
Chromatographic Determination of the Mycotoxin Patulin in 219 Chinese Tea Samples and Implications for Human Health

 , , and
, , and
Abstract
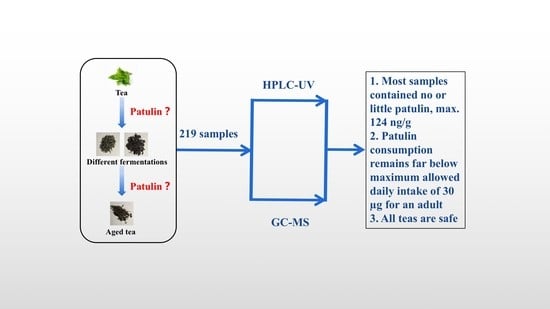
:
1. Introduction
2. Results and Discussion
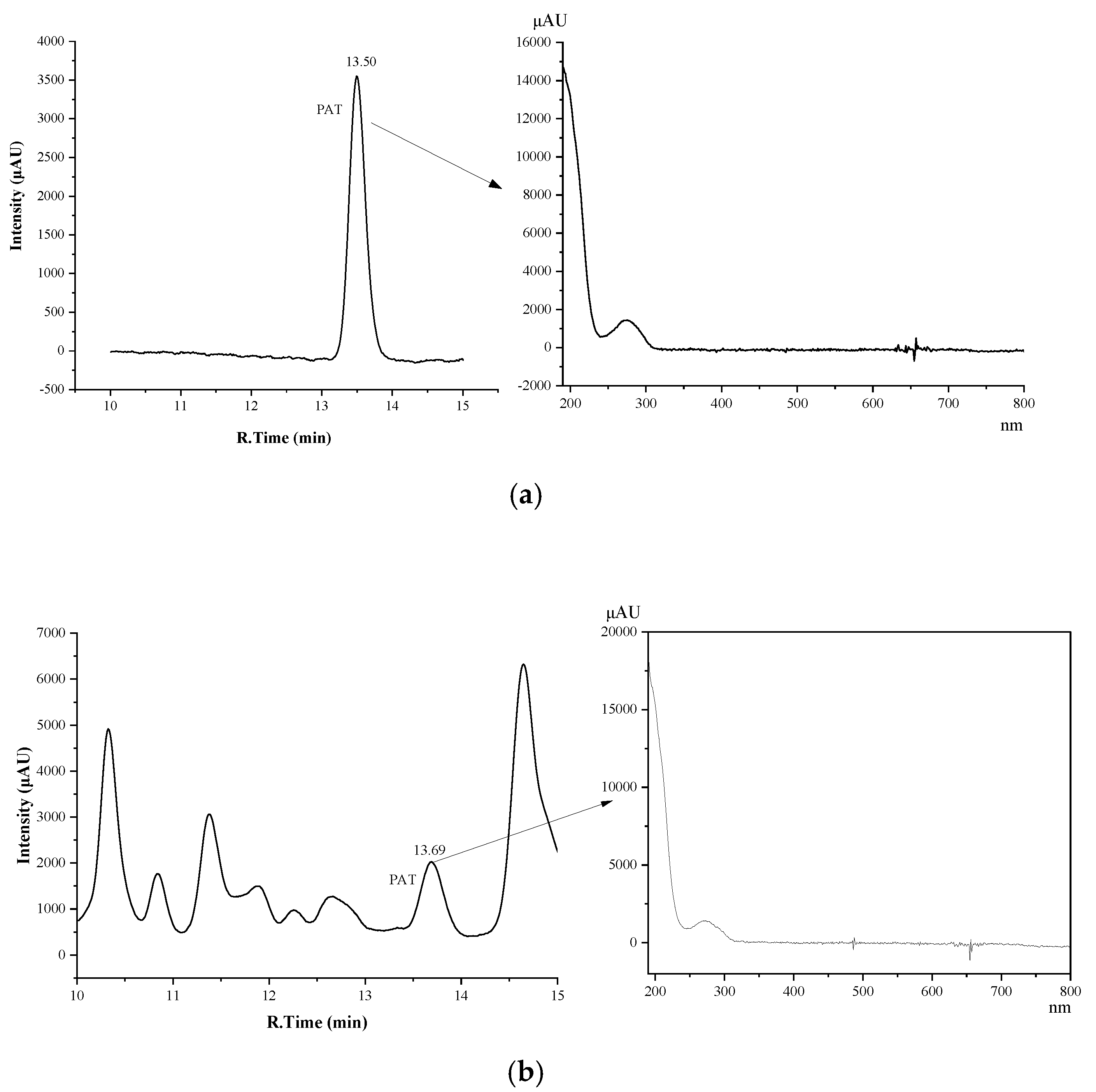
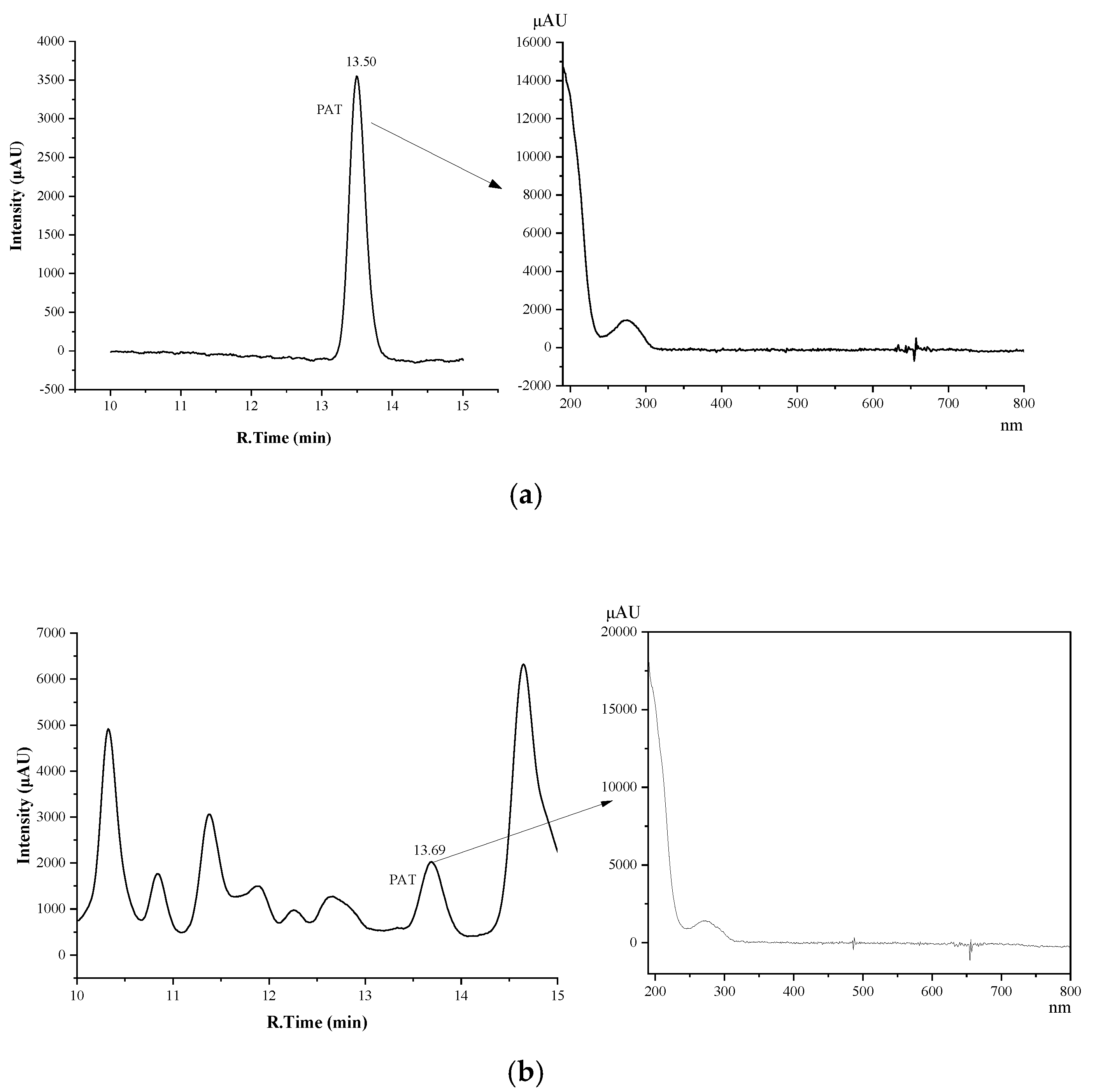
2.1. Development of HPLC-DAD Method for Analysis of PAT in Tea
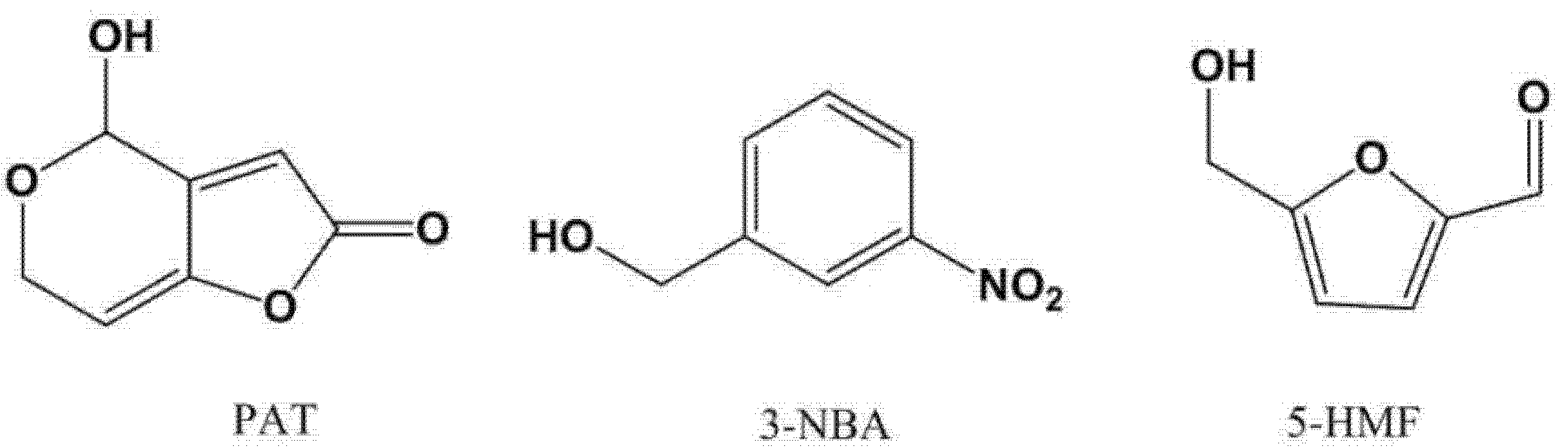
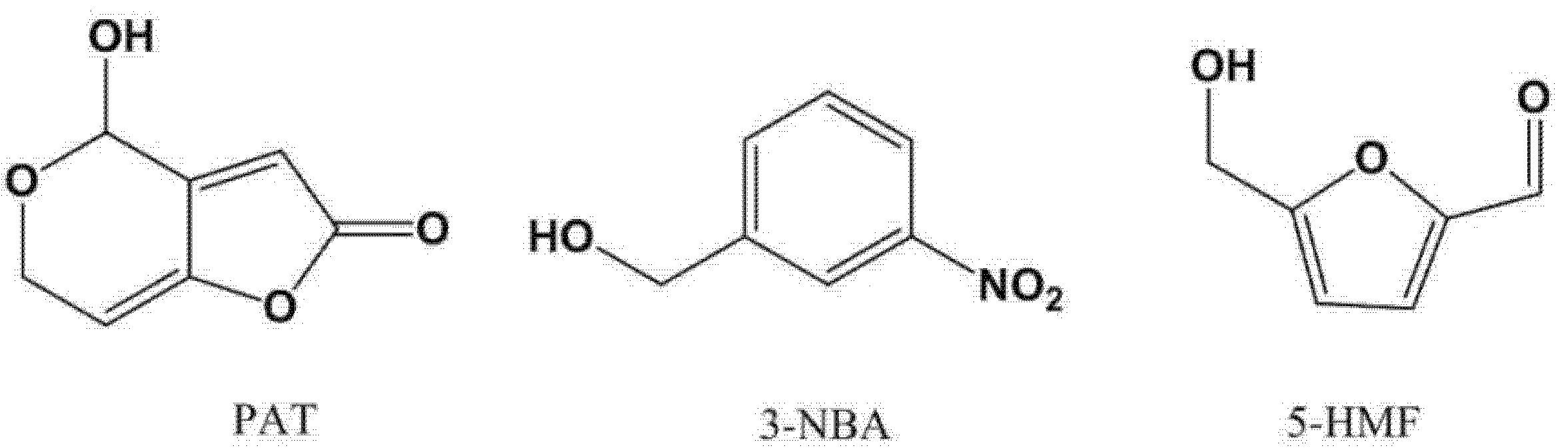
2.2. On-Line Derivatization for GC−MS Analysis
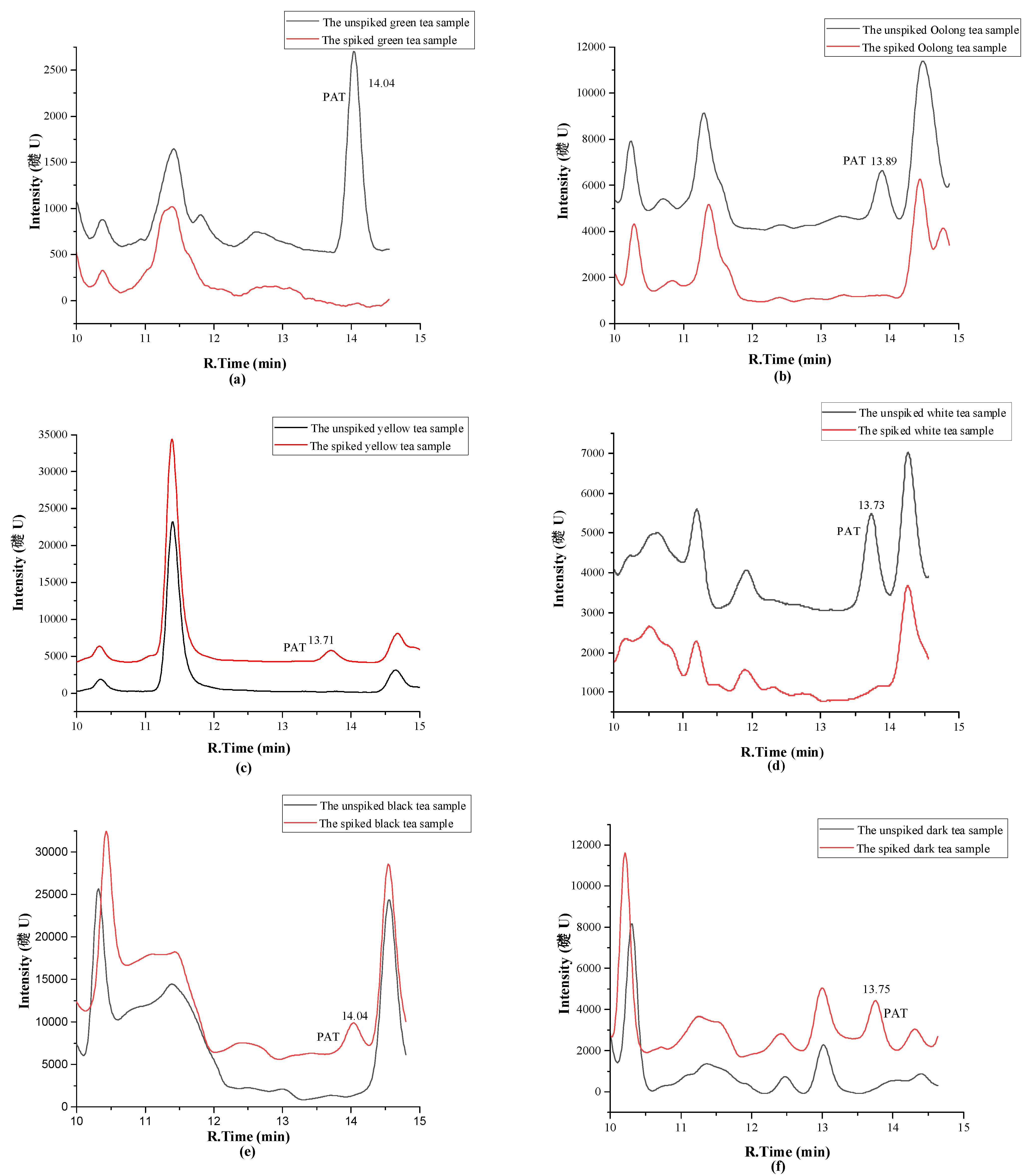
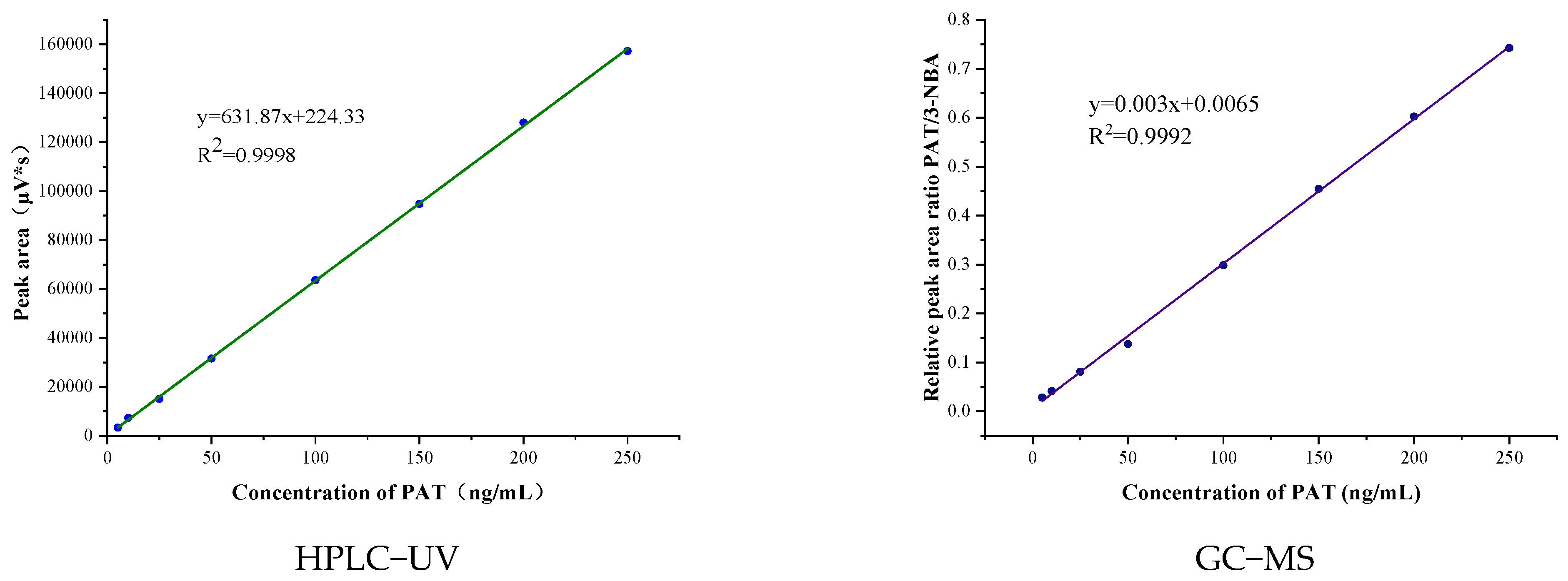
2.3. Method Validation
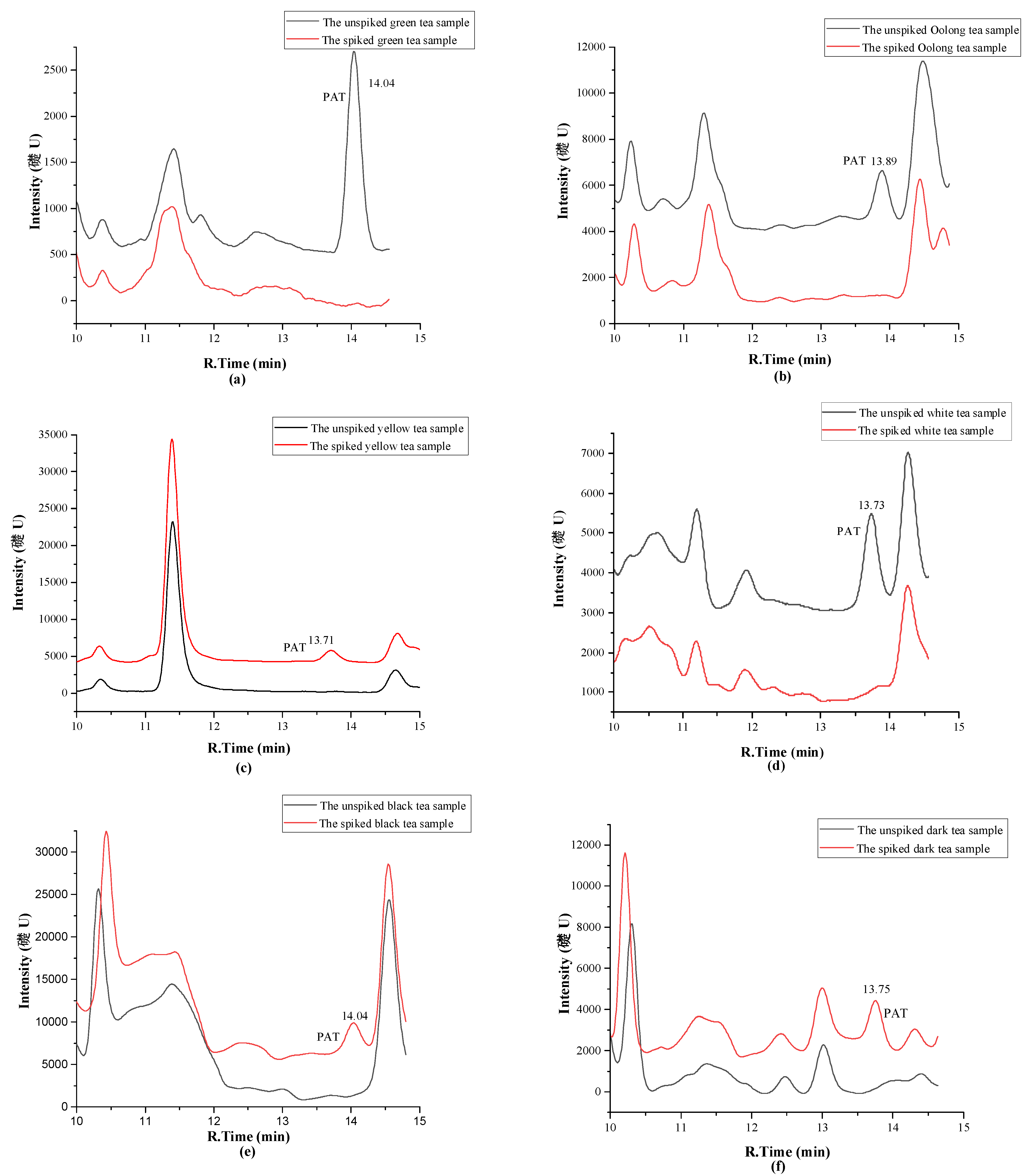
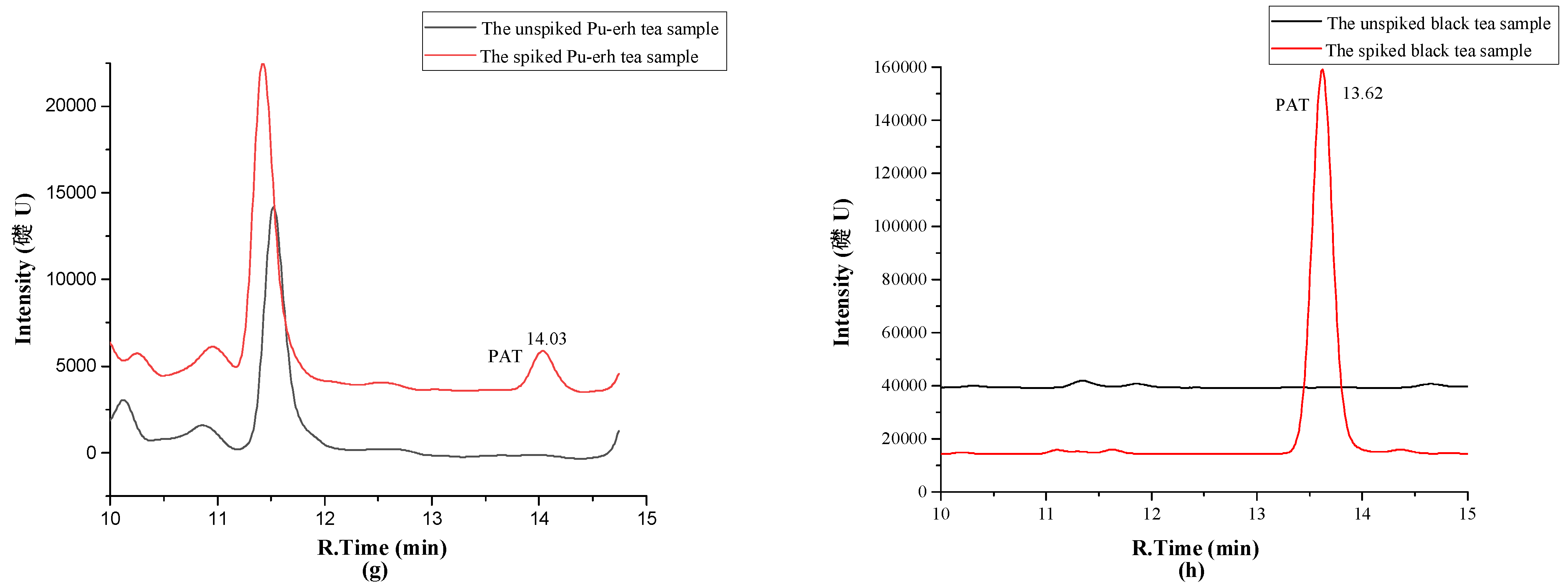
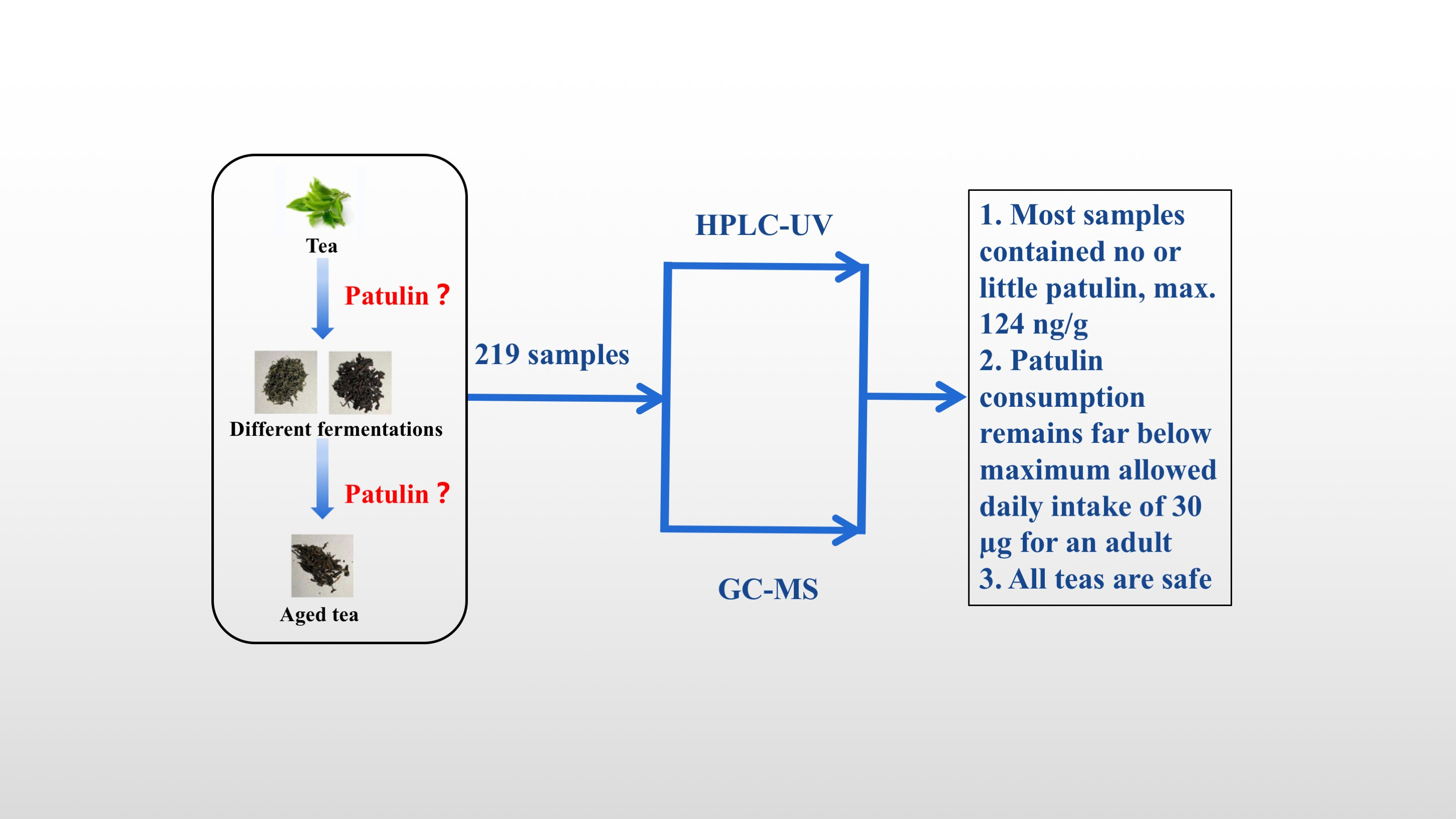
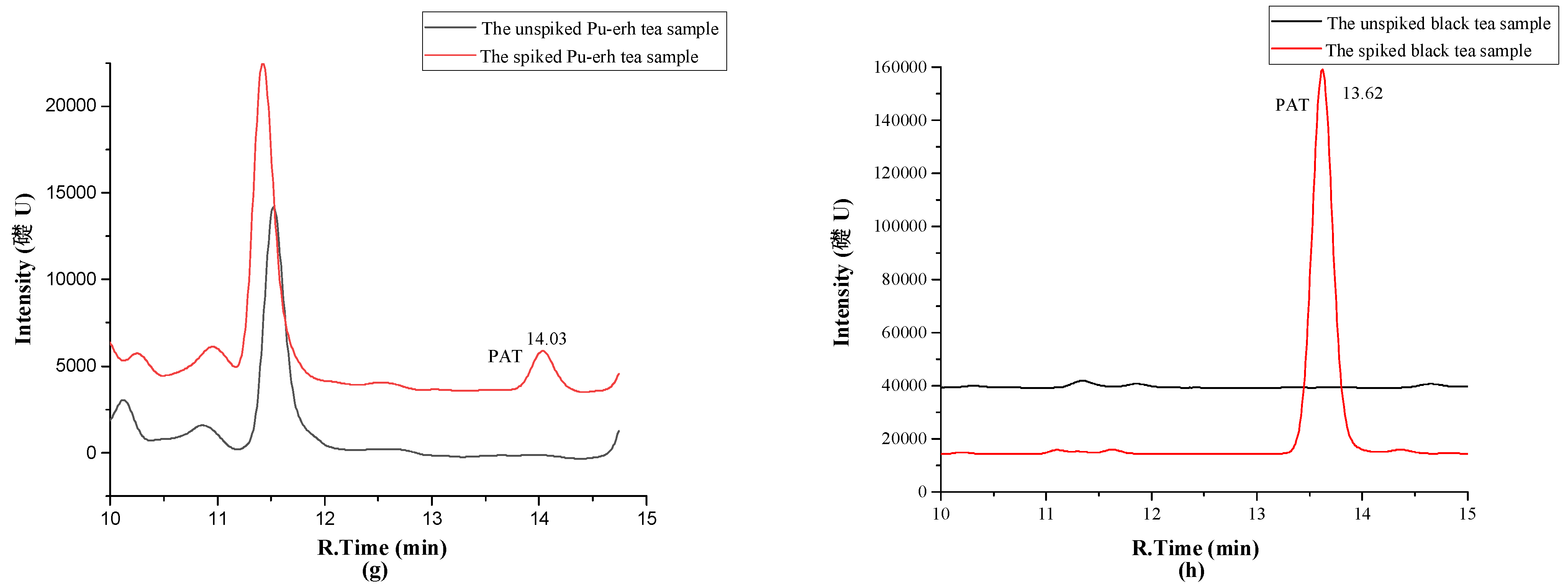
2.4. Investigation of Patulin in Different Chinese Tea Products
3. Materials and Methods
3.1. Materials, Reagents and Chemicals
3.2. HPLC Analysis
3.2.1. Preparation of Standard Solutions
3.2.2. Preparation of Sample Solutions
3.2.3. HPLC Analysis
3.3. Extraction and Pretreatment Experiments
- (1)
- 1.00 g of tea sample was extracted by 2.0 mL of ACN for three times, 100 mg of GCB was added to the pooled ACN extracts to adsorb pigments, after centrifugation the ACN supernatant was dried by a N2 stream, the residue was redissolved in 1.00 mL of H2O, and the solution was analyzed by HPLC after filtration through a 0.22 μm membrane.
- (2)
- 1.00 g of tea sample was extracted by 2.0 mL of ACN for three times, 100 mg of PSA was added to the pooled ACN extracts to adsorb pigments, after centrifugation the ACN supernatant was dried by a N2 stream, the residue was redissolved in 1.00 mL of H2O, and the solution was analyzed by HPLC after filtration through a 0.22 μm membrane.
- (3)
- 1.00 g of tea sample was extracted by 10.0 mL of water for three times. After centrifugation (10,000 rpm for 10 min) of each extract, the three extracts were combined in a separatory funnel. The aqueous solution was extracted three times, each time with 15.0 mL of ethyl acetate. The combined organic solution was washed with 10.0 mL of 1.5% solution of sodium bicarbonate in water, and the aqueous phase was extracted with an additional portion of ethyl acetate (10.0 mL). The organic phases were combined, dried over anhydrous sodium sulfate, and evaporated to dryness in a stream of nitrogen. The residue was dissolved in 1.00 mL of water [30]. The solution was analyzed by HPLC after filtration through a 0.22 μm membrane.
- (4)
- 1.00 g of tea sample was pretreated according to the procedure described in Section 3.2.2.
3.4. On-Line Derivatization for GC−MS Analysis
3.4.1. Preparation of Standard Solutions
3.4.2. Preparation of Sample Solutions
3.4.3. On-line Derivatization
3.4.4. GC−MS Analysis
3.5. Method Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Atkins, D.; Norman, J. Mycotoxins and food safety. Nutr. Food Sci. 1998, 98, 260–266. [Google Scholar] [CrossRef]
- Iman, S.; Ipek, G. The characteristics, occurrence, and toxicological effects of patulin. Food Chem. Toxicol. 2019, 129, 301–311. [Google Scholar]
- International Agency for Research on Cancer (IARC). Agents Classified by the International Agency of Research on Cancer Monographs. 2018. Available online: https://monographs.iarc.fr/wp-content/uploads/2018/09/List_of_Classifications.pdf (accessed on 11 March 2022).
- Pal, S.; Singh, N.; Ansari, K.M. Toxicological effects of patulin mycotoxin on the mammalian system: An overview. Toxicol. Res. 2017, 6, 764–771. [Google Scholar] [CrossRef] [Green Version]
- WHO. Children’s Health and the Environment. 2005. Available online: http://apps.who.int/iris/bitstream/handle/10665/43162/9241562927_eng.pdf?sequence=1&isAllowed=y&ua=1 (accessed on 11 March 2022).
- Mahato, D.K.; Kamle, M.; Sharma, B.; Pandhi, S.; Devi, S.; Dhawan, K.; Selvakumar, R.; Mishra, D.; Kumar, A.; Arora, S.; et al. Patulin in food: A mycotoxin concern for human health and its management strategies. Toxicon 2021, 198, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Babaali, E.; Abbasi, A.; Sarlak, Z. Risks of patulin and its removal procedures: A review. Int. J. Nutr. Sci. 2017, 2, 10–15. [Google Scholar]
- Wright, S.A. Patulin in food. Curr. Opin. Food Sci. 2015, 5, 105–109. [Google Scholar] [CrossRef]
- Vidal, A.; Ouhibi, S.; Ghali, R.; Hedhili, A.; De Saeger, S.; De Boevre, M. The mycotoxin patulin: An updated short review on occurrence, toxicity and analytical challenges. Food Chem. Toxicol. 2019, 129, 249–256. [Google Scholar] [CrossRef]
- Ji, X.; Li, R.; Yang, H.; Qi, P.; Xiao, Y.; Qian, M. Occurrence of patulin in various fruit products and dietary exposure assessment for consumers in China. Food Control 2017, 78, 100–107. [Google Scholar] [CrossRef]
- Liao, Y.; Fu, X.; Zeng, L.; Yang, Z. Strategies for studying in vivo biochemical formation pathways and multilevel distributions of quality or function-related specialized metabolites in tea (Camellia sinensis). Crit. Rev. Food Sci. Nutr. 2020, 62, 429–442. [Google Scholar] [CrossRef]
- Yu, Z.; Yang, Z. Understanding different regulatory mechanisms of proteinaceous and non-proteinaceous amino acid formation in tea (Camellia sinensis) provides new insights into the safe and effective alteration of tea flavor and function. Crit. Rev. Food Sci. Nutr. 2020, 60, 844–858. [Google Scholar] [CrossRef]
- Available online: www.statista.com/statistics/940102/global-tea-consumption/ (accessed on 11 March 2022).
- Li, M.-Y.; Xiao, Y.; Zhong, K.; Bai, J.-R.; Wu, Y.-P.; Zhang, J.-Q.; Gao, H. Characteristics and chemical compositions of Pingwu Fuzhuan brick-tea, a distinctive post-fermentation tea in Sichuan province of China. Int. J. Food Prop. 2019, 22, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, J.; Tan, G.L.; Liu, Z.X.; Li, X.L. Determination of ten mycotoxins in fermented dark tea by QuEChERS-ultra-high-performance liquid chromatography-tandem mass spectrometry. Modern Food Sci. Technol. 2017, 33, 280–288. [Google Scholar]
- Zheng, W.-J.; Wan, X.-C.; Bao, G.-H. Brick dark tea: A review of the manufacture, chemical constituents and bioconversion of the major chemical components during fermentation. Phytochem. Rev. 2015, 14, 499–523. [Google Scholar] [CrossRef]
- Assunção, R.; Twarużek, M.; Kosicki, R.; Viegas, C.; Viegas, S. Drinking Green Tea: Despite the Risks Due to Mycotoxins, Is It Possible to Increase the Associated Health Benefits? Toxins 2021, 13, 119. [Google Scholar] [CrossRef]
- Jianwei, F.; Biao, H.; Caibiao, H.; Hang, W.; Wenjing, L. Simultaneous determination of 16 mycotoxin residues in aged tea by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Food Sci. 2021, 42, 299–305. [Google Scholar]
- Liu, Q.F. Detection of zearalenone, fumonisin, aflatoxin, ochratoxin, deoxynivalenol and T-2 toxin in fermented tea and plant perfume by ELISA. China Tropical Med. 2011, 11, 1381–1382. [Google Scholar]
- Sedova, I.; Kiseleva, M.; Tutelyan, V. Mycotoxins in Tea: Occurrence, Methods of Determination and Risk Evaluation. Toxins 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.; Li, P.; Zhang, Q.; Zhang, W.; Ding, X. Determination for major chemical contaminants in tea (Camellia sinensis) matrices: A review. Food Res. Int. 2013, 53, 649–658. [Google Scholar] [CrossRef]
- Haas, D.; Pfeifer, B.; Reiterich, C.; Partenheimer, R.; Reck, B.; Buzina, W. Identification and quantification of fungi and mycotoxins from Pu-erh tea. Int. J. Food Microbiol. 2013, 166, 316–322. [Google Scholar] [CrossRef]
- Zhou, C.B.; Chen, W.P.; Zhong-Ling, W.U.; Rui-Lu, M.U.; Zhang, M.X. Research on function and safety of Penicillium oxalicum, a preponderant fungus during the fermentative process of Pu’er tea. Food Sci. Tech. 2015, 40, 63–66. [Google Scholar]
- Zhang, Y.; Skaar, I.; Sulyok, M.; Liu, X.; Rao, M.; Taylor, J.W. The Microbiome and Metabolites in Fermented Pu-erh Tea as Revealed by High-Throughput Sequencing and Quantitative Multiplex Metabolite Analysis. PLoS ONE 2016, 11, e0157847. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Fu, S. A Sensitive Gas Chromatography-Mass Spectrometry Method for the Determination of Patulin in Apple Juice. J. AOAC Int. 2012, 95, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Molto, J.; Berrada, H.; Mañes, J. A survey of trichothecenes, zearalenone and patulin in milled grain-based products using GC–MS/MS. Food Chem. 2014, 146, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.; Faria, M.; Fernandes, J. Determination of patulin in apple and quince products by GC–MS using 13C5–7 patulin as internal standard. Food Chem. 2009, 115, 352–359. [Google Scholar] [CrossRef]
- Marsol-Vall, A.; Balcells, M.; Eras, J.; Canela-Garayoa, R. A rapid gas chromatographic injection-port derivatization method for the tandem mass spectrometric determination of patulin and 5-hydroxymethylfurfural in fruit juices. J. Chromatogr. A 2016, 1453, 99–104. [Google Scholar] [CrossRef]
- Moukas, A.; Panagiotopoulou, V.; Markaki, P. Determination of patulin in fruit juices using HPLC-DAD and GC−MSD techniques. Food Chem. 2008, 109, 860–867. [Google Scholar] [CrossRef]
- Rychlik, M.; Schieberle, P. Quantification of the Mycotoxin Patulin by a Stable Isotope Dilution Assay. J. Agric. Food Chem. 1999, 47, 3749–3755. [Google Scholar] [CrossRef]
- Clercq, N.D.; Pamel, E.V.; Coillie, E.V.; Vlaemynck, G.; Devlieghere, F.; Meulenaer, B.D.; Daeseleire, E. Optimization and validation of a method without alkaline clean-up for patulin analysis on apple puree agar medium (APAM) and apple products. Food Anal. Meth. 2016, 9, 370–377. [Google Scholar] [CrossRef]
- Boonzaaijer, G.; Bobeldijk, I.; van Osenbruggen, W. Analysis of patulin in dutch food, an evaluation of a SPE based method. Food Control 2005, 16, 587–591. [Google Scholar] [CrossRef]
- Available online: http://www.meizhengbio.com/131/2398.html (accessed on 17 March 2022).
- Sadhasivam, S.; Barda, O.; Zakin, V.; Reifen, R.; Sionov, E. Rapid Detection and Quantification of Patulin and Citrinin Contamination in Fruits. Molecules 2021, 26, 4545. [Google Scholar] [CrossRef]
- Kharandi, N.; Babri, M.; Azad, J. A novel method for determination of patulin in apple juices by GC–MS. Food Chem. 2013, 141, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Tabata, S.; Iida, K.; Suzuki, J.; Kimura, K.; Ibe, A.; Saito, K. A Quantification and Confirmation Method of Patulin in Apple Juice by GC/MS. Food Hyg. Saf. Sci. (Shokuhin Eiseigaku Zasshi) 2004, 45, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notardonato, I.; Gianfagna, S.; Castoria, R.; Ianiri, G.; De Curtis, F.; Russo, M.V.; Avino, P. Critical review of the analytical methods for determining the mycotoxin patulin in food matrices. Rev. Anal. Chem. 2021, 40, 144–160. [Google Scholar] [CrossRef]
- Nesrin, İ. Occurrence of patulin and 5-hydroxymethylfurfural in apple sour, which is a traditional product of Kastamonu, Turkey. Food Addit. Contam Part A 2019, 36, 952–963. [Google Scholar]
- Heidi, S.R.; Sherri, B.T. Confirmation of patulin and 5-hydroxymethylfurfural in apple juice by gas chromatography/mass spectrometry. J. AOAC Int. 2000, 83, 612–620. [Google Scholar]
- Moss, M.O.; Long, M.T. Fate of patulin in the presence of the yeastSaccharomyces cerevisiae. Food Addit. Contam. 2002, 19, 387–399. [Google Scholar] [CrossRef]
- E Stinson, E.; Osman, S.F.; Huhtanen, C.N.; Bills, D.D. Disappearance of patulin during alcoholic fermentation of apple juice. Appl. Environ. Microbiol. 1978, 36, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Duvivier, W.F.; van Beek, T.A.; Nielen, M.W.F. Critical comparison of mass analyzers for forensic hair analysis by ambient ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 2331–2340. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Recovery (%) | |||
|---|---|---|---|---|
| Method 1 | Method 2 | Method 3 | Method 4 | |
| Green tea | 0.0 | 52.8 ± 10.6 | 67.0 ± 11.8 | 97.6 ± 5.7 |
| Oolong tea | 0.0 | 44.2 ± 9.5 | 55.4 ± 9.6 | 96.5 ± 4.7 |
| Black tea | 0.0 | 40.5 ± 11.8 | 65.4 ± 10.3 | 99.1 ± 4.1 |
| Dark tea | 0.0 | 43.9 ± 11.2 | 62.4 ± 7.8 | 96.5 ± 4.1 |
| On-line | Injector Temp. (°C) | 150 | 200 | 250 | 280 | 300 |
| Peak area (SIM m/z 183) | 0 | 46.4 × 103 ± 1.8 × 103 | 52.0 × 103 ± 1.3 × 103 | 57.7 × 103 ± 0.6 × 103 | 57.8 × 103 ± 0.7 × 103 | |
| Off-line | Heating time at 80 °C (min) (Inj. Temp. 280 °C) | 10 | 20 | 30 | 40 | 50 |
| Peak area (SIM m/z 183) | 57.9 × 103 ± 1.2 × 103 | 57.4 × 103 ± 1.1 × 103 | 55.4 × 103 ± 1.2 × 103 | 52.3 × 103 ± 1.7 × 103 | 52.1 × 103 ± 1.8 × 103 |
| Sample | Conc. (ng/g) | Recovery (%) | |
|---|---|---|---|
| GC−MS | HPLC−UV | ||
| Green tea | 10 | 89.7 ± 10.5 | 95.2 ± 4.6 |
| 50 | 96.9 ± 8.4 | 97.6 ± 5.7 | |
| 100 | 94.3 ± 5.2 | 98.1 ± 3.4 | |
| Oolong tea | 10 | 92.1 ± 3.4 | 97.4 ± 3.8 |
| 50 | 94.9 ± 7.5 | 96.5 ± 4.7 | |
| 100 | 98.6 ± 5.1 | 101.2 ± 4.4 | |
| Black tea | 10 | 102.6 ± 8.7 | 95.5 ± 3.6 |
| 50 | 99.2 ± 7.3 | 99.1 ± 4.1 | |
| 100 | 92.4 ± 4.4 | 98.7 ± 4.4 | |
| Dark tea | 10 | 95.6 ± 5.7 | 96.8 ± 3.3 |
| 50 | 89.9 ± 8.2 | 96.5 ± 4.1 | |
| 100 | 93.3 ± 6.1 | 102.5 ± 2.7 | |
| Nr of Samples | Average PAT Concentration (ng/g) | Standard Deviation | Minimum and Maximum Conc. | |
|---|---|---|---|---|
| Green tea | 88 | 1.3 | 3.3 | ND-22.8 |
| Yellow tea | 7 | 0.41 | 1.0 | ND-2.9 |
| White tea | 13 | 3.3 | 3.6 | ND-10.9 |
| Oolong tea | 21 | 1.5 | 4.7 | ND-21.4 |
| Pu-erh tea | 17 | 1.2 | 2.2 | ND-6.7 |
| Dark tea | 38 | 4.6 | 20.1 | ND-124 |
| Black tea | 35 | 6.6 | 14.2 | ND-68.5 |
| No. | Sample Name | Fermentation Degree | Starting Year of Aging | Region | Place of Origin | Content (ng/g) (HPLC−UV/GC−MS) |
|---|---|---|---|---|---|---|
| 2 | Black tea | Completely fermented (Jingjunmei) | 2017 | East China | Fujian | 43.4/42.2 |
| 4 | Black tea | Completely fermented | 2018 | East China | Fujian | 68.5/64.2 |
| 6 | Black tea | Completely fermented | 2016 | East China | Fujian | 11.2/12.3 |
| 7 | Black tea | Completely fermented (Zhenshanxiaozhong) | 2017 | East China | Fujian | 39.6/37.4 |
| 37 | Green tea | Non-fermented | 2018 | Central China | Hunan | 13.8/11.2 |
| 38 | Green tea | Non-fermented (YYS008) | 2021 | Central China | Hunan | 22.8/23.8 |
| 125 | Oolong tea | Partially fermented | 2018 | East China | Fujian | 21.4/20.7 |
| 127 | White tea | Partially fermented | 2010 | East China | Fujian | 10.9/10.4 |
| 165 | Dark tea | Post-fermented (YYS024) | 2021 | Central China | Hunan | 26.6/24.1 |
| 166 | Dark tea | Post-fermented (YYS025) | 2021 | Central China | Hunan | 124/112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Liu, C.; Luo, S.; Zhu, S.; Tang, S.; Zeng, H.; Qin, Y.; Ma, M.; Zeng, D.; van Beek, T.A.; et al. Chromatographic Determination of the Mycotoxin Patulin in 219 Chinese Tea Samples and Implications for Human Health. Molecules 2022, 27, 2852. https://doi.org/10.3390/molecules27092852
Li H, Liu C, Luo S, Zhu S, Tang S, Zeng H, Qin Y, Ma M, Zeng D, van Beek TA, et al. Chromatographic Determination of the Mycotoxin Patulin in 219 Chinese Tea Samples and Implications for Human Health. Molecules. 2022; 27(9):2852. https://doi.org/10.3390/molecules27092852
Chicago/Turabian StyleLi, Hai, Candi Liu, Shurong Luo, Sijie Zhu, Shan Tang, Huimei Zeng, Yu Qin, Ming Ma, Dong Zeng, Teris A. van Beek, and et al. 2022. "Chromatographic Determination of the Mycotoxin Patulin in 219 Chinese Tea Samples and Implications for Human Health" Molecules 27, no. 9: 2852. https://doi.org/10.3390/molecules27092852
APA StyleLi, H., Liu, C., Luo, S., Zhu, S., Tang, S., Zeng, H., Qin, Y., Ma, M., Zeng, D., van Beek, T. A., Wang, H., & Chen, B. (2022). Chromatographic Determination of the Mycotoxin Patulin in 219 Chinese Tea Samples and Implications for Human Health. Molecules, 27(9), 2852. https://doi.org/10.3390/molecules27092852