
External Electric Field Effect on the Strength of σ-Hole Interactions: A Theoretical Perspective in Like⋯Like Carbon-Containing Complexes

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results and Discussion
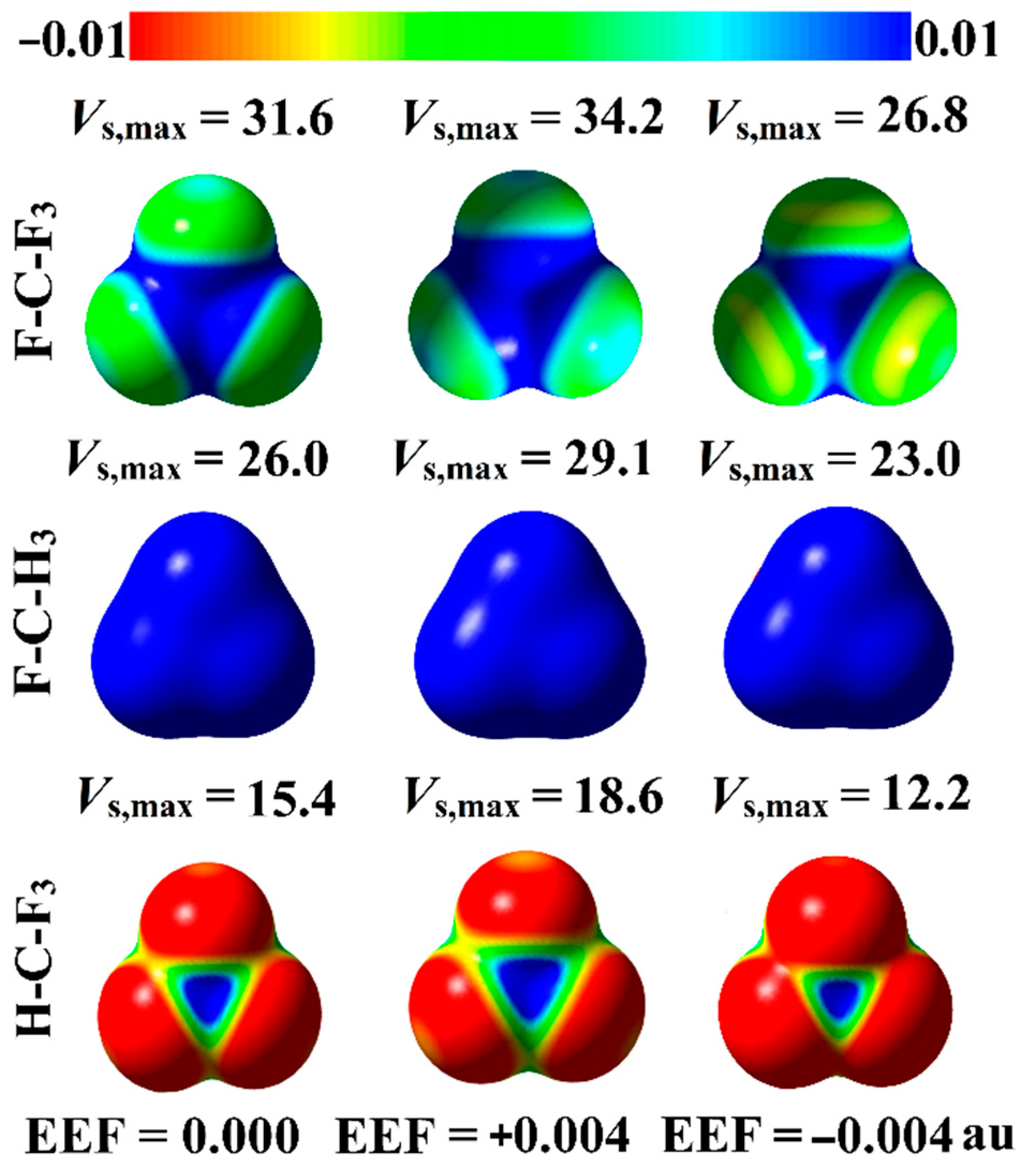
2.1. MEP and Vs,max Calculations
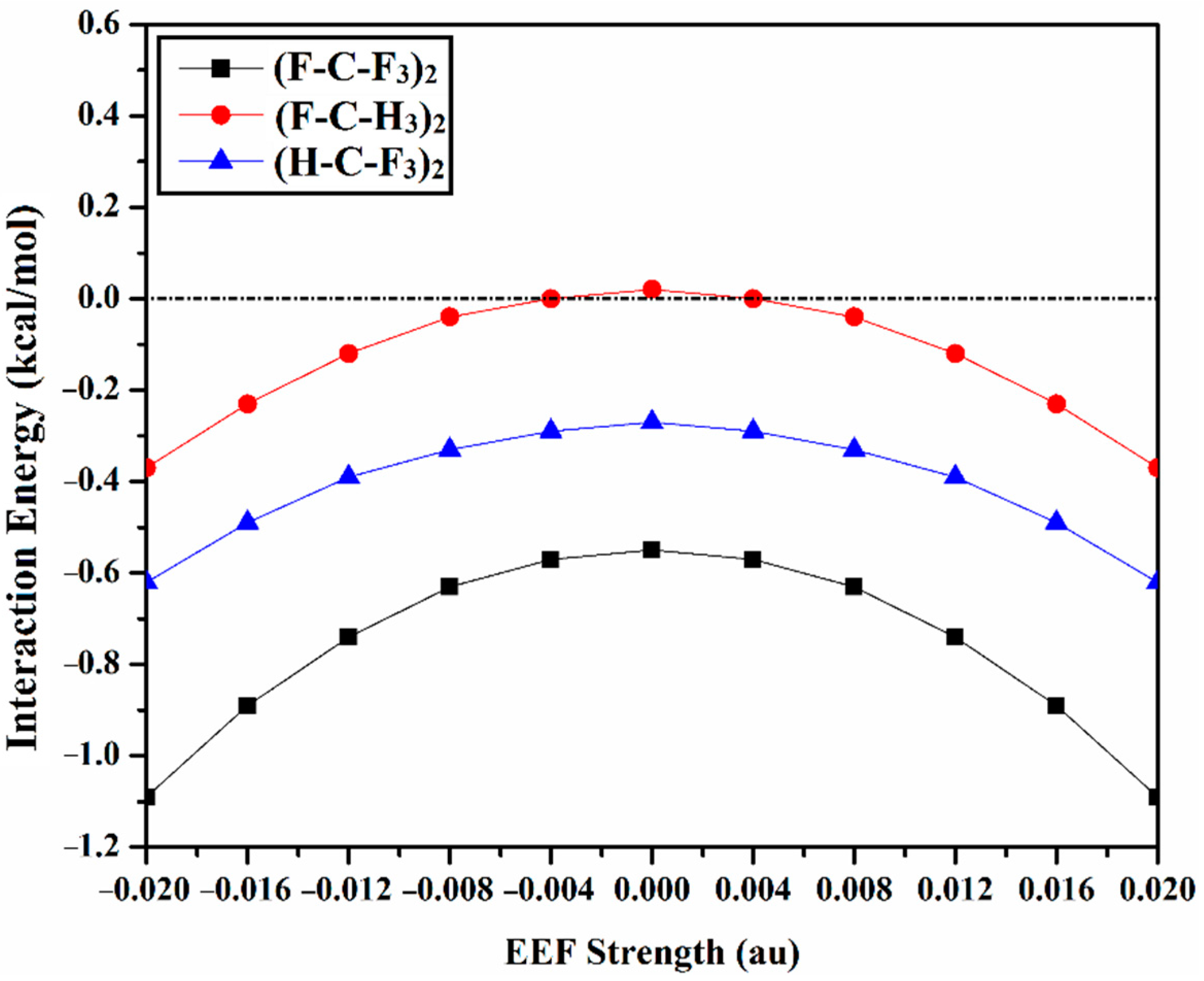
2.2. Interaction Energy
2.3. QTAIM Analysis
2.4. NCI Analysis
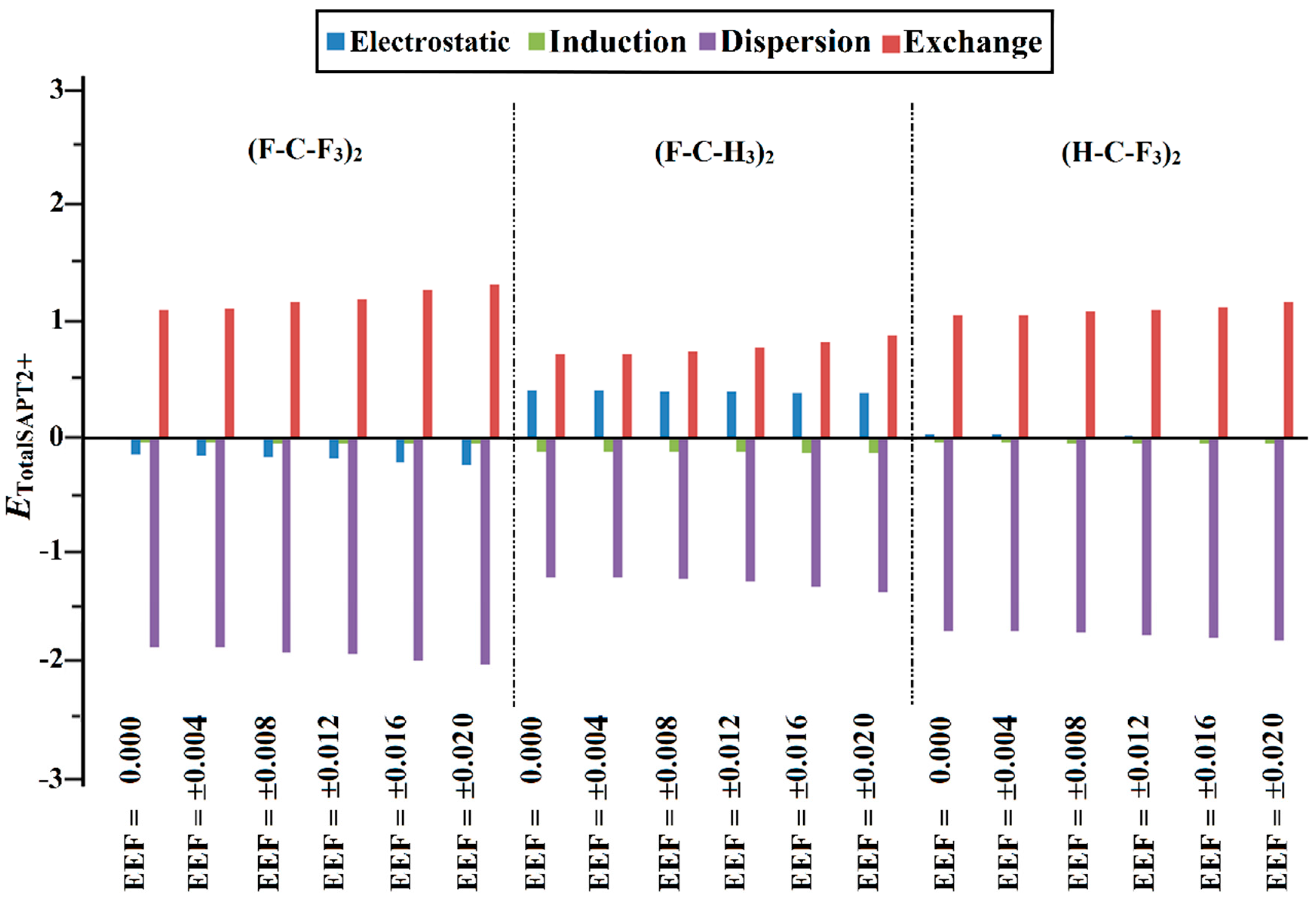
2.5. SAPT Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The halogen bond in the design of functional supramolecular materials: Recent advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hamdani, Y.S.; Tkatchenko, A. Understanding non-covalent interactions in larger molecular complexes from first principles. J. Chem. Phys. 2019, 150, 10901. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, L.; Miranda-Rojas, S.; Mendizabal, F. Noncovalent interactions in inorganic supramolecular chemistry based in heavy metals. Quantum chemistry point of view. Int. J. Quantum Chem. 2019, 119, e25675. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Y.; Zhu, W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discov. 2012, 7, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Hasb, A.A.M.; Mekhemer, G.A.H. Role and nature of halogen bonding in inhibitor···receptor complexes for drug discovery: Casein kinase-2 (CK2) inhibition as a case study. Theor. Chem. Acc. 2018, 137, 38–47. [Google Scholar] [CrossRef]
- Fish, R.H.; Jaouen, G. Bioorganometallic chemistry: Structural diversity of organometallic complexes with bioligands and molecular recognition studies of several supramolecular hosts with biomolecules, alkali-metal ions, and organometallic pharmaceuticals. Organometallics 2003, 22, 2166–2177. [Google Scholar] [CrossRef]
- Mazik, M. Molecular recognition of carbohydrates by acyclic receptors employing noncovalent interactions. Chem. Soc. Rev. 2009, 38, 935–956. [Google Scholar] [CrossRef]
- Schalley, C.A. Supramolecular chemistry goes gas phase: The mass spectrometric examination of noncovalent interactions in host–guest chemistry and molecular recognition. Int. J. Mass Spectrom. 2000, 194, 11–39. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, 5–9 September 2005. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The sigma-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Systematic elucidation of factors that influence the strength of tetrel bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional noncovalent interaction. Int. J. Quantum Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bond-sigma-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. Sigma-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Concha, M.C.; Lane, P.; Hobza, P.; Politzer, P. Blue shifts vs red shifts in sigma-hole bonding. J. Mol. Model. 2008, 14, 699–704. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Tetrel bonding interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Non-covalent sp(3) carbon bonding with ArCF3 is analogous to CH-pi interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, G.; Bauza, A.; Amini, M.; Molins, E.; Mague, J.T.; Frontera, A. On the importance of tetrel bonding interactions in lead(ii) complexes with (iso)nicotinohydrazide based ligands and several anions. Dalton Trans. 2016, 45, 10708–10716. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A. Theoretical study on the dual behavior of XeO3 and XeF4 toward aromatic rings: Lone pair-π versus aerogen-π interactions. ChemPhysChem 2015, 16, 3625–3630. [Google Scholar] [CrossRef]
- Scheiner, S. Competition between a tetrel and halogen bond to a common Lewis acid. J. Phys. Chem. A 2021, 125, 308–316. [Google Scholar] [CrossRef]
- Frontera, A. Tetrel bonding interactions involving carbon at work: Recent advances in crystal engineering and catalysis. C J. Carbon Res. 2020, 6, 60. [Google Scholar] [CrossRef]
- Karim, A.; Schulz, N.; Andersson, H.; Nekoueishahraki, B.; Carlsson, A.C.; Sarabi, D.; Valkonen, A.; Rissanen, K.; Grafenstein, J.; Keller, S.; et al. Carbon’s three-center, four-electron tetrel bond, treated experimentally. J. Am. Chem. Soc. 2018, 140, 17571–17579. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C···π (X = F, Cl, Br, CN) carbon bond. J. Phys. Chem. A 2014, 118, 10081–10089. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M.; Soliman, M.E.S.; Moustafa, M.F.; Al-Fahemi, J.H.; El-Mageed, H.R.A. On the potentiality of X-T-X3 compounds (T = C, Si, and Ge, and X = F, Cl, and Br) as tetrel- and halogen-bond donors. ACS Omega 2021, 6, 19330–19341. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The ditetrel bond: Noncovalent bond between neutral tetrel atoms. Phys. Chem. Chem. Phys. 2020, 22, 16606–16614. [Google Scholar] [CrossRef] [PubMed]
- Grabarz, A.; Michalczyk, M.; Zierkiewicz, W.; Scheiner, S. Noncovalent bonds between tetrel atoms. ChemPhysChem 2020, 21, 1934–1944. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Mohammadian-Sabet, F. Pnicogen–pnicogen interactions in O2XP:PH2Y complexes (X = H, F, CN; Y = H, OH, OCH3, CH3, NH2). Chem. Phys. Lett. 2015, 638, 122–127. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the pnicogen bond in complexes involving group Va elements N, P, and As. J. Phys. Chem. A 2015, 119, 1642–1656. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Shehata, M.N.I.; Soliman, M.E.S.; Moustafa, M.F.; El-Mageed, H.R.A.; Moussa, N.A.M. Unusual chalcogen···chalcogen interactions in like···like and unlike Y=C=Y···Y=C=Y complexes (Y = O, S, and Se). Phys. Chem. Chem. Phys. 2022, 24, 3386–3399. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, N.; Yan, N.N.; Wang, T.Y.; Zhang, Y.T.; Song, Q.; Li, H.J. Adsorption of small hydrocarbons on pristine, N-doped and vacancy graphene by DFT study. Appl. Surf. Sci. 2020, 515, 146028. [Google Scholar] [CrossRef]
- Sanchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular weak interactions in HTeXH dimers (X = O, S, Se, Te): Hydrogen bonds, chalcogen-chalcogen contacts and chiral discrimination. ChemPhysChem 2012, 13, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Metrangolo, P.; Resnati, G. Type II halogen···halogen contacts are halogen bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef]
- Niyas, M.A.; Ramakrishnan, R.; Vijay, V.; Sebastian, E.; Hariharan, M. Anomalous halogen-halogen interaction assists radial chromophoric assembly. J. Am. Chem. Soc. 2019, 141, 4536–4540. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Moussa, N.A.M. Unconventional type III halogen···halogen interactions: A quantum mechanical elucidation of σ-hole···σ-hole and di-σ-hole interactions. ACS Omega 2020, 5, 21824–21835. [Google Scholar] [CrossRef] [PubMed]
- Muruganathan, M.; Sun, J.; Imamura, T.; Mizuta, H. Electrically tunable van der Waals interaction in graphene–molecule complex. Nano Lett. 2015, 15, 8176–8180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabi, A.A.; Matta, C.F. Effects of external electric fields on double proton transfer kinetics in the formic acid dimer. Phys. Chem. Chem. Phys. 2011, 13, 13738–13748. [Google Scholar] [CrossRef] [PubMed]
- Calvaresi, M.; Martinez, R.V.; Losilla, N.S.; Martinez, J.; Garcia, R.; Zerbetto, F. Splitting CO2 with electric fields: A computational investigation. J. Phys. Chem. Lett. 2010, 1, 3256–3260. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Saad, S.M.A.; Al-Fahemi, J.H.; Mekhemer, G.A.H.; Ahmed, S.A.; Shawky, A.M.; Moussa, N.A.M. External electric field effects on the σ-hole and lone-pair hole interactions of group V elements: A comparative investigation. RSC Adv. 2021, 11, 4022–4034. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Kamel, A.A.K.; Soliman, M.E.S.; Moustafa, M.F.; El-Mageed, H.R.A.; Taha, F.; Mohamed, L.A.; Moussa, N.A.M. Effect of external electric field on tetrel bonding interactions in (FTF3···FH) complexes (T = C, Si, Ge, and Sn). ACS Omega 2021, 6, 25476–25485. [Google Scholar] [CrossRef] [PubMed]
- Foroutan-Nejad, C.; Marek, R. Potential energy surface and binding energy in the presence of an external electric field: Modulation of anion–π interactions for graphene-based receptors. Phys. Chem. Chem. Phys. 2014, 16, 2508–2514. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, J.; Li, Q.; Li, W. Some measures for making a traditional halogen bond be chlorine-shared or ion-pair one in FCl•NH3 complex. Mol. Phys. 2016, 114, 3643–3649. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mohamed, Y.A.M.; Abuelliel, H.A.A.; Rady, A.S.S.M.; Soliman, M.E.S.; Ahmed, M.N.; Mohamed, L.A.; Moussa, N.A.M. σ-hole interactions of tetrahedral group IV–VIII lewis acid centers with lewis bases: A comparative study. ChemistrySelect 2021, 6, 11856–11864. [Google Scholar] [CrossRef]
- Weiner, P.K.; Langridge, R.; Blaney, J.M.; Schaefer, R.; Kollman, P.A. Electrostatic potential molecular surfaces. Proc. Natl. Acad. Sci. USA 1982, 79, 3754–3758. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Panini, P.; Gonnade, R.G.; Chopra, D. Experimental and computational analysis of supramolecular motifs involving Csp2(aromatic)–F and CF3 groups in organic solids. New J. Chem. 2016, 40, 4981–5001. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mahmoud, A.H.M.; Moussa, N.A.M. Comparative investigation of ±σ–hole interactions of carbon-containing molecules with Lewis bases, acids and di-halogens. Chem. Pap. 2020, 74, 3569–3580. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Ahmed, O.A.M.; Moussa, N.A.M.; El-Taher, S.; Moustafa, H. Comparative investigation of interactions of hydrogen, halogen and tetrel bond donors with electron-rich and electron-deficient π-systems. RSC Adv. 2019, 9, 32811–32820. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation-theory approach to intermolecular potential-energy surfaces of van der Waals complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. 1994, 100, 2975–2988. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.A. Molecular mechanical perspective on halogen bonding. J. Mol. Model. 2012, 18, 4625–4638. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen bonding: A halogen-centered noncovalent interaction yet to be understood. Inorganics 2019, 7, 40–102. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.K.; Karthikeyan, S.; Ramanathan, V. Tuning the C-H···Pi interaction by different substitutions in benzene-acetylene complexes. J. Chem. Theory Comput. 2012, 8, 1935–1942. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Nziko Vde, P.; Scheiner, S. Comparison of pi-hole tetrel bonding with sigma-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. PSI4: An open-source ab initio electronic structure program. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 14101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 94106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EEF a (au) | (F-C-F3)2 | (F-C-H3)2 | (H-C-F3)2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| D b | E1 | E2 | D b | E1 | E2 | D b | E1 | E2 | |
| 0.000 | 3.88 | −0.55 | −0.87 | 3.45 | 0.02 | 0.01 | 4.00 | −0.27 | −0.31 |
| ±0.004 | 3.88 | −0.57 | −0.89 | 3.45 | 0.00 | −0.16 | 4.00 | −0.29 | −0.35 |
| ±0.008 | 3.87 | −0.63 | −1.00 | 3.44 | −0.04 | −0.18 | 4.00 | −0.33 | −0.42 |
| ±0.012 | 3.86 | −0.74 | −1.21 | 3.43 | −0.12 | −0.26 | 3.99 | −0.39 | −0.54 |
| ±0.016 | 3.84 | −0.89 | −1.58 | 3.41 | −0.23 | −0.38 | 3.98 | −0.49 | −0.73 |
| ±0.020 | 3.83 | −1.09 | −1.88 | 3.39 | −0.37 | −0.53 | 3.97 | −0.62 | −0.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Moussa, N.A.M.; Kamel, A.A.K.; Shehata, M.N.I.; Ahmed, M.N.; Taha, F.; Abourehab, M.A.S.; Shawky, A.M.; Elkaeed, E.B.; Soliman, M.E.S. External Electric Field Effect on the Strength of σ-Hole Interactions: A Theoretical Perspective in Like⋯Like Carbon-Containing Complexes. Molecules 2022, 27, 2963. https://doi.org/10.3390/molecules27092963
Ibrahim MAA, Moussa NAM, Kamel AAK, Shehata MNI, Ahmed MN, Taha F, Abourehab MAS, Shawky AM, Elkaeed EB, Soliman MES. External Electric Field Effect on the Strength of σ-Hole Interactions: A Theoretical Perspective in Like⋯Like Carbon-Containing Complexes. Molecules. 2022; 27(9):2963. https://doi.org/10.3390/molecules27092963
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Nayra A. M. Moussa, Afnan A. K. Kamel, Mohammed N. I. Shehata, Muhammad Naeem Ahmed, Fouad Taha, Mohammed A. S. Abourehab, Ahmed M. Shawky, Eslam B. Elkaeed, and Mahmoud E. S. Soliman. 2022. "External Electric Field Effect on the Strength of σ-Hole Interactions: A Theoretical Perspective in Like⋯Like Carbon-Containing Complexes" Molecules 27, no. 9: 2963. https://doi.org/10.3390/molecules27092963