, aromatic), 127.94 (CH, aromatic), 129.40 (CH, aromatic), 132.76 (CH, aromatic), 134.74 (CH, aromatic), 136.68 (C, aromatic), 141.27 (C, aromatic), 143.88 (C, aromatic), 175.97 (C, COO), 199.34 (C, CO); analysis for C16H12Br2O3, calculated [M − H]− (m/z) = 410.9060 (100.0%), 408.9080 (51.4%), 412.9039 (48.6%), 411.9094 (9.7%), 413.9073 (8.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 410.9046 (100.0%), 408.9067 (50.8%), 412.9029 (50.3%), 411.9080 (17.4%), 413.9062 (10.1%), δ [ppm] = −3.4.
4-(2′,5′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6h
Reagents of 5h (25 mg, 0.06 mmol), TFA (1.0 mL) and H2O (1.0 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6h (Rf = 0.42). After column chromatography, a white solid 6h was obtained in 67% yield (17 mg, 0.04 mmol). m.p.: 77–80 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 2:1) δ 2.73 (t, J = 6.5 Hz, 2H, CH2), 3.33 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H,-OCH3), 7.19 (d, J = 8.0 Hz, 1H, aromatic), 7.47 (dd, J = 8.5, 2.0 Hz, 2H, aromatic) 7.51 (dd, J = 8.0, 2.0 Hz, 1H, aromatic), 7.82 (d, J = 2.0 Hz, 1H, aromatic), 8.02 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 2:1) δ 27.46 (CH2), 32.95 (CH2), 121.75 (C-Br, aromatic), 122.24 (C-Br, aromatic), 127.39 (CH, aromatic), 129.12 (CH, aromatic), 130.29 (CH, aromatic), 131.53 (CH, aromatic), 134.99 (CH, aromatic), 135.36 (C, aromatic), 139.98 (C, aromatic), 144.46 (C, aromatic), 174.79 (C, COO), 198.32 (C, CO); analysis for C16H12Br2O3, calculated [M − H]− (m/z) = 410.9060 (100.0%), 408.9080 (51.4%), 412.9039 (48.6%), 411.9094 (9.7%), 413.9073 (8.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 410.9043 (100.0%), 408.9065 (49.8%), 412.9030 (52.0%), 411.9087 (25.8%), 413.9068 (14.5%), δ [ppm] = −4.1.
4-(2′,4′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6i
Reagents of 5i (45 mg, 0.11 mmol), TFA (1.5 mL) and H2O (0.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6i (Rf = 0.32). After column chromatography, a white solid 6i was obtained in 69% yield (31 mg, 0.076 mmol). m.p.: 108–110 °C.
1H NMR (500 MHz, CDCl3) δ 2.74 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 7.37 (dd, J = 8.5 Hz, J = 2.0 Hz, 1H, aromatic), 7.44 (d, J = 2.0 Hz, 1H, aromatic), 7.48 (d, J = 8.5 Hz, 2H, aromatic), 7.54 (d, J = 8.0 Hz, 1H, aromatic), 8.03 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 28.62 (CH2), 34.21 (CH2), 121.52 (C-Br, aromatic), 121.95 (C-Br, aromatic), 128.58 (CH, aromatic), 130.30 (CH, aromatic), 133.06 (CH, aromatic), 134.25 (CH, aromatic), 135.37 (CH, aromatic), 136.82 (CH, aromatic), 144.07 (C, aromatic), 145.37 (C, aromatic), 175.98 (C, COO), 199.51 (C, CO); analysis for C16H12Br2O3, calculated [M − H]− (m/z) = 410.9060 (100.0%), 408.9080 (51.4%), 412.9039 (48.6%), 411.9094 (9.7%), 413.9073 (8.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 410.9062 (100.0%), 408.9071 (43.5%), 412.9036 (55.3%), 411.9074 (13.4%), 413.9054 (9.4%), δ [ppm] = 0.5.
4-(3′,5′-dibromo-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6j
Reagents of 5j (85 mg, 0.20 mmol), TFA (1.0 mL) and H2O (1.0 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 3 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6j (Rf = 0.34). After column chromatography, a white solid 6j was obtained in 60% yield (48 mg, 0.12 mmol). m.p.: 181–183 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.74 (t, J = 6.5 Hz, 2H, CH2), 3.30 (t, J = 6.5 Hz, 2H, CH2), 7.60 (dd, J = 8.5, 2.0 Hz, 2H, aromatic), 7.65 (s, 3H, aromatic), 8.02 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.25 (CH2), 33.73 (CH2), 123.68 (C-Br, aromatic), 127.62 (CH, aromatic), 129.04 (CH, aromatic), 129.32 (CH, aromatic), 133.76 (CH, aromatic), 136.38 (C, aromatic), 143.23 (C, aromatic), 143.63 (C, aromatic), 174.04 (C, COO), 198.44 (C, CO); analysis for C16H12Br2O3, calculated [M − H]− (m/z) = 410.9060 (100.0%), 408.9080 (51.4%), 412.9039 (48.6%), 411.9094 (9.7%), 413.9073 (8.4%), 409.9114 (4.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 410.9021 (100.0%), 408.9042 (50.6%), 412.9003 (46.7%), 411.9054 (16.1%), 413.9029 (12.4%), 409.9078 (8.6%), δ [ppm] = −9.5.
4-(4′-acetyl-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6k
Reagents of 5k (100 mg, 0.32 mmol), TFA (1.5 mL) and H2O (1.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 3 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6j (Rf = 0.34). After column chromatography, a white solid 6k was obtained in 88% yield (83 mg, 0.28 mmol). m.p.: 203–205 °C.
1H NMR (500 MHz, DMSO-d6) δ 2.60 (t, J = 6.5 Hz, 5H, Ar–COCH2), 3.28 (t, J = 6.5 Hz, 2H, CH2), 7.89 (d, J = 8.0 Hz, 4H, aromatic), 8.05 (d, J = 8.5 Hz, 2H, aromatic), 8.08 (d, J = 8.5 Hz, 2H, aromatic), 12.14 (s, 1H, -COOH); 13C NMR (125 MHz, DMSO-d6) δ 26.74 (CH3, COCH3), 27.85 (CH2), 33.17 (CH2), 127.19 (CH, aromatic), 127.23 (CH, aromatic), 128.57 (CH, aromatic), 128.289 (CH, aromatic), 135.91 (C, aromatic), 136.28 (C, aromatic), 143.13 (C, aromatic), 173.72 (C, COOH), 197.45 (C, CO), 198.03 (C, CO); analysis for C18H16O4, calculated [M − H]− (m/z) = 295.0970 (100.0%), 296.1009 (19.5%), 297.1043 (1.8%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 295.0968 (100.0%), 296.1003 (18.3%), 297.1032 (2.6%), δ [ppm] = −0.8.
4-(4′-hydroxy-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid
6l [25]Reagents of 5l (74 mg, 0.26 mmol), TFA (3.0 mL) and H2O (2.0 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.68) and formation of the product 6l (Rf = 0.26). After column chromatography, a white solid 6l was obtained in 81% yield (56 mg, 0.21 mmol). 217–220 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.30 (t, J = 6.5 Hz, 2H, CH2), 6.87 (d, J = 8.5 Hz, 2H, aromatic), 7.46 (d, J = 8.5 Hz, 2H, aromatic), 7.61 (d, J = 8.5 Hz, 2H, aromatic), 7.96 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.73 (CH2), 33.94 (CH2), 116.49 (CH, aromatic), 127.03 (CH, aromatic), 128.91 (CH, aromatic), 129.19 (CH, aromatic), 131.76 (C, aromatic), 135.14 (C, aromatic), 146.77 (C, aromatic), 158.34 (C-OH, aromatic), 174.59 (C, COO), 199.32 (C, CO); analysis for C14H14O4, calculated [M − H]− (m/z) = 269.0814 (100.0%), 270.0853 (17.3%), 271.0886 (1.4%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 269.0810 (100.0%), 270.0843 (17.5%), 271.0868 (2.0%), δ [ppm] = −1.4.
4-(2′,4′-dihydroxy-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6m
Reagents of 5m (36 mg, 0.12 mmol), a solution of aqueous THF (1.2 mL, 75% vol) were used. The gold solution was cooled at 0 °C and LiOH (10 mg, 0.42 mmol, 3.5 eq) was added. After 10 min, the ice bath was removed and the reaction was allowed for 30 min and it turned dark green. A further 30 min reaction indicated the consumption of the starting material (Rf = 0.70) and formation of the product 6m (Rf = 0.38) from TLC (CH3OH/CH2Cl2 = 1/9). It was treated with cationic exchange resin (H+) and the solution turned orange. Followed by filtration, concentration of the filtrate and column chromatography (CH3OH/CH2Cl2 = 1/9) of the residue, a white solid 6m was obtained in 50% yield (18 mg, 0.06 mmol). m.p.: 210–212 °C.
1H NMR (500 MHz, CD3OD) δ 2.70 (t, J = 6.5 Hz, 2H, CH2), 3.32 (t, J = 6.5 Hz, 2H, CH2), 6.39 (d, J = 8.0 Hz, 2H, aromatic), 7.14 (d, J = 8.0 Hz, 1H, aromatic), 7.66 (d, J = 8.5 Hz, 2H, aromatic), 7.97 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CD3OD) δ 29.06 (CH2), 34.36 (CH2), 104.11 (CH, aromatic), 108.51 (CH, aromatic), 120.29 (C, aromatic), 128.78 (CH, aromatic), 130.20 (CH, aromatic), 132.25 (CH, aromatic), 135.32 (C, aromatic), 145.94 (C, aromatic), 156.83 (C-OH, aromatic), 1589.82 (C-OH, aromatic), 176.77 (C, COOH), 200.36 (C, CO); analysis for C16H14O5, calculated [M − H]− (m/z) = 285.0768 (100.0%), 286.0802 (17.3%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 285.0764 (100%), 286.0783 (19.5%), δ [ppm] = −1.4.
4-(4′-nitro-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6n
Reagents of 5n (100 mg, 0.32 mmol), TFA (2.5 mL) and H2O (1.0 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 4 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.98) and formation of the product 6n (Rf = 0.40). After column chromatography, a white solid 6n was obtained in 72% yield (70 mg, 0.23 mmol). m.p.: 157–160 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.73 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 7.76 (d, J = 8.5 Hz, 2H, aromatic), 7.82 (d, J = 9.0 Hz, 2H, aromatic), 8.08 (d, J = 8.5 Hz, 2H, aromatic), 8.29 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.63 (CH2), 34.26 (CH2), 124.80 (CH, aromatic), 128.36 (CH, aromatic), 128.86 (CH, aromatic), 129.53 (CH, aromatic), 137.36 (C, aromatic), 144.04 (C, aromatic), 146.96 (C, aromatic), 148.45 (C-NO2, aromatic), 176.01 (C, COO), 199.44 (C, CO); analysis for C16H13NO5, calculated [M − H]− (m/z) = 298.0716 (100.0%), 299.0755 (17.3%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 298.0719 (100.0%), 299.0690 (23.9%), δ [ppm] = 1.2.
4-(4′-amino-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 6o
Reagents of 5o (77 mg, 0.27 mmol), TFA (1.5 mL) and H2O (1.5 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 1 h and TLC (CH3OH/CH2Cl2 = 1/19) indicated the consumption of the starting material (Rf = 0.80) and formation of the product 6o (Rf = 0.10). After concentration under reduced pressure, the cooled CH2Cl2 was added. The mixture was filtered through suction. Additional cooled CH2Cl2 was added to wash the solid. The solid was then concentrated under reduced pressure to afford a cream-colored solid 6o in 74% yield (53 mg, 0.20 mmol). m.p.: 150–153 °C.
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.72 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 7.14 (dd, J = 8.5, 2.5 Hz, 2H, aromatic), 7.61 (d, J = 8.5 Hz, 2H, aromatic), 7.66 (dd, J = 8.5, 1.5 Hz, 2H, aromatic), 8.02 (d, J = 8.0 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.65 (CH2), 34.08 (CH2), 120.21 (CH, aromatic), 127.29 (CH, aromatic), 128.12 (CH, aromatic), 129.12 (CH, aromatic), 129.38 (CH, aromatic), 135.78 (C, aromatic), 145.23 (C, aromatic), 145.85 (C, aromatic), 176.06 (C, COO), 199.55 (C, CO); analysis for C16H15NO3, calculated [M − H]− (m/z) = 268.0979 (100.0%), 269.1013 (17.3%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 268.0960 (74.1%), 269.1041 (15.9%), δ [ppm] = −7.1.
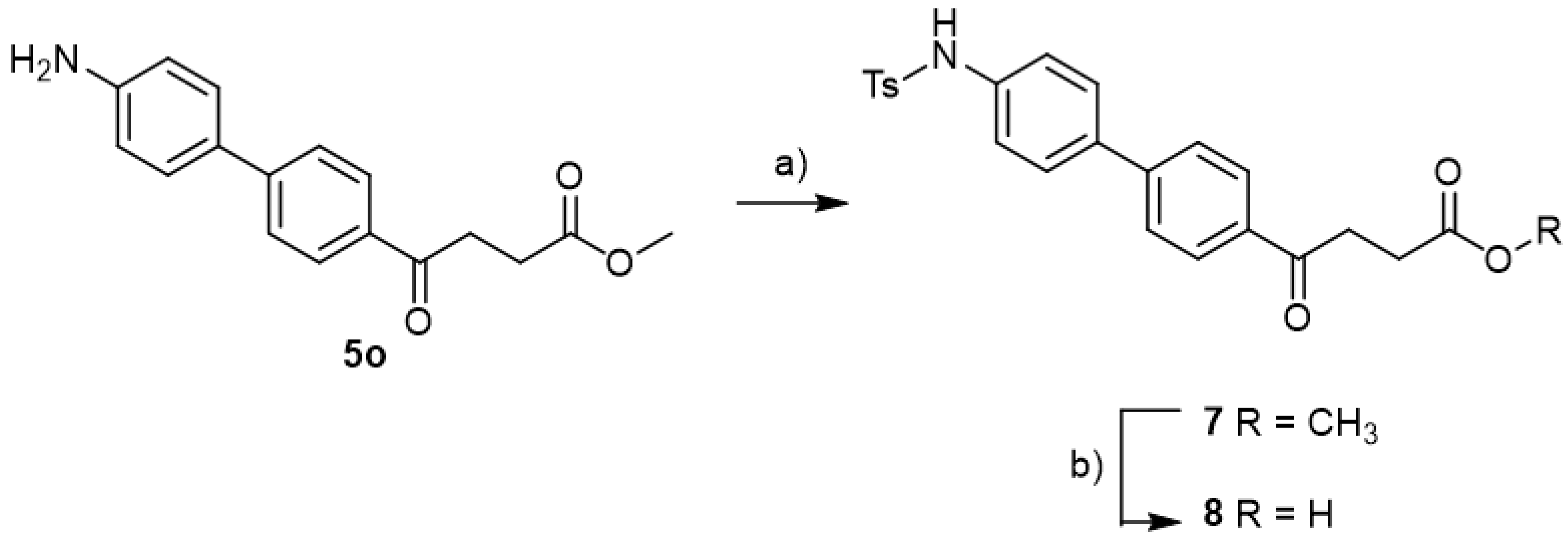
methyl 4-(4′-((4-methylphenyl)sulfonamido)-[1,1′-biphenyl]-4-yl)-4-oxobutanoate 7
![Molecules 27 02850 i033]()
Compound 5o (30 mg, 0.11 mmol, 1 eq) was co-distilled using toluene and CH2Cl2 three times followed by drying under high vacuo for 30 min. Dissolving by CH2Cl2 (0.5 mL) and adding pyridine (17 mg, 0.22 mmol, 2 eq), the mixture was cooled to 0 °C. p-TsCl (42 mg, 0.22 mmol, 2 eq) was added and the stirring was allowed for 10 min. Followed by removing the ice bath, the mixture turned from orange to pink during the next 30 min at rt. TLC (EtOAc/CH2Cl2 = 1/39) indicated the consumption of 5o (Rf = 0.4) and formation of the product 7 (Rf = 0.32). After a further 17 h reaction, 1N HCl(aq) (4.5 mL) was added followed by partitioning using CH2Cl2 (15 mL). Following washing with sat. NaCl(aq) (3 mL) and collecting the organic layer, the aqueous layer was back extracted using CH2Cl2 (15 mL) twice. Combining the organic layers and drying over MgSO4, the mixture was filtered through gravitational filtration. The filtrate was concentrated under reduced pressure for subsequent flash chromatography using eluents (EtOAc/CH2Cl2 = 1:49) to afford white powder (88 mg). The analytic sample (58 mg) was crystallized from toluene to give white powder 7 in 48% yield (29 mg, 0.07 mmol). m.p.: 174–178 °C.
1H NMR (500 MHz, CDCl3) δ 2.46 (s, 3H, CH3, Htosyl), 2.78 (t, J = 6.5 Hz, 2H, CH2), 3.34 (t, J = 6.5 Hz, 2H, CH2), 3.70 (s, 3H, -OCH3), 6.76 (bs, 1H, -NH), 7.14 (d, J = 8.5 Hz, 2H, aromatic), 7.23 (d, J = 8.5 Hz, 2H, aromatic), 7.48 (d, J = 8.0 Hz, 2H, aromatic, Htosyl), 7.58 (d, J = 8.5 Hz, 2H, aromatic), 7.68 (d, J = 8.0 Hz, 2H, aromatic, Htosyl), 8.00 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3) δ 21.53 (CH3, Ctosyl), 28.05 (CH2), 33.42 (CH2), 51.86 (CH3, O-CH3), 121.58 (CH, aromatic), 126.87 (CH, aromatic), 127.28 (CH, aromatic), 128.14 (CH, aromatic), 128.68 (CH, aromatic), 129.74 (CH, aromatic), 135.27 (C, aromatic), 136.21 (C, aromatic), 136.61 (C, aromatic), 136.75 (C, aromatic), 144.06 (C, aromatic), 144.69 (C, aromatic), 173.42 (C, COO), 197.54 (C, CO); analysis for C24H23NO5S, calculated [M + Na]+ (m/z) = 460.1189 (100.0%), 461.1223 (26.0%), ESI-Q-TOF HR-ESI-MS found: [M + Na]+ = 460.1187 (65.4%), 461.1216 (18.2%), δ [ppm] = −0.4.
4-(4′-((4-methylphenyl)sulfonamido)-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 8
Reagents of 7 (17 mg, 0.04 mmol) and a solution of aqueous THF (0.6 mL, 75% v/v) were used. The gold solution was cooled at 0 °C and LiOH (2.9 mg, 0.12 mmol, 3.0 eq) was added. After 10 min, the ice bath was removed and the reaction was allowed for 1 h. TLC (CH3OH/CH2Cl2 = 1:19) indicated the consumption of the starting material 7 (Rf = 0.84) and formation of the product 8 (Rf = 0.30). It was treated with cationic exchange resin (strong H+). Followed by filtration and concentration of the filtrate, a white solid 8 was obtained in 75% yield (12.8 mg, 0.03 mmol). m.p.: 165–167 °C.
1H NMR (500 MHz, CD3OD/CDCl3 = 1:1) δ 2.33 (s, 3H, CH3, Htosyl), 2.71 (t, J = 6.5 Hz, 2H, CH2), 3.30 (t, J = 6.5 Hz, 2H, CH2), 7.17 (d, J = 8.5 Hz, 2H, aromatic), 7.22 (d, J = 8.0 Hz, 2H, aromatic, Htosyl), 7.47 (d, J = 8.5 Hz, 2H, aromatic), 7.59 (d, J = 8.5 Hz, 2H, aromatic), 7.66 (d, J = 8.0 Hz, 2H, Htosyl), 8.00 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CD3OD/CDCl3 = 1:1) δ 21.56 (CH3, Ctosyl), 28.63 (CH2), 33.99 (CH2), 121.74 (CH, aromatic), 127.44 (CH, aromatic), 127.74 (CH, aromatic), 128.54 (CH, aromatic), 129.30 (CH, aromatic), 130.23 (CH, aromatic), 135.72 (C, aromatic), 136.43 (C, aromatic), 137.38 (C, aromatic), 138.63 (C, aromatic), 144.50 (C, aromatic), 145.88 (C, aromatic), 174.72 (C, COO), 199.35 (C, CO); analysis for C23H21NO5S, calculated [M − H]− (m/z) = 422.1068 (100.0%), 423.1101 (24.9%), ESI-Q-TOF HR-ESI-MS found: [M − H]− = 422.1070 (100.0%), 423.1098 (26.9%), δ [ppm] = 0.5.
p-hydroxymethyl fenbufen methyl ester 12
![Molecules 27 02850 i035]()
To a flask (250 mL) was added the bromo compound 2 (500 mg, 1.8 mmol, 1 eq) into THF (8 mL). The borono compound, 4-boronopinacol phenylmethanol (464 mg, 2.0 mmol, 1.1 eq), Pd(PPh3)4 (104 mg, 0.09 mmol, 0.05 eq) and aqueous K2CO3 (4.5 g, 32.4 mmol, 18 eq) in H2O (1 mL) were added, sequentially. The reaction was allowed at a gentle-reflux condition for 4 h (one drop per second). TLC (acetone/n-hexane = 3:7) indicated the consumption of the starting material (Rf = 0.56) and formation of the product 12 (Rf = 0.20). The mixture was partitioned between EtOAc (30 mL) and aqueous saline (satd., 15 mL × 3). The organic layer was collected, dried over Na2SO4, filtered through a celite pad. The filtrate was concentrated and chromatographed using eluents acetone/n-hexane in a gradient mode from 3:7 to 4:6 to afford a white solid which formed a transparent yellow solid under a high vacuum. The yield was 38% (205 mg). 1H-NMR (500 MHz, CDCl3) δ 2.73 (t, J = 6.7 Hz, 2H, HAliphatic), 3.21 (t, J = 6.7 Hz, 2H, HAliphatic), 3.65 (s, 3H, HOCH3), 4.70 (s, 2H, CH2), 7.47 (d, J = 8.5 Hz, 2H, HAr), 7.62 (d, J = 8.0 Hz, 2H, HAr), 7.68 (d, J = 8.5 Hz, 2H, HAr), 8.06 (d, J = 8.5 Hz, 2H, HAr); 13C-NMR (125 MHz, CDCl3) δ 28.00 (aliphatic, CH2), 33.40 (aliphatic, CH2), 51.85 (OCH3, CH3), 64.84 (OH-CH2, CH2), 127.14 (Ar, CH), 127.38 (Ar, CH), 127.49 (Ar, CH), 128.45 (Ar, CH), 128.54 (Ar, CH), 128.63 (Ar, CH), 128.72, 131.95 (Ar, CH), 131.97 (Ar, CH), 132.02 (Ar, CH), 132.10 (Ar, CH), 135.21 (Ar, C), 139.06 (Ar, C), 141.09 (Ar, C), 145.51 (Ar, C), 173.42 (CO-OCH3, C), 197.65 (Ar-CO, C); analysis for C18H18O4, calculated [M + Na]+ (m/z) = 321.1 (100.0%), 322.1 (19.5%), 323.1 (1.8%), ESI+MS Q-TOF found: [M + Na]+ = 321.2 (7.01%), 322.2 (1.33%), m.p. 117–119 °C. Elem. anal. for C and H, calculated C: 72.47%, H: 6.08%, found C: 72.41%, H: 6.14%.
4-(4′-(hydroxymethyl)-[1,1′-biphenyl]-4-yl)-4-oxobutanoic acid 11
Reagents of 12 (72 mg, 0.24 mmol), TFA (3 mL) and H2O (3 mL) were used. The procedure followed that described for preparing 6a. Reaction was allowed for 1 h and TLC (CH3OH/CH2Cl2 = 1/9) indicated the consumption of the starting material 12 (Rf = 0.86) and formation of the product 11 (Rf = 0.54). After column chromatography, a white solid 11 was obtained in 85% yield (58 mg, 0.2 mmol).
1H NMR (500 MHz, CDCl3/CD3OD = 1:1) δ 2.39 (t, J = 6.5 Hz, 2H, CH2), 2.96 (t, J = 6.5 Hz, 2H, CH2), 4.32 (s, 2H, CH2OH), 7.09 (d, J = 8.0 Hz, 2H, aromatic), 7.26 (d, J = 8.0 Hz, 2H, aromatic), 7.35 (d, J = 8.5 Hz, 2H, aromatic), 7.69 (d, J = 8.5 Hz, 2H, aromatic); 13C NMR (125 MHz, CDCl3/CD3OD = 1:1) δ 28.74 (CH2), 34.15 (CH2), 64.52 (CH2), 127.72 (CH, aromatic), 127.78 (CH, aromatic), 128.09 (CH, aromatic), 129.27 (CH, aromatic), 136.05 (C, aromatic), 139.45 (C, aromatic), 142.32 (C, aromatic), 146.54 (C, aromatic), 176.00 (C, COO), 199.65 (C, CO); analysis for C17H16O4, calculated [M + H]+ (m/z) = 285.1121 (100.0%), 286.1155 (18.4%), [M + Na]+ (m/z) = 307.0941 (100.0%), 308.0974 (18.4%), ESI-Q-TOF HR-ESI-MS found: [M + H]+ (m/z) = 285.1124 (11.7%), 286.1154 (2.0%),δ [ppm] = 1.1, [M + Na]+ (m/z) = 307.0943 (12.5%), 308.0973 (2.1%), δ [ppm] = 0.7.
























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


