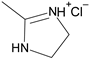
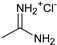
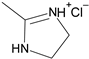
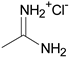
1. Introduction
Dipeptidyl peptidase III (DPP III; EC 3.4.14.4; formerly: dipeptidyl arylamidase III, dipeptidyl aminopeptidase III) is a proteolytic enzyme that catalyzes the hydrolytic cleavage of dipeptides sequentially from the N-termini of its peptide substrates. It was discovered in extracts of bovine pituitary gland as the third in a series of dipeptidyl arylamidases through the hydrolysis of synthetic substrate Arg-Arg-2-naphthylamide [
1]. The enzyme was further purified and biochemically characterized from the cytosolic fraction of several human and animal tissues and lower eukaryotes [
2]. Studies in vitro have demonstrated that DPP III prefers the Arg-Arg-arylamide among synthetic substrates, but displays a relatively broad specificity towards peptide substrates, optimally sized from tetra- to octa-peptides [
1,
3]. A high affinity of DPP III was shown for biologically active peptides, anigotensins, enkephalins and endomorphins [
3,
4,
5]. Investigation with general protease inhibitors revealed that DPP III is a metallopeptidase sensitive to thiol-blocking agents [
2]. In addition, earlier biochemical and histochemical research of DPP III activity and protein content showed its broad tissue distribution, and enhanced level in endometrial and ovarian malignancies [
6,
7].
Structure–activity relationship study of DPP III began when the genes encoding rat and human enzymes were cloned and their amino acid sequences deduced [
2,
8]. The experimental evidence on the presence of one Zn
2+ ion per molecule of human placental and rat recombinant enzyme, together with low dissociation constant (250 fM), proved that DPP III is a zinc-metallopeptidase [
8]. The flow of data on primary protein structures, due to the whole genome sequencing, similarity search and multiple sequence alignment, led to the discovery of bacterial DPPs III and recognition of the unique DPP III family, also named metallopeptidase family M49 in the MEROPS database [
9]. The M49 family is defined with five evolutionarily conserved regions, including motifs HEXXGH and EEXR(K)AE(D) that contain the residues important for zinc binding and catalytic activity [
8,
9,
10,
11]. The next breakthrough in the research of DPP III was a resolution of the crystal structure of yeast and human DPP III [
12,
13] which enabled the application of computational methods and study of protein flexibility and catalytic mechanism [
14,
15,
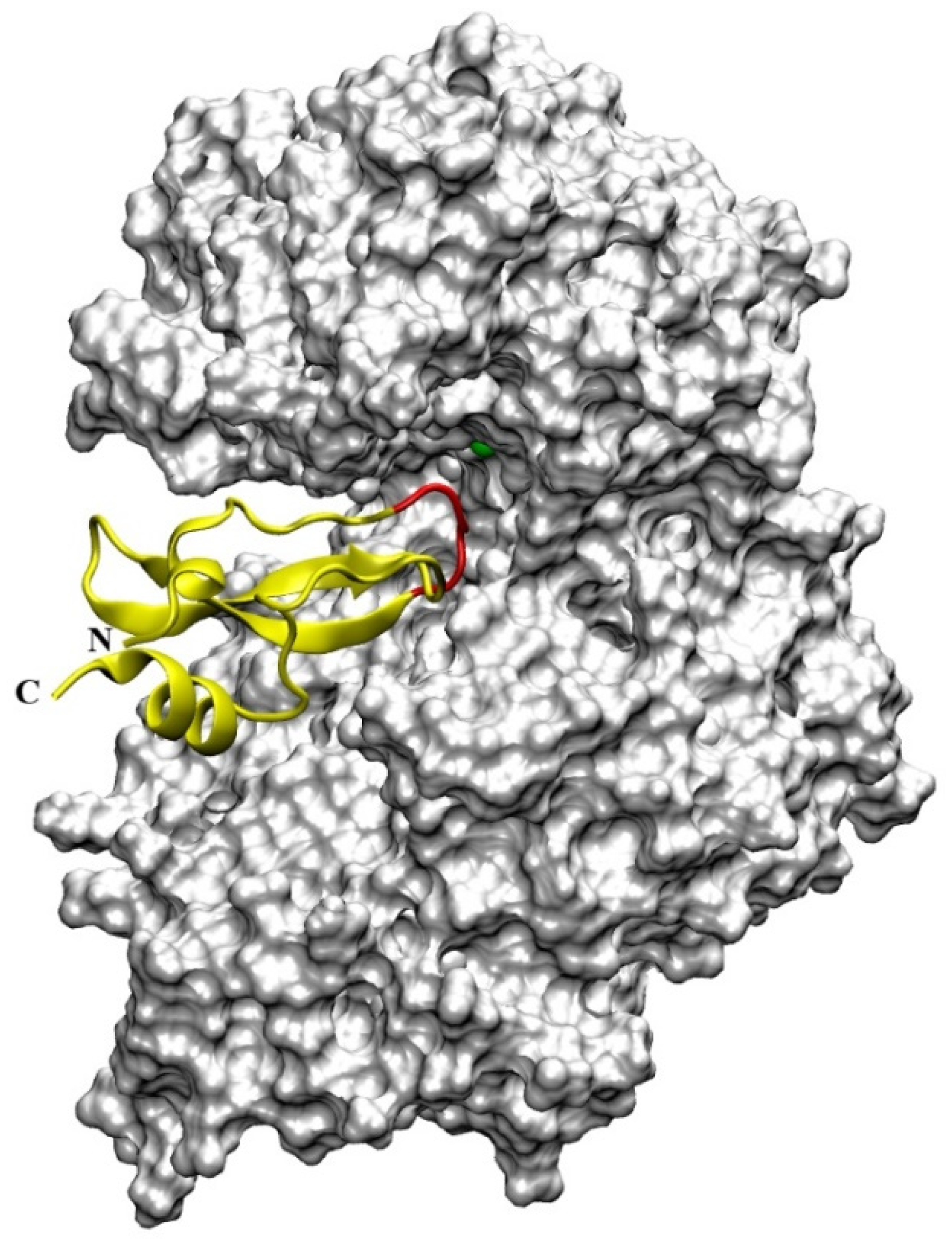
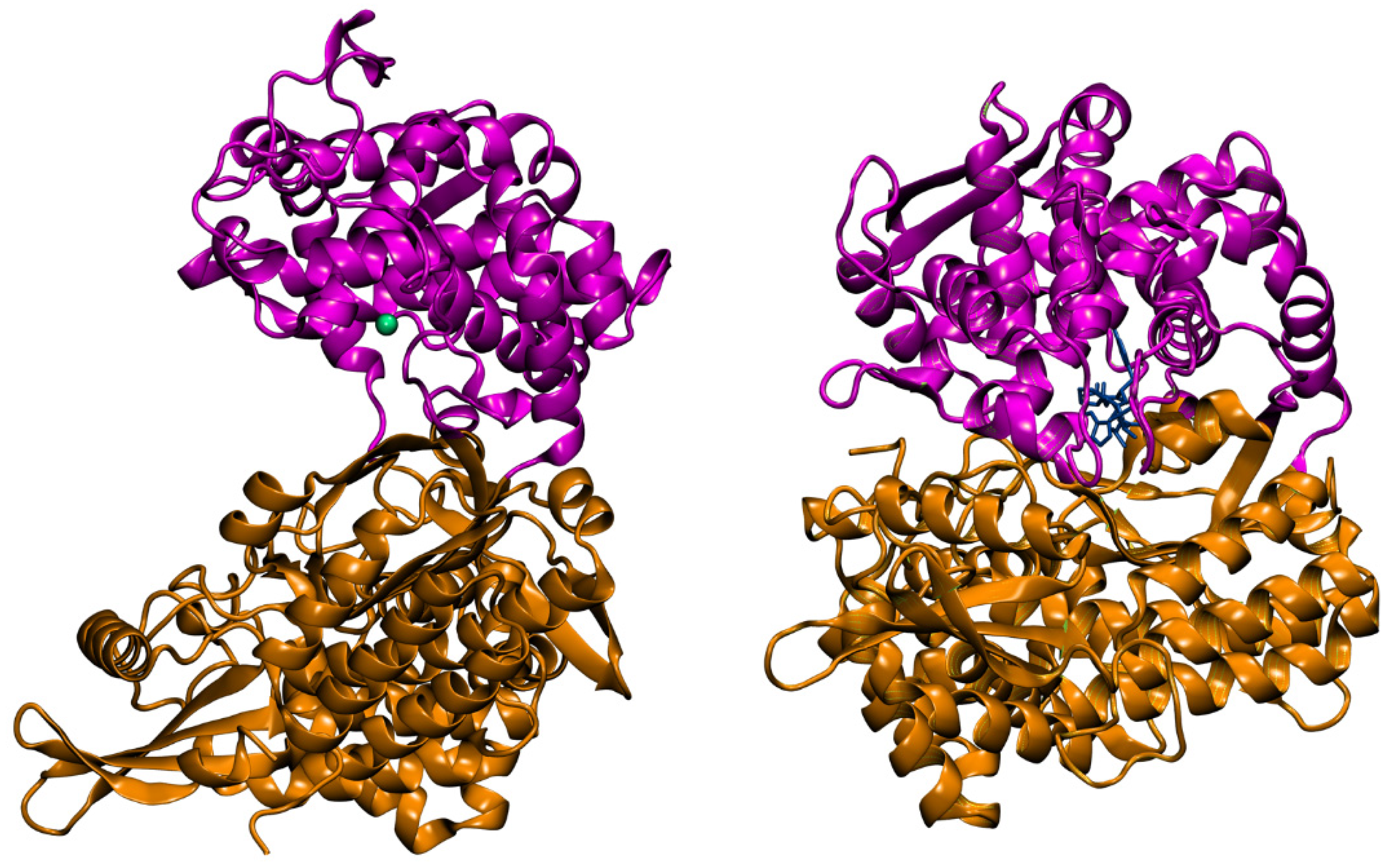
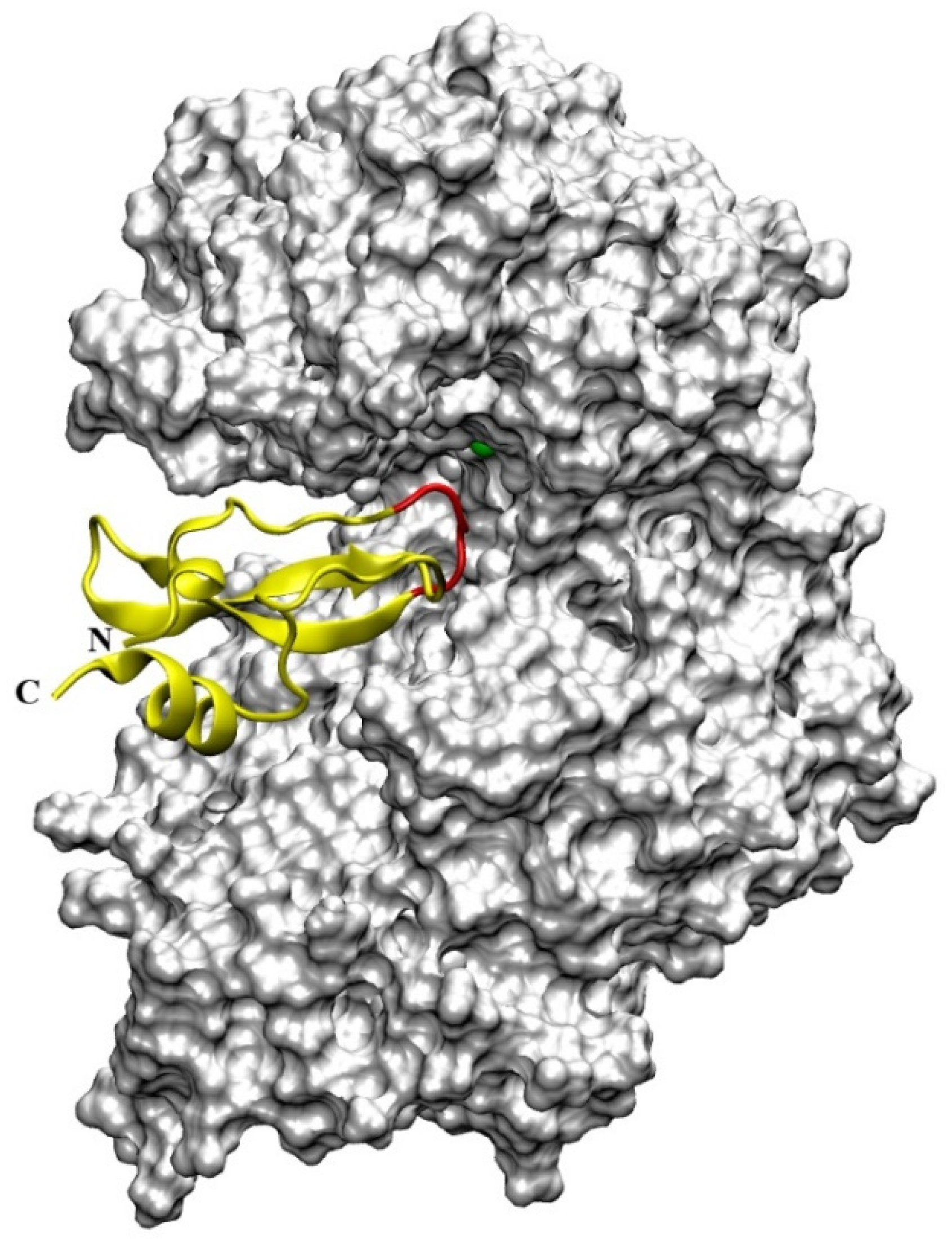
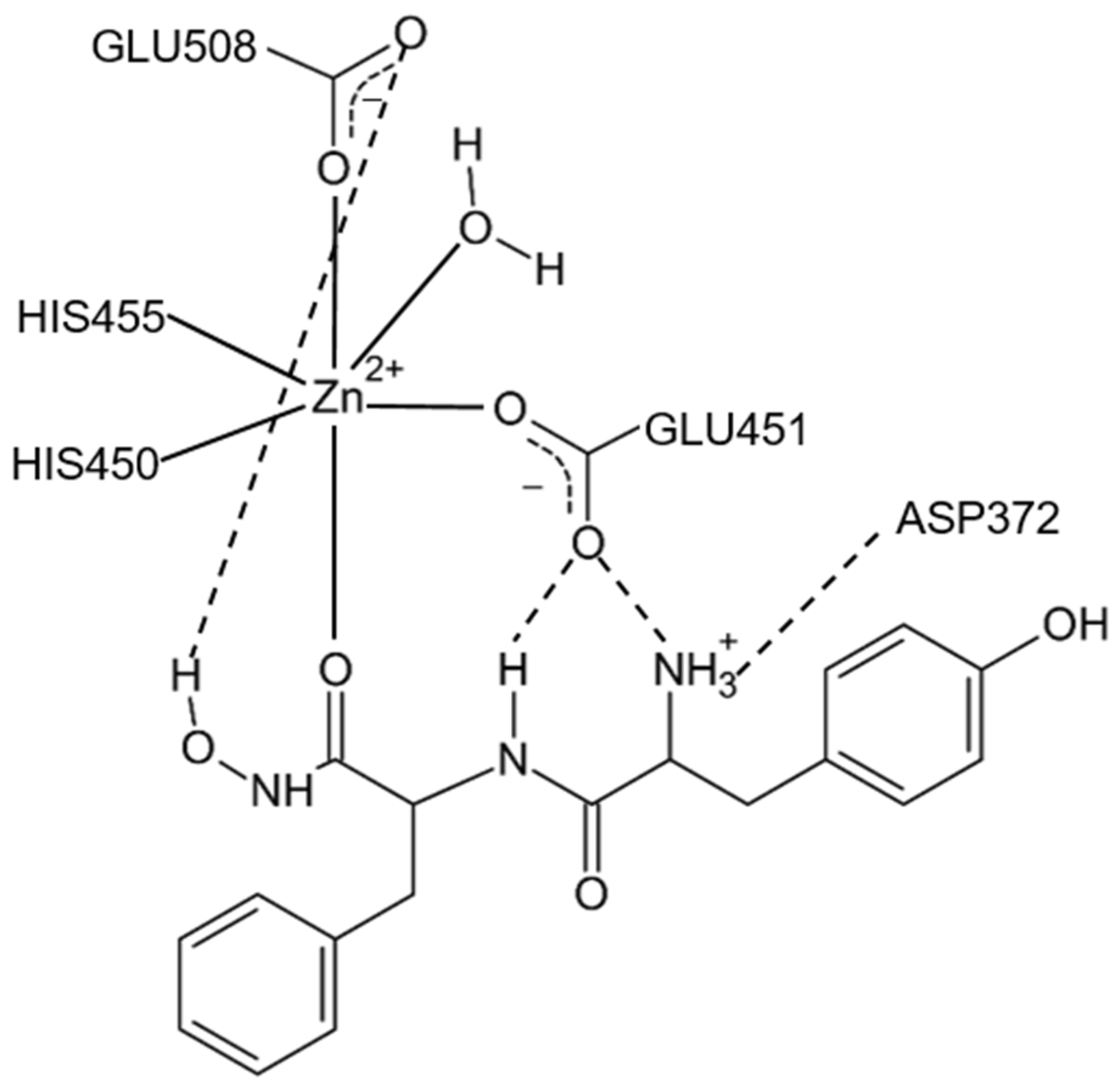
16]. The crystal structure of DPP III features an elongated protein molecule with two lobe-like domains separated by a wide cleft (
Figure 1). The zinc-binding site and the catalytically important residues are located in the upper lobe, which is mostly α-helical. The lower lobe, besides having α-helices, contains a smaller β-sheet portion (a five-stranded β-barrel). The two domains are connected by a number of loop regions. The first 3-D structure of human DPP III in complex with pentapeptide revealed a huge domain motion resulting in the complete closure of the cleft around the bound peptide substrate [
13] (
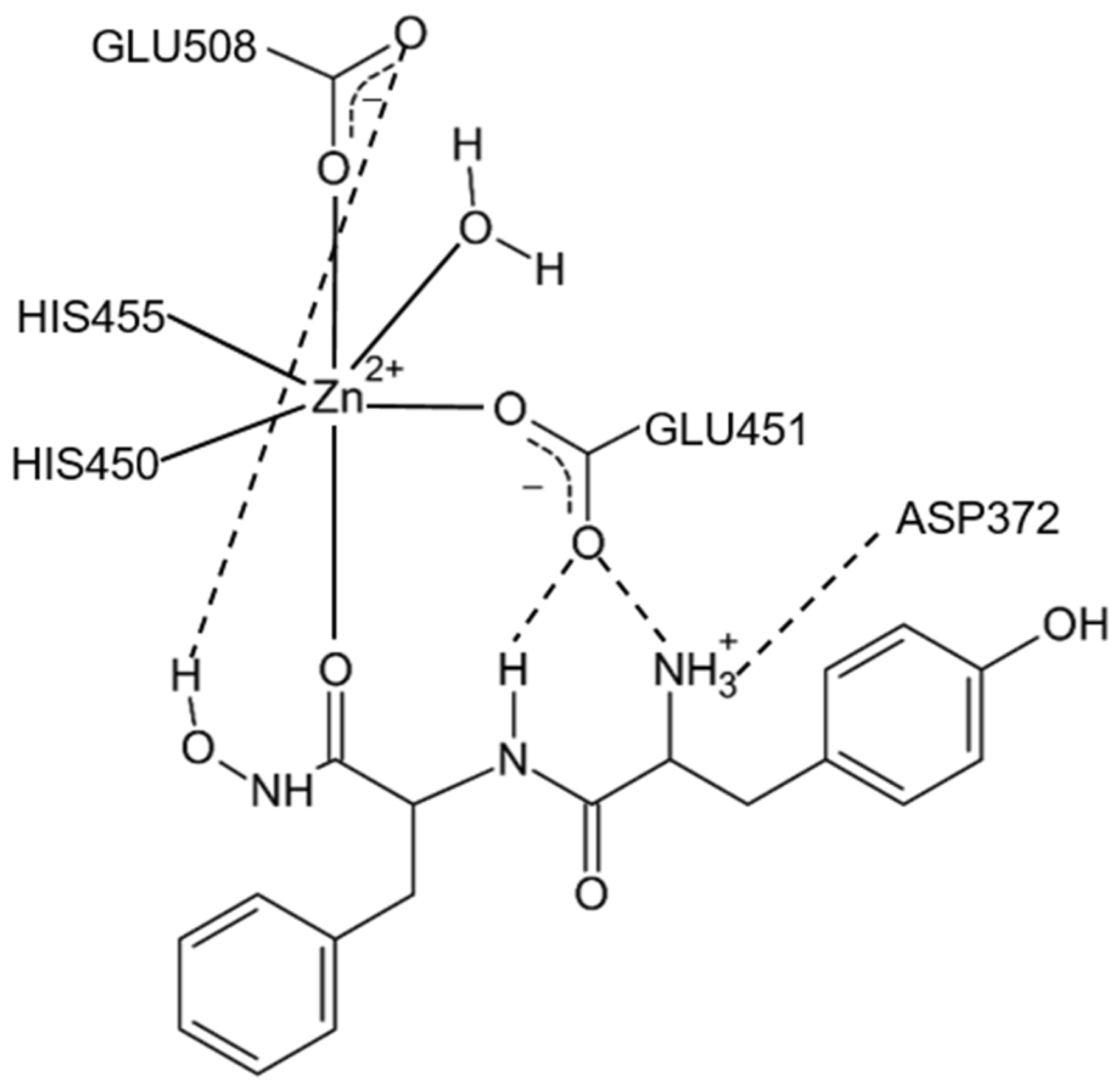
Figure 1). The active-site zinc ion is coordinated by the two histidine residues that belong to the conserved HEXXGH motif, a second glutamic acid residue from the conserved sequence motif EEXR(K)AE(D) and by the water molecule. In the structure of human DPP III, zinc-ligands are His450, His455 and Glu508. The Glu from the first zinc-binding motif (Glu451 in human DPP III) is proposed to act as a general base activating the water molecule which attacks the scissile peptide bond [
12,
16].
Direct evidence about the biological functions of DPP III was elusive for a long time, partly due to the lack of specific inhibitors. Metal chelating agents and sulfhydryl blocking reagents are known non-specific inhibitors of DPP III from various tissue sources [
2]. Inhibition with EDTA and 1,10-phenanthroline is consistent with the metallopeptidase nature of DPP III. Among the sulfhydryl reagents, the most potent inhibitors are organomercurial compounds PCMS, PCMB and pHMB, effective in micromolar concentration. Strong inhibition caused by some thiol reagents and reversal arising from the addition of thiol compounds suggested that SH-group(s) are essential for DPP III catalytic activity. However, mutational analyses and 3-D structure elucidation did not support this hypothesis [
12,
13,
17]. Comparative biochemical studies have shown that DPP III sensitivity to thiol blockers varies with species: e.g., all tested reagents inhibited the rat erythrocyte enzyme with higher potency than the human erythrocyte enzyme [
18]. Rat DPP III was hyperreactive to organomercurial pHMB whose IC
50 was 3 nM, and the same reagent produced 50% inhibition of human DPP III activity at about 1µM concentration [
18,
19]. Reactive cysteine residues, responsible for DPP III inactivation by sulfhydryl reagents, were identified by site-directed mutagenesis studies conducted for the rat (Cys176), yeast (Cys518, Cys639), human (Cys19, Cys176, Cys654) and bacterial enzyme (Cys450) [
17,
20,
21,
22]. Interestingly, while reactive cysteine is a part of the active–site motif HEC450LGH of the bacterial enzyme, Cys176, responsible for inactivation of human and rat DPP III by the organomercurial compound, is a residue 44 Å apart from the catalytic center of the ligand-free human DPP III. Modification of Cys176 probably hinders the formation of the closed active site upon substrate binding.
Participation in the final steps of intracellular protein catabolism was suggested for DPP III based on its cytosolic localization, wide distribution and broad specificity for oligopeptides. The revival of research interest in this enzyme in the last decade confirmed the suspected role of DPP III in post-proteasomal cleavage of peptides [
23] and revealed several more specific roles. Most important is DPP III’s role in the degradation of biologically active peptides of the renin–angiotensin system—angiotensin II to IV, and angiotensin (1–7)—and endogenous defense against oxidative stress [
24,
25,
26]. Participation of DPP III in oxidative stress regulation in mammalian cells does not depend on its enzymatic activity but is related to its ability to bind to the ubiquitin ligase Keap1, a constituent of the Keap1–Nrf2 signaling pathway, the cell’s main defense mechanism against environmental toxins and carcinogens. The molecular mechanism of interaction of DPP III-Keap1 has been recently reported [
27].
DPP III expression is dysregulated in several cancers [
7,
26,
28]. In ovarian primary carcinomas, DPP III activity correlates with tumor aggressiveness [
29]. Overexpression of DPP III was shown to be correlated with poor prognosis of human breast cancer patients [
30] and colorectal cancer patients [
28]. According to recent findings, the pathophysiological role of DPP III in malignant growth seems to be connected to its interaction with Keap1, leading to upregulation of the Keap1-Nrf2 pathway [
26,
30,
31] and with cyclin-dependent kinase 1 (CDK1) [
28]. Most recent studies using the luminometric immunoassay method have shown that DPP III is elevated in the plasma of septic patients, and that the highest levels of circulating DPP III are present in non-survivor septic shock patients [
32]. Therapeutic potential of circulating DPP III inhibition by the specific antibody Procizumab in the restoration of altered cardiac function during sepsis is under investigation in preclinical sepsis models [
32,
33].
DPP III in vitro hydrolyzes pentapeptides Leu- and Met-enkephalin at the Gly2-Gly3 bond as so-called enkephalinase B, a peptidase at first isolated from the rat brain membranes by detergent treatment [
34]. However, the identity of enkephalinase B and DPP III is not yet proven by biochemical and/or structural data. Also, there is no conclusive evidence on the role of DPP III in the endogenous pain-modulatory system, although this enzyme in high concentrations colocalizes in the neurons of the rat spinal dorsal horn with endogenous opioid peptides enkephalins and endomorphins [
35]. Earlier findings indicated an association of DPP III with cataract formation [
36] and, more recently, with influenza A virus infection [
37]. Obviously, elucidation of the (patho)physiological relevance of DPP III, especially of the human enzyme, requires further investigations, for which specific and effective inhibitors would be necessary. In this review we present an historical perspective and the most current strategies for the discovery or development of DPP III inhibitors, with special emphasis on small molecules and their mechanism of inhibitory action. Inhibitor potency is expressed as the IC
50 value, defined as the concentration of an inhibitor which causes 50% reduction of enzyme activity, or by the inhibition constant K
i, the equilibrium dissociation constant of the enzyme-inhibitor complex.
6. Conclusions
This review encompassed a whole range of natural and synthetic (low molecular mass) compounds with experimentally proven in vitro inhibitory activity toward mammalian DPP III. Whenever known, the molecular basis for observed inhibition was explained.
Although DPP III belongs to the metallopeptidase class of proteolytic enzymes, it is inhibited with a low micromolar concentration of some sulfhydryl reagents due to the reactivity of its cysteine residues and high flexibility of its 3-D structure.
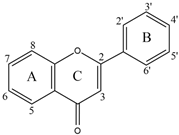
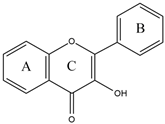
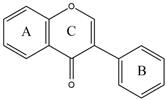
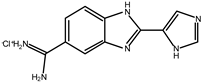
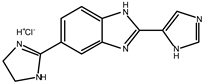
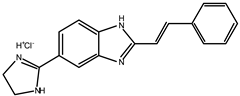
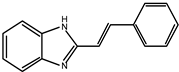
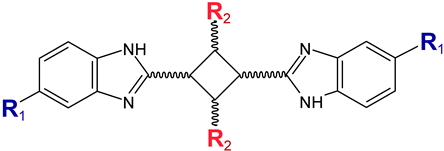
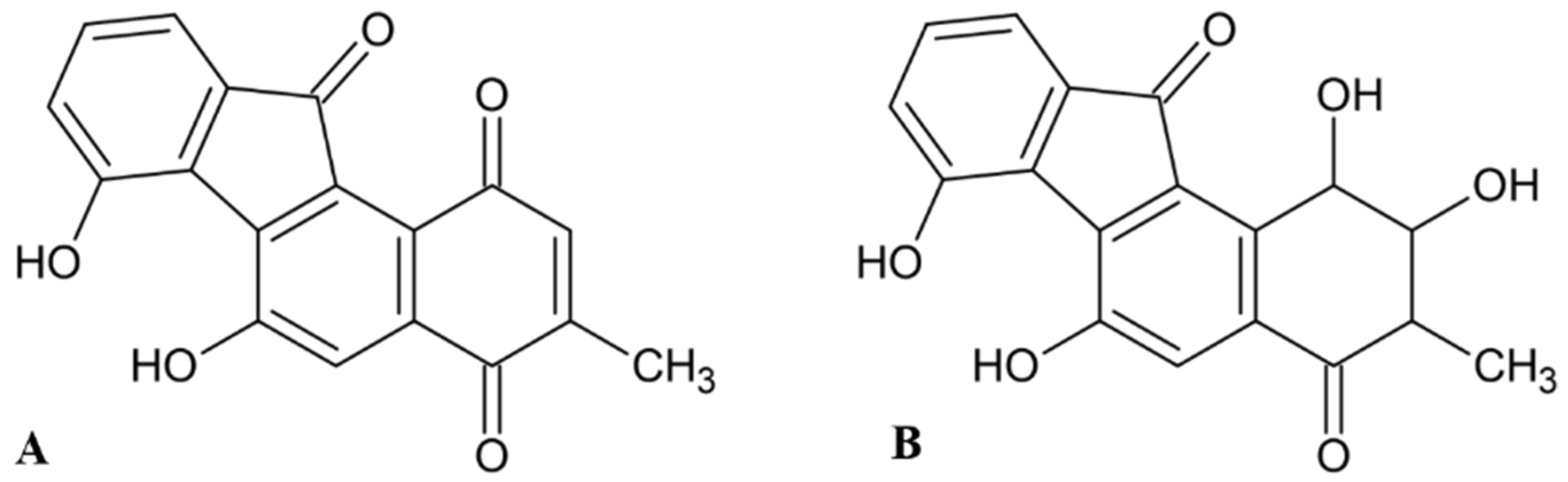
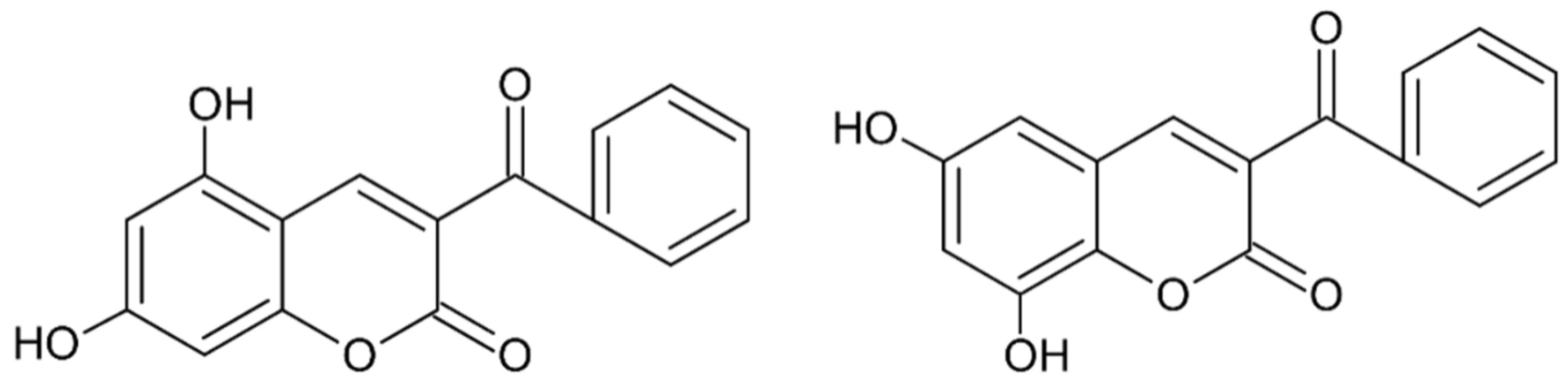
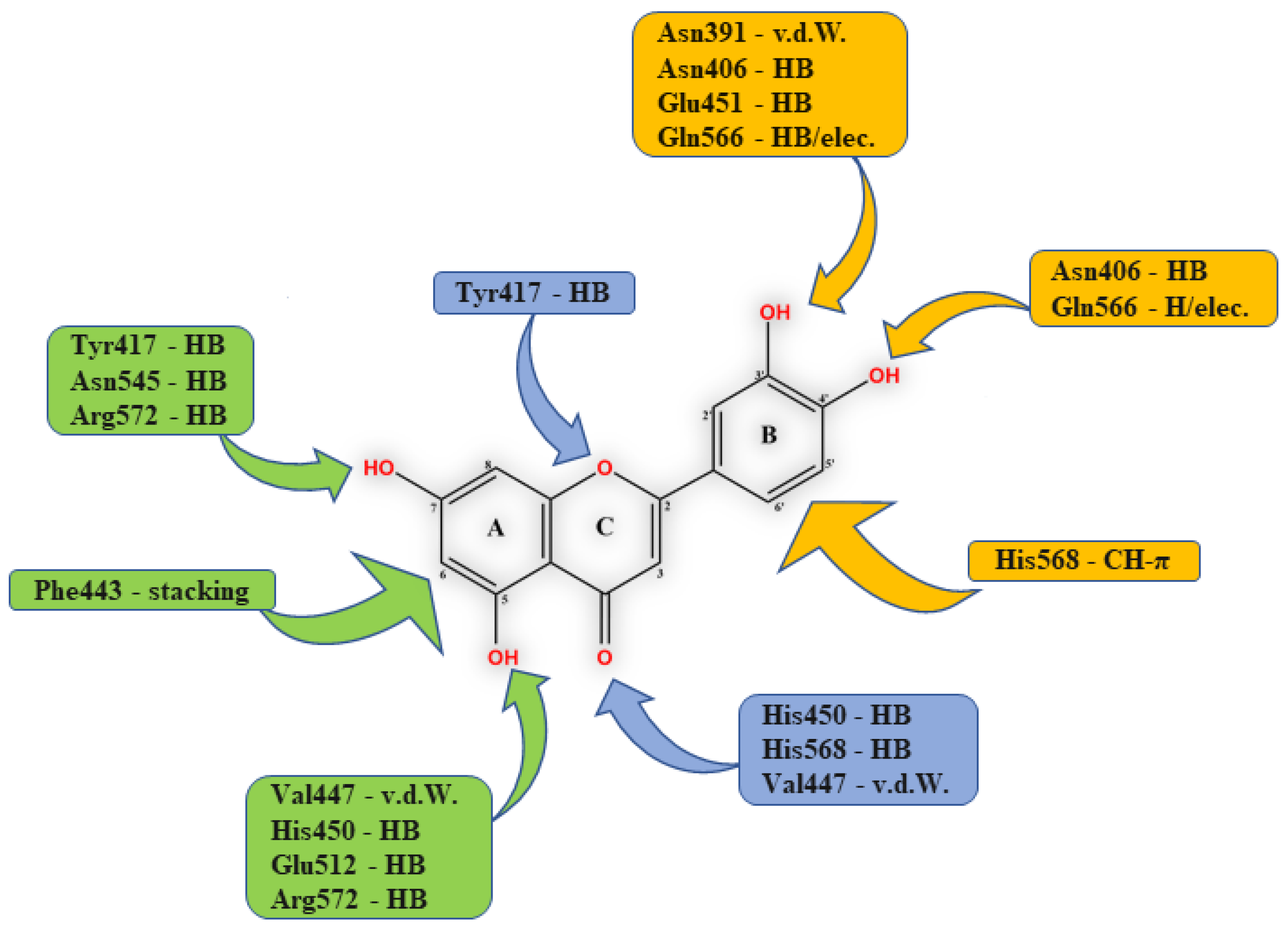
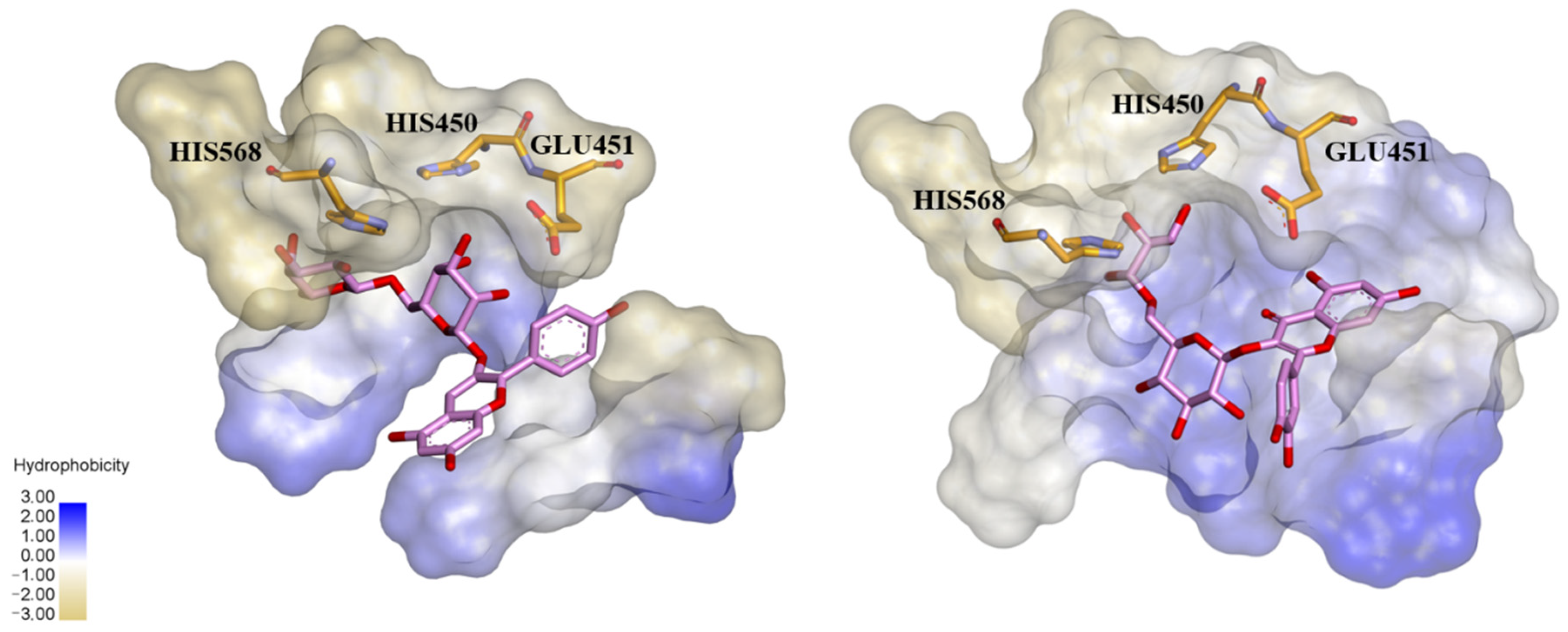
Early searches for specific inhibitors of DPP III activity, based on the screening of microbial culture filtrates, yielded several low molecular mass secondary metabolites, with propioxatin A and B as the most potent compounds (Ki: 13.0 nM and 5.6 nM). Up to now, propioxatin B is one of the strongest inhibitors of DPP III yet reported. DPP III inhibitors were further recognized among polyphenolic compounds (flavonoids and their glycosides), and coumarin and benzimidazole derivatives. Natural polypeptide aprotinin is also on the list of DPP III inhibitors in vitro (Ki: 11.7 µM).
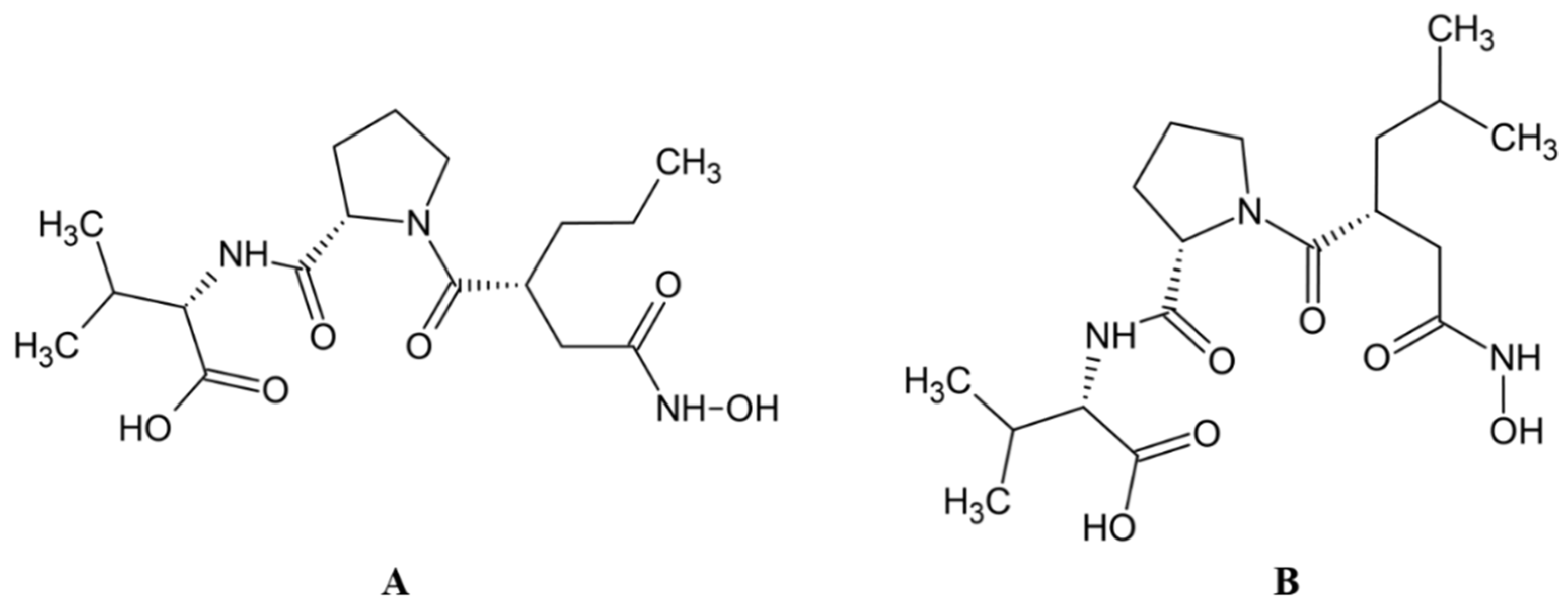
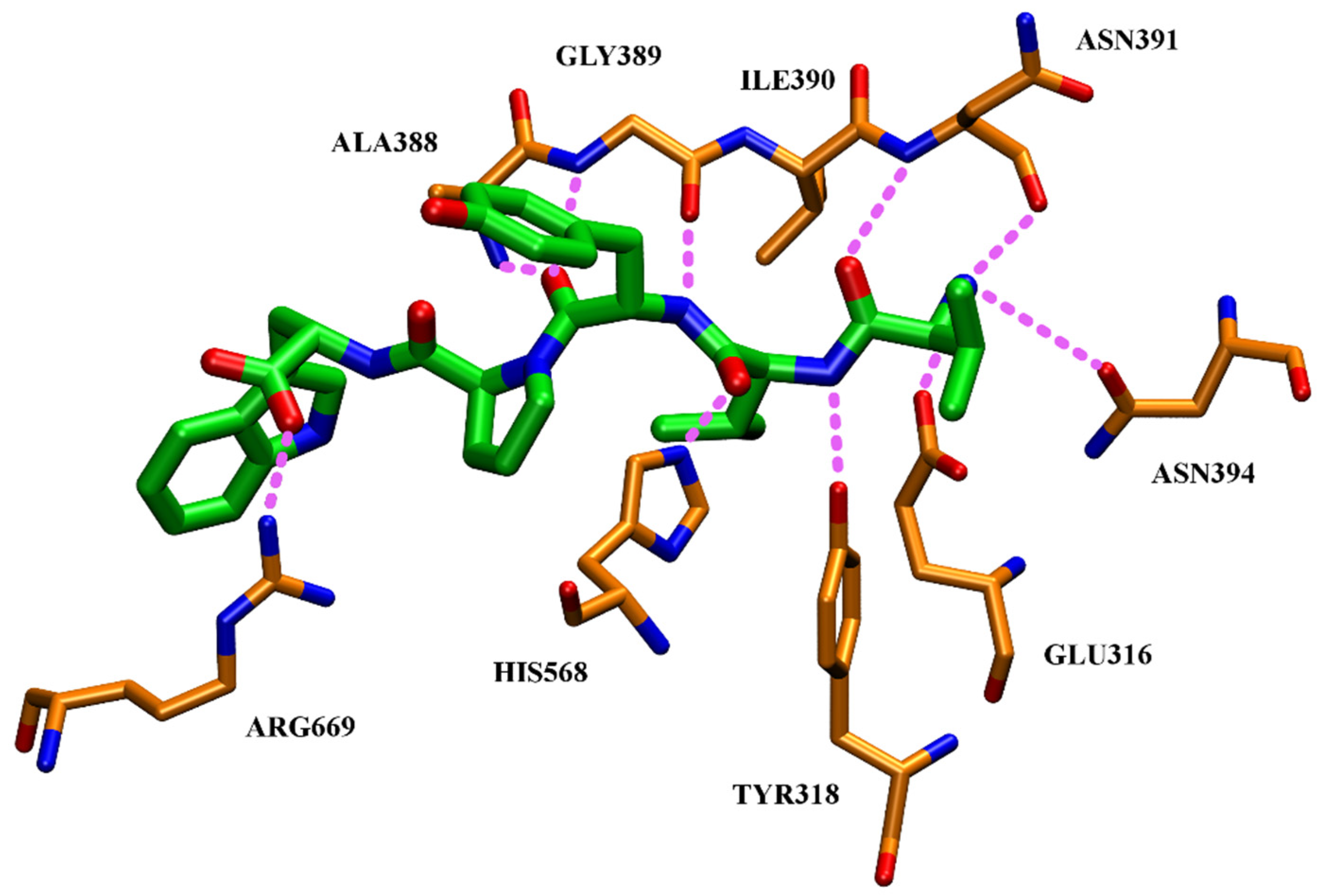
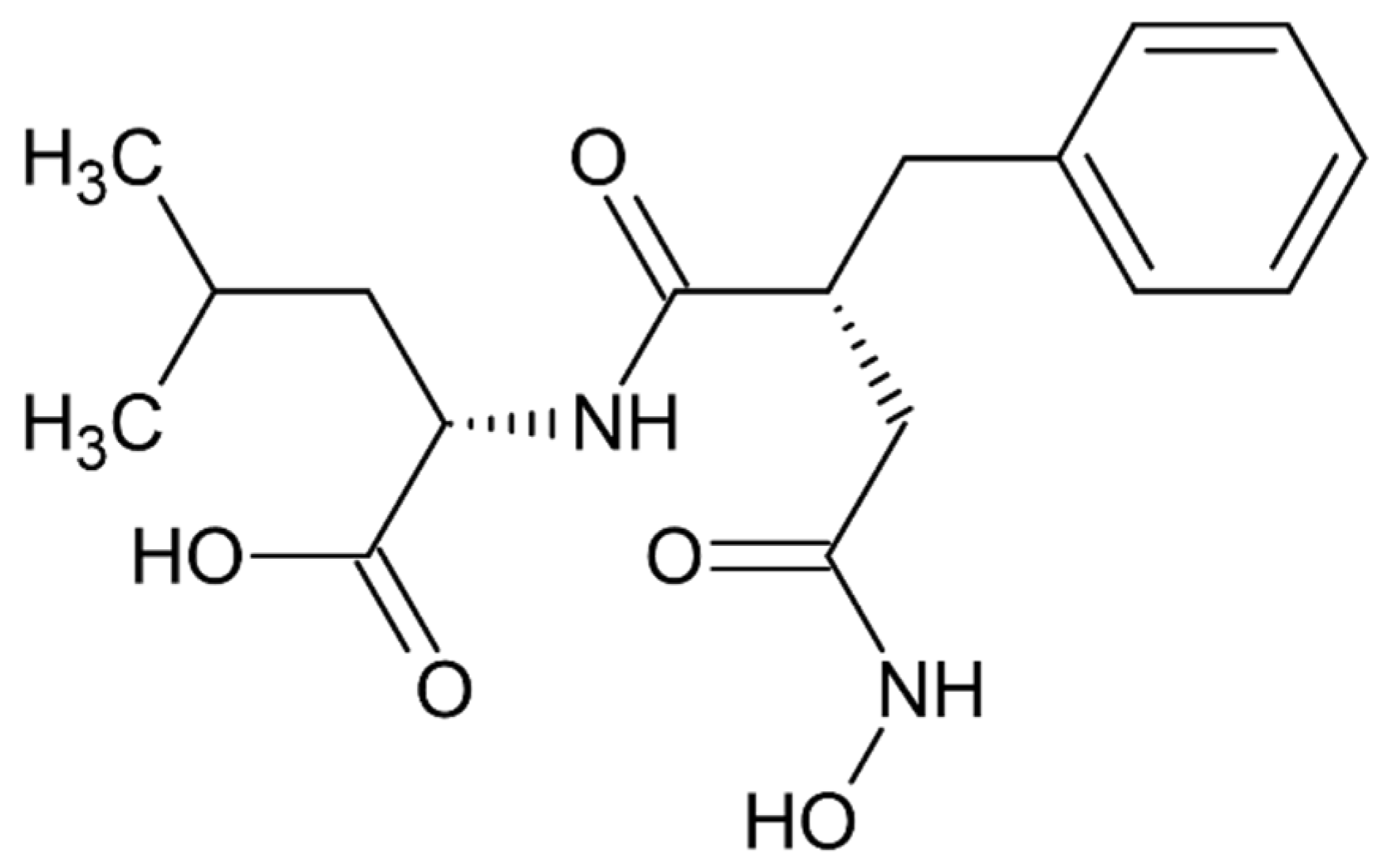
High inhibitory potential for DPP III was revealed in DPP III substrate analogues, dipeptidyl hydroxamic acids (Ki values in nM range), and in oligopeptides from the hemorphin group (valorphin, tynorphin). However, the latter are actually “slow” substrates of DPP III in vitro, susceptible to rapid degradation in vivo by aminopeptidases. Recently, the crystal structures of human DPP III, ligand-free and in complex, have become accessible allowing, combined with computational methods, the elucidation of inhibition mechanisms and design of improved, more specific, inhibitors. Thus resolved 3-D structure of the human DPP III-tynorphin complex enabled the design of the first transition-state peptidomimetics inhibitors, effective in the low micromolar range and resistant to proteolytic attack by DPP III. In addition, the combination of experimental and in silico study facilitated the discovery of new molecules with improved inhibitory activity, as was indicated by the QSAR analysis of coumarin derivatives.
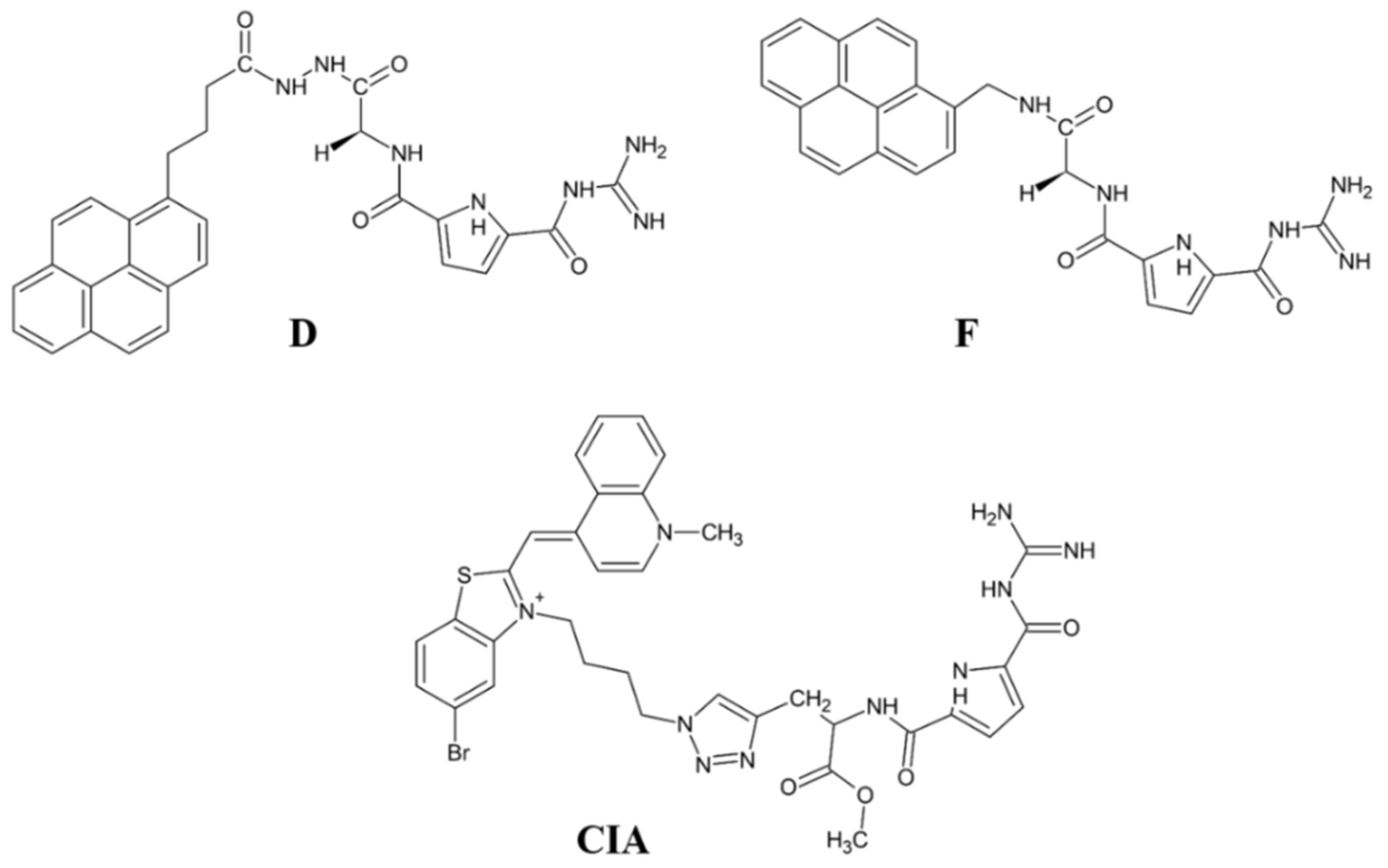
A new direction in the field is the development of fluorescent inhibitor for monitoring the DPP III activity. The results obtained with the pyrene- or cyanine-guanidiniocarbonyl-pyrrole conjugates are promising in this respect, as these compounds show strong fluorescence upon binding to DPP III and potent inhibition of this enzyme (Ki value 0.2–0.3 µM).
Most recently overexpressed DPP III emerges as a potential drug target in several human pathologies, like cancer progression, sepsis and septic shock. To date, no inhibitors of DPP III have been evaluated in clinical trials. However, DPP III-blocking therapy has been shown to improve outcomes in preclinical sepsis models where inhibition of circulating DPP III by a specific antibody was obtained. To reveal new, or to prove yet indicated (patho)physiological, functions of DPP III, selective and strong inhibitors are needed. A successful example is the use of the highly potent inhibitor JMV-390 (IC50: 1.4 nM) to reduce intracellular DPP III activity which helped identification of DPP III as the angiotensin (1–7) degrading enzyme in human renal epithelial cells.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}